
Beneficial Antioxidant Effects of Coenzyme Q10 in In Vitro and In Vivo Models of CDKL5 Deficiency Disorder

 , ,
, ,  , , , ,
, , , ,  ,
,  and
and 
Abstract
1. Introduction
2. Results
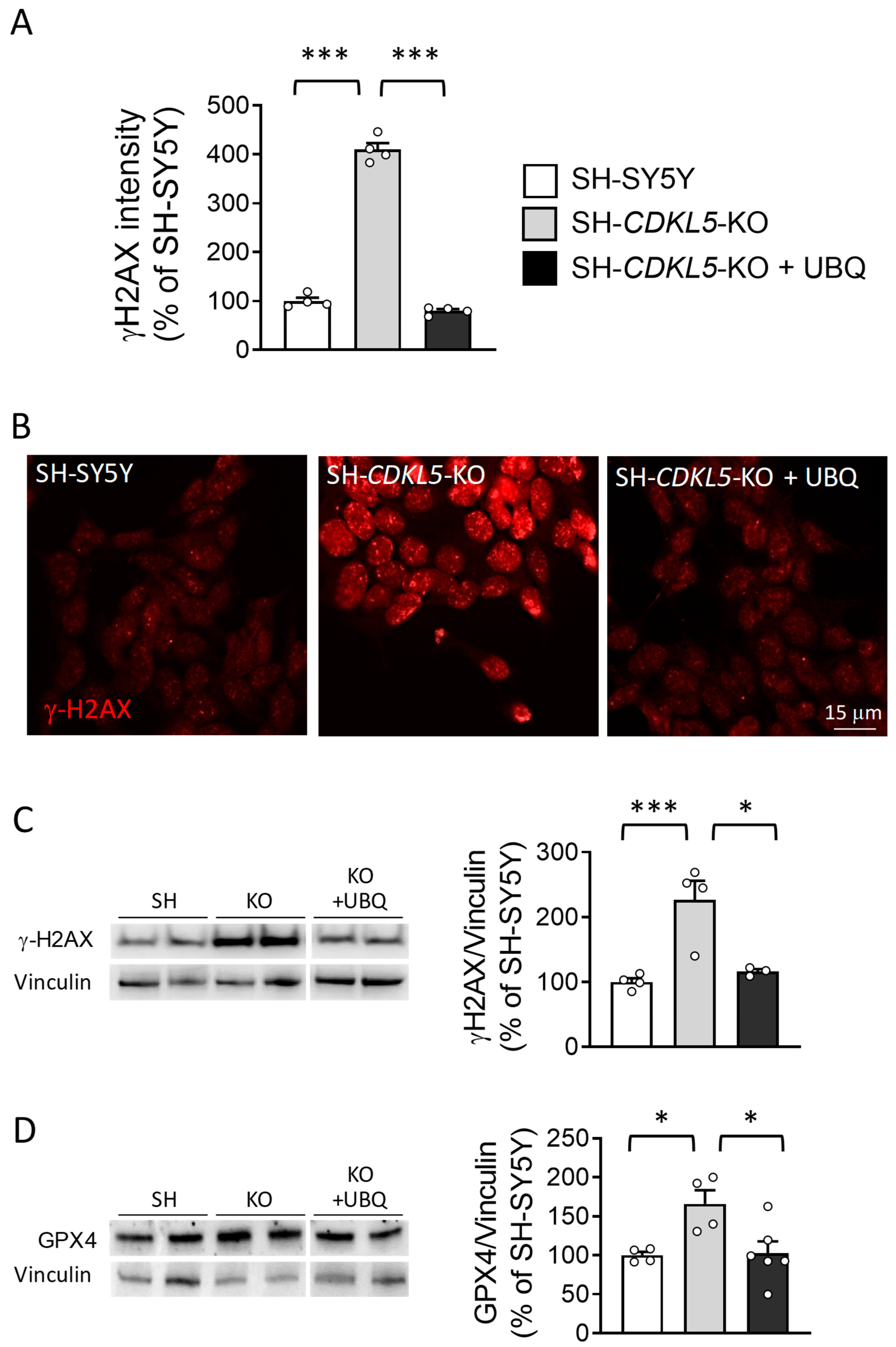
2.1. Treatment with UBQ Has a Protective Effect Against Increased Oxidative Stress in a Human Cellular Model of CDKL5 Deficiency
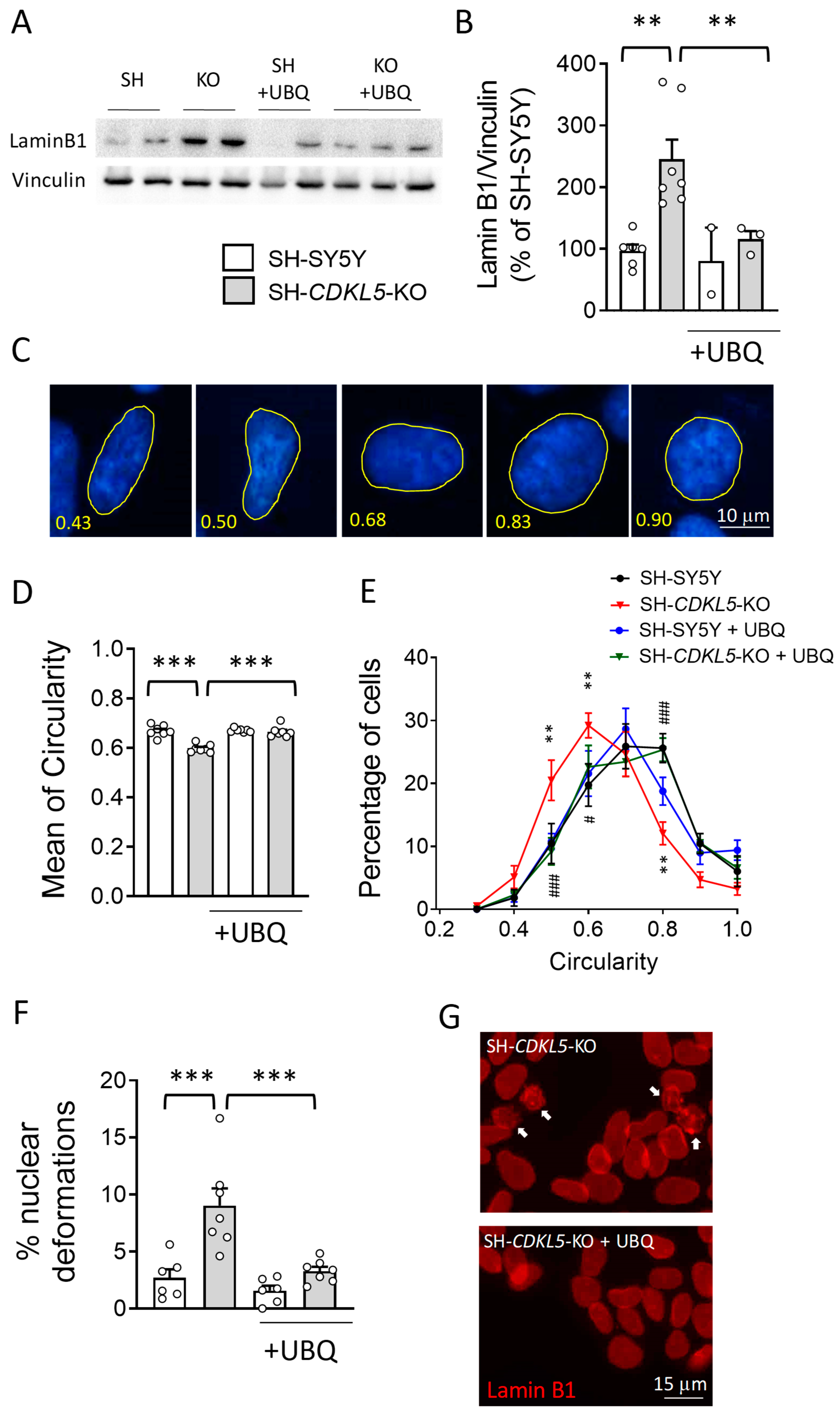
2.2. Treatment with UBQ Restores Lamin B1 Levels and Nuclear Shape in a Human Cellular Model of CDKL5 Deficiency
2.3. Treatment with UBQ Restores Biological Markers Associated with DNA Damage in a Human Cellular Model of CDKL5 Deficiency
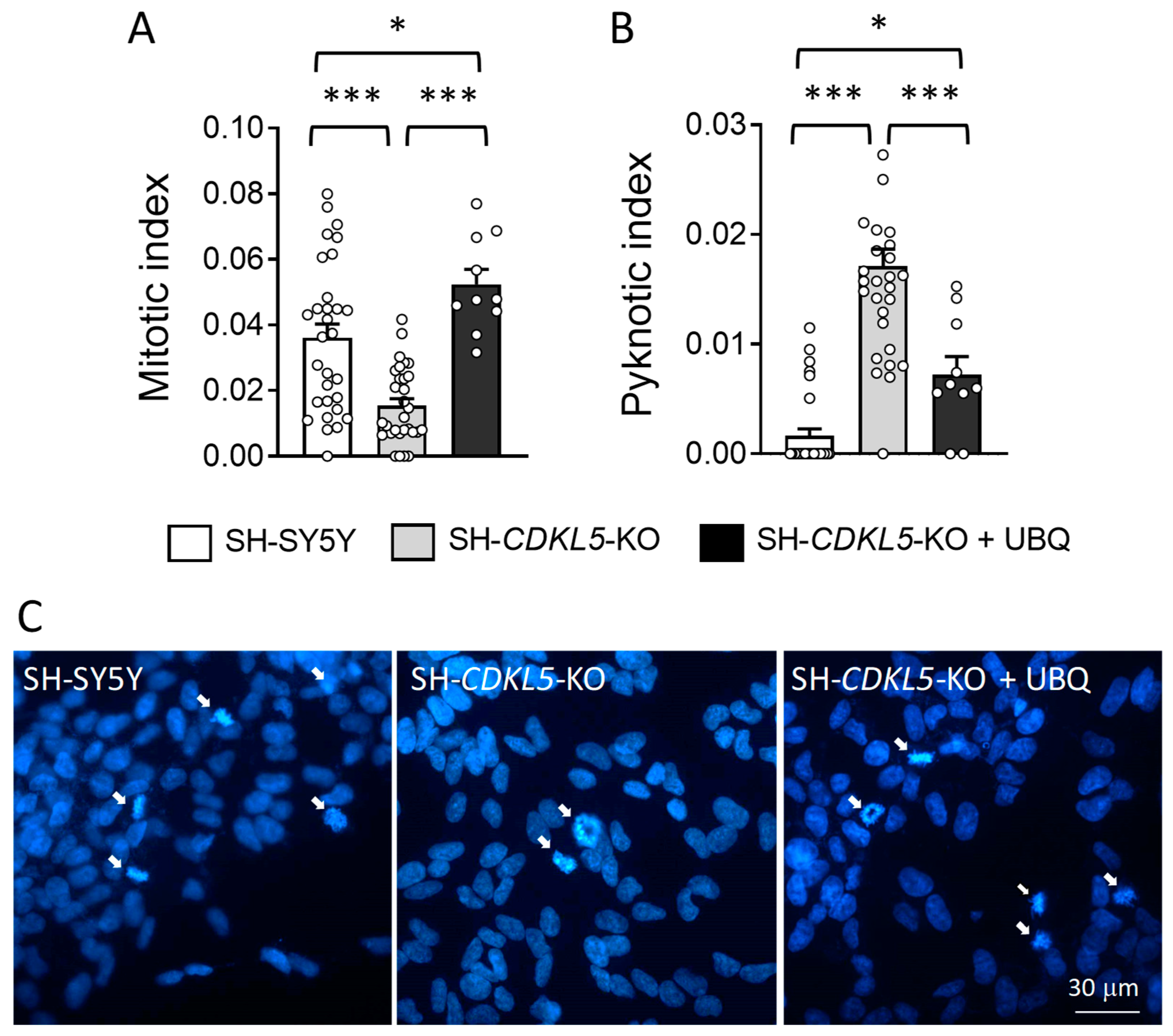
2.4. Treatment with UBQ Restores Neuronal Proliferation and Survival of a Human Cellular Model of CDKL5 Deficiency
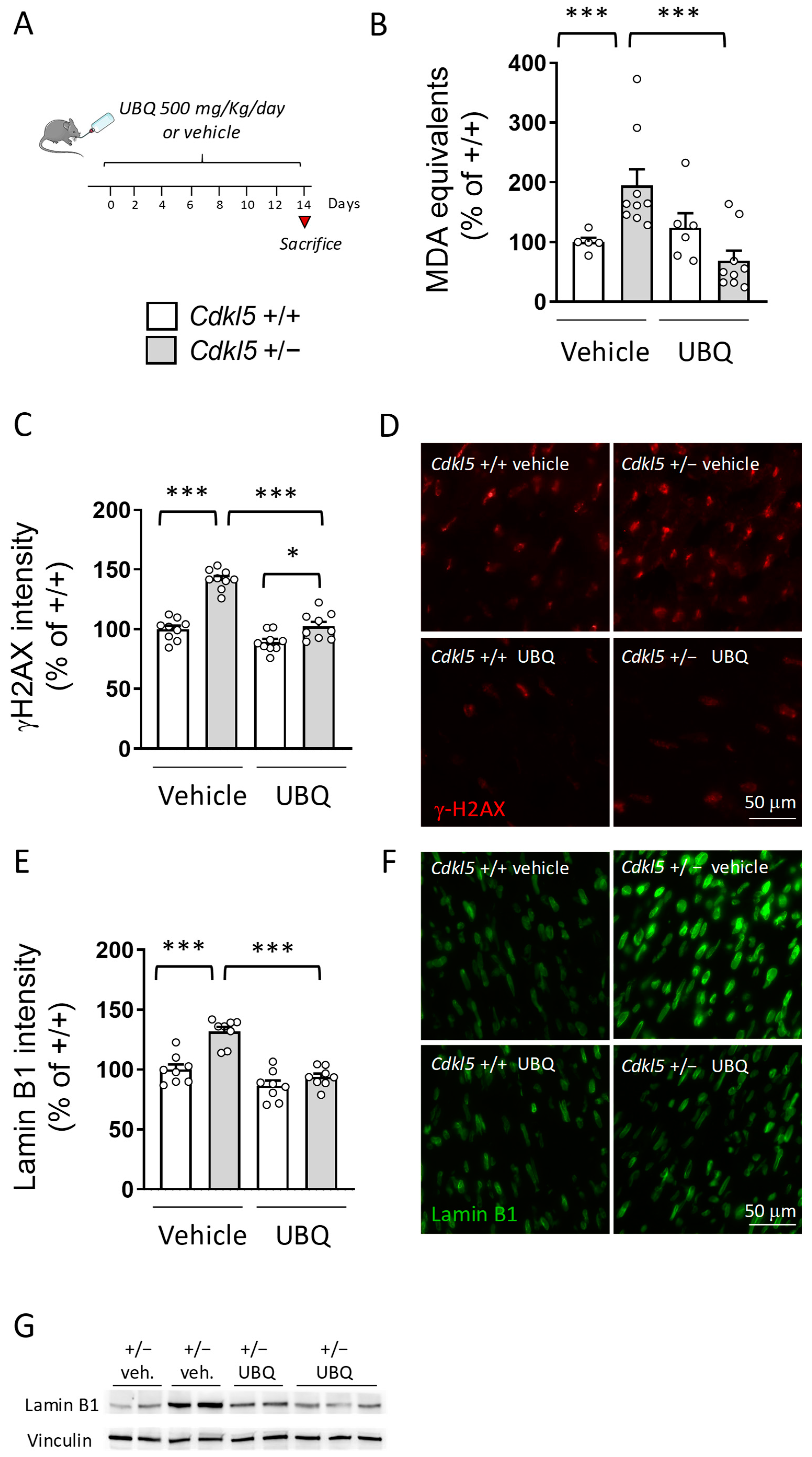
2.5. Treatment with UBQ Decreases ROS Production in the Hearts of Cdkl5 +/− Mice
3. Discussion
3.1. Increased ROS Cellular Levels in CDKL5-Deficient Cells
3.2. Treatment with CoQ10 Prevents ROS Cellular Accumulation and DNA Damage in CDKL5-Deficient Cells
3.3. Coenzyme Q10 Supplementation Prevents Nuclear DNA Damage and Restores Lamin B1 Levels in the Cdkl5 +/− Heart
4. Materials and Methods
4.1. Cell Lines and Treatments
4.2. Measurement of ROS
4.3. Mitochondrial Oxygen Consumption Assay
4.4. Immunocytochemistry
4.5. Apoptotic and Mitotic Index
4.6. Circularity Index Evaluation
4.7. Nuclear Deformation Analysis
4.8. Glutathione Assay
4.9. Superoxide Determination
4.10. Animal Husbandry
4.11. In Vivo Treatments
4.12. Heart Dissection
4.13. γH2AX and Lamin B1 Immunohistochemistry
4.14. Cdkl5 mRNA Detection
4.15. Image Acquisition and Measurements
4.16. Quantification of CoQ9 and CoQ10 Levels in Plasma and Heart Homogenates
4.17. Measurement of Lipid Peroxidation
4.18. Western Blot Analysis
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef]
- Demarest, S.T.; Olson, H.E.; Moss, A.; Pestana-Knight, E.; Zhang, X.; Parikh, S.; Swanson, L.C.; Riley, K.D.; Bazin, G.A.; Angione, K.; et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019, 60, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.; Pestana-Knight, E.M.; Olson, H.E.; Downs, J.; Marsh, E.D.; Kaufmann, W.E.; Partridge, C.A.; Leonard, H.; Gwadry-Sridhar, F.; Frame, K.E.; et al. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr. Neurol. 2019, 97, 38–42. [Google Scholar] [CrossRef]
