
Mechanism of Action and Interaction of Garlic Extract and Established Therapeutics in Prostate Cancer

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Effect of GE on the Proliferation of Healthy Prostate Epithelial and PCa Cell Lines
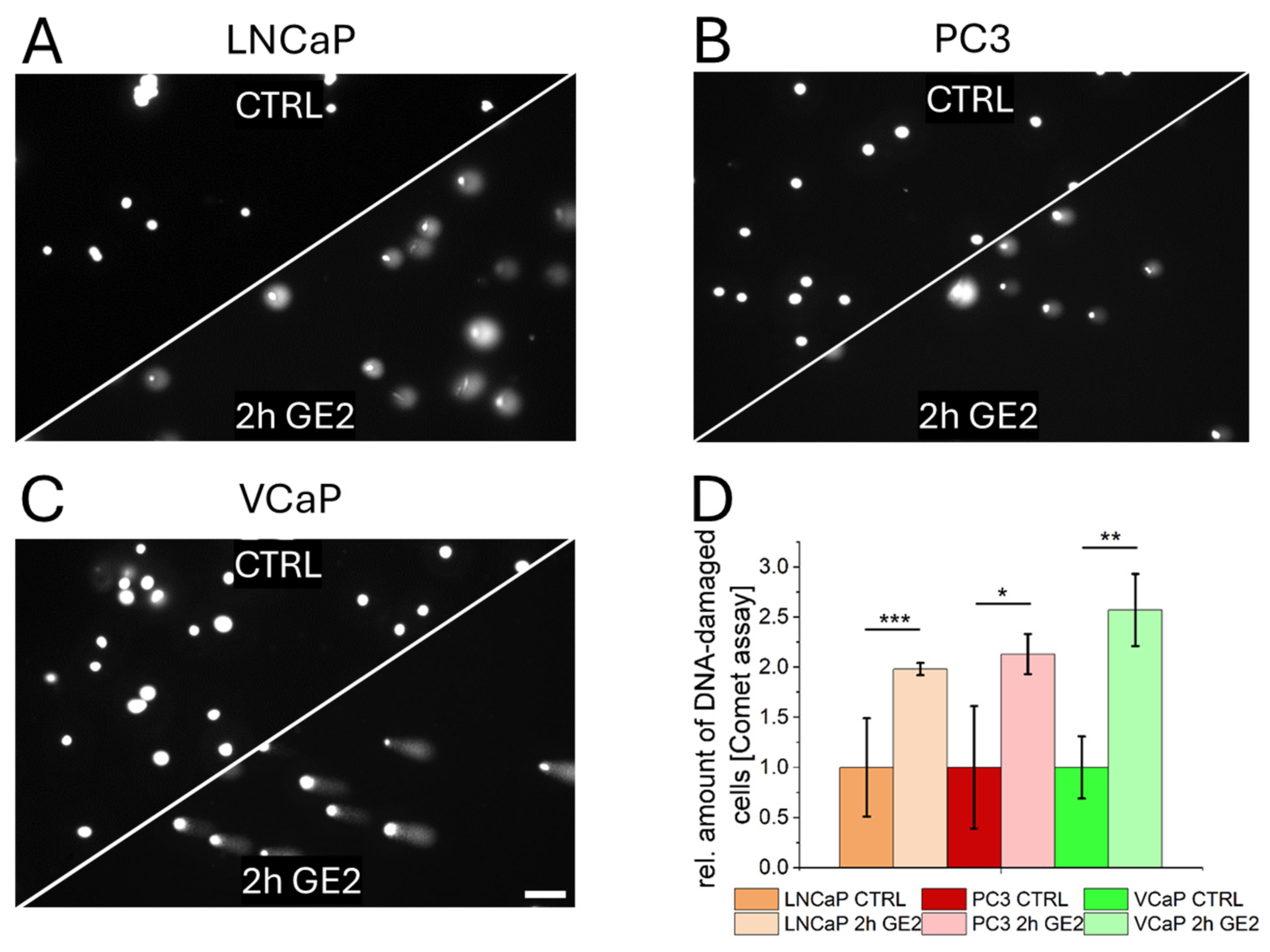
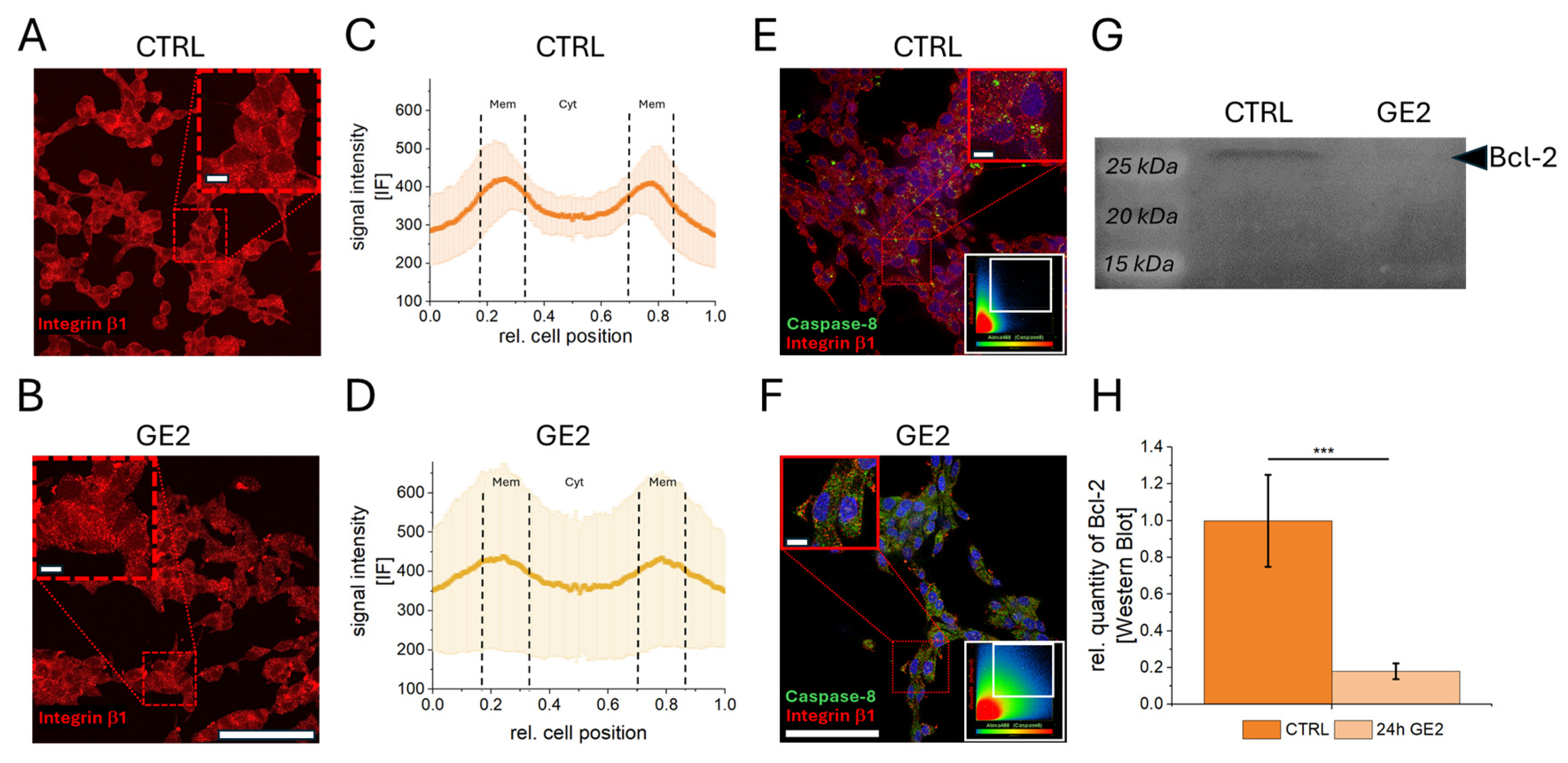
2.2. Mechanisms of Action of GE on PCa Cells