- Lindy, A.S.; Stosser, M.B.; Butler, E.; Downtain-Pickersgill, C.; Shanmugham, A.; Retterer, K.; Brandt, T.; Richard, G.; McKnight, D.A. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018, 59, 1062–1071. [Google Scholar] [CrossRef]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef]
- Jakimiec, M.; Paprocka, J.; Smigiel, R. CDKL5 Deficiency Disorder-A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Bertani, I.; Rusconi, L.; Bolognese, F.; Forlani, G.; Conca, B.; De Monte, L.; Badaracco, G.; Landsberger, N.; Kilstrup-Nielsen, C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006, 281, 32048–32056. [Google Scholar] [CrossRef]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhu, Y.C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 2010, 30, 12777–12786. [Google Scholar] [CrossRef]
- Wang, I.T.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [PubMed]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; et al. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Fuchs, C.; Rimondini, R.; Viggiano, R.; Trazzi, S.; De Franceschi, M.; Bartesaghi, R.; Ciani, E. Inhibition of GSK3beta rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 2015, 82, 298–310. [Google Scholar] [CrossRef]
- Fuchs, C.; Gennaccaro, L.; Trazzi, S.; Bastianini, S.; Bettini, S.; Lo Martire, V.; Ren, E.; Medici, G.; Zoccoli, G.; Rimondini, R.; et al. Heterozygous CDKL5 Knockout Female Mice Are a Valuable Animal Model for CDKL5 Disorder. Neural. Plast 2018, 2018, 9726950. [Google Scholar] [CrossRef]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S.; et al. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.C.; Xiong, Z.Q. Molecular and Synaptic Bases of CDKL5 Disorder. Dev. Neurobiol. 2019, 79, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E.; Gross, C.; Calza, L.; Bartesaghi, R.; Ciani, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Trazzi, S.; Fuchs, C.; Galvani, G.; Medici, G.; Gennaccaro, L.; Tassinari, M.; Ciani, E. Increased DNA Damage and Apoptosis in CDKL5-Deficient Neurons. Mol. Neurobiol. 2020, 57, 2244–2262. [Google Scholar] [CrossRef]
- Loi, M.; Bastianini, S.; Candini, G.; Rizzardi, N.; Medici, G.; Papa, V.; Gennaccaro, L.; Mottolese, N.; Tassinari, M.; Uguagliati, B.; et al. Cardiac Functional and Structural Abnormalities in a Mouse Model of CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2023, 24, 5552. [Google Scholar] [CrossRef]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Pizzo, R.; Alvente, S.; Berteotti, C.; Lupori, L.; Sagona, G.; Galvani, G.; Gurgone, A.; et al. Age-Related Cognitive and Motor Decline in a Mouse Model of CDKL5 Deficiency Disorder is Associated with Increased Neuronal Senescence and Death. Aging Dis. 2021, 12, 764–785. [Google Scholar] [CrossRef]
- Vigli, D.; Rusconi, L.; Valenti, D.; La Montanara, P.; Cosentino, L.; Lacivita, E.; Leopoldo, M.; Amendola, E.; Gross, C.; Landsberger, N.; et al. Rescue of prepulse inhibition deficit and brain mitochondrial dysfunction by pharmacological stimulation of the central serotonin receptor 7 in a mouse model of CDKL5 Deficiency Disorder. Neuropharmacology 2019, 144, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Chaabane, L.; Butti, C.; De Palma, C.; Aimar, P.; Salio, C.; Vignoli, A.; Giustetto, M.; Landsberger, N.; Frasca, A. In vivo magnetic resonance spectroscopy in the brain of Cdkl5 null mice reveals a metabolic profile indicative of mitochondrial dysfunctions. J. Neurochem. 2021, 157, 1253–1269. [Google Scholar] [CrossRef]
- Pecorelli, A.; Belmonte, G.; Meloni, I.; Cervellati, F.; Gardi, C.; Sticozzi, C.; De Felice, C.; Signorini, C.; Cortelazzo, A.; Leoncini, S.; et al. Alteration of serum lipid profile, SRB1 loss, and impaired Nrf2 activation in CDKL5 disorder. Free Radic. Biol. Med. 2015, 86, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Ciccoli, L.; Signorini, C.; Leoncini, S.; Giardini, A.; D’Esposito, M.; Filosa, S.; Hayek, J.; De Felice, C.; Valacchi, G. Increased levels of 4HNE-protein plasma adducts in Rett syndrome. Clin. Biochem. 2011, 44, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, S.; De Felice, C.; Signorini, C.; Zollo, G.; Cortelazzo, A.; Durand, T.; Galano, J.M.; Guerranti, R.; Rossi, M.; Ciccoli, L.; et al. Cytokine Dysregulation in MECP2- and CDKL5-Related Rett Syndrome: Relationships with Aberrant Redox Homeostasis, Inflammation, and omega-3 PUFAs. Oxid. Med. Cell Longev. 2015, 2015, 421624. [Google Scholar] [CrossRef]
- Cortelazzo, A.; de Felice, C.; Leoncini, S.; Signorini, C.; Guerranti, R.; Leoncini, R.; Armini, A.; Bini, L.; Ciccoli, L.; Hayek, J. Inflammatory protein response in CDKL5-Rett syndrome: Evidence of a subclinical smouldering inflammation. Inflamm. Res. 2017, 66, 269–280. [Google Scholar] [CrossRef]
- Hayashi, M.; Miyata, R.; Tanuma, N. Oxidative stress in developmental brain disorders. Adv. Exp. Med. Biol. 2012, 724, 278–290. [Google Scholar] [CrossRef]
- Hayashi, M. Oxidative stress in developmental brain disorders. Neuropathology 2009, 29, 1–8. [Google Scholar] [CrossRef]
- De Felice, C.; Ciccoli, L.; Leoncini, S.; Signorini, C.; Rossi, M.; Vannuccini, L.; Guazzi, G.; Latini, G.; Comporti, M.; Valacchi, G.; et al. Systemic oxidative stress in classic Rett syndrome. Free Radic. Biol. Med. 2009, 47, 440–448. [Google Scholar] [CrossRef]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Grosser, E.; Hirt, U.; Janc, O.A.; Menzfeld, C.; Fischer, M.; Kempkes, B.; Vogelgesang, S.; Manzke, T.U.; Opitz, L.; Salinas-Riester, G.; et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis. 2012, 48, 102–114. [Google Scholar] [CrossRef]
- Muller, M. Disturbed redox homeostasis and oxidative stress: Potential players in the developmental regression in Rett syndrome. Neurosci. Biobehav. Rev. 2019, 98, 154–163. [Google Scholar] [CrossRef]
- Di Pierro, D.; Ciaccio, C.; Sbardella, D.; Tundo, G.R.; Bernardini, R.; Curatolo, P.; Galasso, C.; Pironi, V.; Coletta, M.; Marini, S. Effects of oral administration of common antioxidant supplements on the energy metabolism of red blood cells. Attenuation of oxidative stress-induced changes in Rett syndrome erythrocytes by CoQ10. Mol. Cell Biochem. 2020, 463, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40, 207–218. [Google Scholar] [CrossRef]
- Pastor-Maldonado, C.J.; Suarez-Rivero, J.M.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Munuera-Cabeza, M.; Suarez-Carrillo, A.; Talaveron-Rey, M.; Sanchez-Alcazar, J.A. Coenzyme Q(10): Novel Formulations and Medical Trends. Int. J. Mol. Sci. 2020, 21, 8432. [Google Scholar] [CrossRef]
- Tiano, L.; Padella, L.; Santoro, L.; Carnevali, P.; Principi, F.; Bruge, F.; Gabrielli, O.; Littarru, G.P. Prolonged coenzyme Q10 treatment in Down syndrome patients: Effect on DNA oxidation. Neurobiol. Aging 2012, 33, e621–e628. [Google Scholar] [CrossRef]
- Cucinotta, F.; Ricciardello, A.; Turriziani, L.; Mancini, A.; Keller, R.; Sacco, R.; Persico, A.M. Efficacy and Safety of Q10 Ubiquinol with Vitamins B and E in Neurodevelopmental Disorders: A Retrospective Chart Review. Front. Psychiatry 2022, 13, 829516. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007, 7, S78–S88. [Google Scholar] [CrossRef] [PubMed]
- Petrangolini, G.; Ronchi, M.; Frattini, E.; De Combarieu, E.; Allegrini, P.; Riva, A. A New Food-grade Coenzyme Q10 Formulation Improves Bioavailability: Single and Repeated Pharmacokinetic Studies in Healthy Volunteers. Curr. Drug Deliv. 2019, 16, 759–767. [Google Scholar] [CrossRef]
- Rizzardi, N.; Liparulo, I.; Antonelli, G.; Orsini, F.; Riva, A.; Bergamini, C.; Fato, R. Coenzyme Q10 Phytosome Formulation Improves CoQ10 Bioavailability and Mitochondrial Functionality in Cultured Cells. Antioxidants 2021, 10, 927. [Google Scholar] [CrossRef]
- Drobnic, F.; Fonts, S.; Garcia-Alday, I.; Petrangolini, G.; Riva, A.; Frattini, E.; Allegrini, P.; Togni, S.; Vitale, J. Efficacy of artichoke and ginger extracts with simethicone to treat gastrointestinal symptoms in endurance athletes: A pilot study. Minerva Gastroenterol. 2022, 68, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Yoshihiro, K.; Hasko, G.; Pacher, P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 2007, 358, 203–208. [Google Scholar] [CrossRef]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Imam, N.; Lopez, B.; Bertrand, P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012, 31, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Gruenbaum, Y.; Moir, R.D.; Shumaker, D.K.; Spann, T.P. Nuclear lamins: Building blocks of nuclear architecture. Genes Dev. 2002, 16, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Davidovich, M.; Nissim-Rafinia, M.; Wiesel-Motiuk, N.; Bar, D.Z.; Barkan, R.; Meshorer, E.; Gruenbaum, Y. Nuclear lamins: Key regulators of nuclear structure and activities. J. Cell Mol. Med. 2009, 13, 1059–1085. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Hyun, Y.J.; Park, J.E.; Shilnikova, K.; Zhen, A.X.; Kang, H.K.; Koh, Y.S.; Jeong, Y.J.; et al. Luteolin induces apoptotic cell death via antioxidant activity in human colon cancer cells. Int. J. Oncol. 2017, 51, 1169–1178. [Google Scholar] [CrossRef]
- Rehfeldt, S.C.H.; Silva, J.; Alves, C.; Pinteus, S.; Pedrosa, R.; Laufer, S.; Goettert, M.I. Neuroprotective Effect of Luteolin-7-O-Glucoside against 6-OHDA-Induced Damage in Undifferentiated and RA-Differentiated SH-SY5Y Cells. Int. J. Mol. Sci. 2022, 23, 2914. [Google Scholar] [CrossRef]
- Naia, L.; Pinho, C.M.; Dentoni, G.; Liu, J.; Leal, N.S.; Ferreira, D.M.S.; Schreiner, B.; Filadi, R.; Fao, L.; Connolly, N.M.C.; et al. Neuronal cell-based high-throughput screen for enhancers of mitochondrial function reveals luteolin as a modulator of mitochondria-endoplasmic reticulum coupling. BMC Biol. 2021, 19, 57. [Google Scholar] [CrossRef]
- Fielder, E.; von Zglinicki, T.; Jurk, D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J. Alzheimers Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Andreyev, A.Y.; Zhang, S.F.; Starkova, N.N.; Korneeva, M.; Syromyatnikov, M.; Popov, V.N. Scavenging of H2O2 by mouse brain mitochondria. J. Bioenerg. Biomembr. 2014, 46, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Alcala-Vida, R.; Garcia-Forn, M.; Castany-Pladevall, C.; Creus-Muncunill, J.; Ito, Y.; Blanco, E.; Golbano, A.; Crespi-Vazquez, K.; Parry, A.; Slater, G.; et al. Neuron type-specific increase in lamin B1 contributes to nuclear dysfunction in Huntington’s disease. EMBO Mol. Med. 2021, 13, e12105. [Google Scholar] [CrossRef]
- Khatau, S.B.; Hale, C.M.; Stewart-Hutchinson, P.J.; Patel, M.S.; Stewart, C.L.; Searson, P.C.; Hodzic, D.; Wirtz, D. A perinuclear actin cap regulates nuclear shape. Proc. Natl. Acad. Sci. USA 2009, 106, 19017–19022. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Liu, P.Z.; Banigan, E.J.; Almassalha, L.M.; Backman, V.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 2018, 29, 220–233. [Google Scholar] [CrossRef]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef]
- Vergnes, L.; Peterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef]
- Frost, B. Alzheimer’s disease: An acquired neurodegenerative laminopathy. Nucleus 2016, 7, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, N.J.; Skalak, S.H.; Whitehead, A.J.; Hocker, J.D.; Beri, P.; Vogler, G.; Hum, B.; Wang, M.; Lakatta, E.G.; Ren, B.; et al. Age-dependent Lamin changes induce cardiac dysfunction via dysregulation of cardiac transcriptional programs. Nat. Aging 2023, 3, 17–33. [Google Scholar] [CrossRef]
- Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef]
- Takahashi, M.; Takahashi, K. Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria. Antioxidants 2019, 8, 61. [Google Scholar] [CrossRef]
- Crane, F.L.; Sun, I.L.; Crowe, R.A.; Alcain, F.J.; Low, H. Coenzyme Q10, plasma membrane oxidase and growth control. Mol. Aspects Med. 1994, 15, s1–s11. [Google Scholar] [CrossRef]
- McCarthy, S.; Somayajulu, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol. Appl. Pharmacol. 2004, 201, 21–31. [Google Scholar] [CrossRef]
- Tomasetti, M.; Littarru, G.P.; Stocker, R.; Alleva, R. Coenzyme Q10 enrichment decreases oxidative DNA damage in human lymphocytes. Free Radic. Biol. Med. 1999, 27, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Del Pozo-Cruz, J.; Sanchez-Cuesta, A.; Cortes-Rodriguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef]
- Sohal, R.S.; Kamzalov, S.; Sumien, N.; Ferguson, M.; Rebrin, I.; Heinrich, K.R.; Forster, M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006, 40, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Llanos-Gonzalez, E.; Alcain, F.J. The Use of Coenzyme Q10 in Cardiovascular Diseases. Antioxidants 2021, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. Int. J. Mol. Sci. 2021, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Bowry, V.W.; Frei, B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc. Natl. Acad. Sci. USA 1991, 88, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Mine, Y.; Okamoto, T. Extracellular coenzyme Q(10) (CoQ(10)) is reduced to ubiquinol-10 by intact Hep G2 cells independent of intracellular CoQ(10) reduction. Arch. Biochem. Biophys. 2019, 672, 108067. [Google Scholar] [CrossRef]
- Aaseth, J.; Alexander, J.; Alehagen, U. Coenzyme Q(10) supplementation—In ageing and disease. Mech. Ageing Dev. 2021, 197, 111521. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q biosynthesis inhibition induces HIF-1alpha stabilization and metabolic switch toward glycolysis. FEBS J. 2021, 288, 1956–1974. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; De Groef, L. Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Sci. Rep. 2017, 7, 1576. [Google Scholar] [CrossRef]
- Matias, I.; Diniz, L.P.; Damico, I.V.; Araujo, A.P.B.; Neves, L.D.S.; Vargas, G.; Leite, R.E.P.; Suemoto, C.K.; Nitrini, R.; Jacob-Filho, W.; et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 2022, 21, e13521. [Google Scholar] [CrossRef] [PubMed]
- Diquigiovanni, C.; Rizzardi, N.; Kampmeier, A.; Liparulo, I.; Bianco, F.; De Nicolo, B.; Cataldi-Stagetti, E.; Cuna, E.; Severi, G.; Seri, M.; et al. Mutant SPART causes defects in mitochondrial protein import and bioenergetics reversed by Coenzyme Q. Open Biol. 2023, 13, 230040. [Google Scholar] [CrossRef] [PubMed]
- Medici, G.; Tassinari, M.; Galvani, G.; Bastianini, S.; Gennaccaro, L.; Loi, M.; Mottolese, N.; Alvente, S.; Berteotti, C.; Sagona, G.; et al. Expression of a Secretable, Cell-Penetrating CDKL5 Protein Enhances the Efficacy of Gene Therapy for CDKL5 Deficiency Disorder. Neurotherapeutics 2022, 19, 1886–1904. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Ikenoya, S.; Yuzuriha, T.; Katayama, K. Simultaneous determination of reduced and oxidized ubiquinones. Methods Enzymol. 1984, 105, 147–155. [Google Scholar] [CrossRef]
- Reilly, C.A.; Aust, S.D. Measurement of lipid peroxidation. Curr. Protoc. Toxicol. 2001, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Delanty, N.; Dichter, M.A. Antioxidant therapy in neurologic disease. Arch. Neurol. 2000, 57, 1265–1270. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Q9 (pmol/mL Plasma) | |||
|---|---|---|---|
| Cdkl5 +/+ | Cdkl5 +/− | p | |
| vehicle | 178.9 ± 13.3 (n = 6) | 192.9 ± 20.3 (n = 9) | n.s. |
| UBQ | 471.3 ± 136.6 (n = 7) | 232.7 ± 27.1 (n = 10) | * |
| p | ** | n.s. | |
| Q10 (pmol/mL plasma) | |||
| Cdkl5 +/+ | Cdkl5 +/− | p | |
| vehicle | 68.9 ± 16.9 (n = 5) | 37.9 ± 2.3 (n = 5) | n.s. |
| UBQ | 126.0 ± 9.2 (n = 5) | 42.6 ± 9. 8 (n = 8) | * |
| p | * | n.s. | |
| Q9 (pmol/mg Protein) | |||
|---|---|---|---|
| Cdkl5 +/+ | Cdkl5 +/− | p | |
| vehicle | 644.1 ± 74.6 (n = 6) | 712.2 ± 64.4 (n = 10) | n.s. |
| UBQ | 574.9 ± 75.5 (n = 7) | 754.5 ± 54.4 (n = 10) | n.s. |
| p | n.s. | n.s. | |
| Q10 (pmol/mg protein) | |||
| Cdkl5 +/+ | Cdkl5 +/− | p | |
| vehicle | 83.3 ± 9.5 (n = 6) | 95.5 ± 9 (n = 10) | n.s. |
| UBQ | 75.1 ± 11.2 (n = 7) | 100.6 ± 10.2 (n = 10) | n.s. |
| p | n.s. | n.s. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loi, M.; Valenti, F.; Medici, G.; Mottolese, N.; Candini, G.; Bove, A.M.; Trebbi, F.; Pincigher, L.; Fato, R.; Bergamini, C.; et al. Beneficial Antioxidant Effects of Coenzyme Q10 in In Vitro and In Vivo Models of CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2025, 26, 2204. https://doi.org/10.3390/ijms26052204
Loi M, Valenti F, Medici G, Mottolese N, Candini G, Bove AM, Trebbi F, Pincigher L, Fato R, Bergamini C, et al. Beneficial Antioxidant Effects of Coenzyme Q10 in In Vitro and In Vivo Models of CDKL5 Deficiency Disorder. International Journal of Molecular Sciences. 2025; 26(5):2204. https://doi.org/10.3390/ijms26052204
Chicago/Turabian StyleLoi, Manuela, Francesca Valenti, Giorgio Medici, Nicola Mottolese, Giulia Candini, Angelica Marina Bove, Federica Trebbi, Luca Pincigher, Romana Fato, Christian Bergamini, and et al. 2025. "Beneficial Antioxidant Effects of Coenzyme Q10 in In Vitro and In Vivo Models of CDKL5 Deficiency Disorder" International Journal of Molecular Sciences 26, no. 5: 2204. https://doi.org/10.3390/ijms26052204
APA StyleLoi, M., Valenti, F., Medici, G., Mottolese, N., Candini, G., Bove, A. M., Trebbi, F., Pincigher, L., Fato, R., Bergamini, C., Trazzi, S., & Ciani, E. (2025). Beneficial Antioxidant Effects of Coenzyme Q10 in In Vitro and In Vivo Models of CDKL5 Deficiency Disorder. International Journal of Molecular Sciences, 26(5), 2204. https://doi.org/10.3390/ijms26052204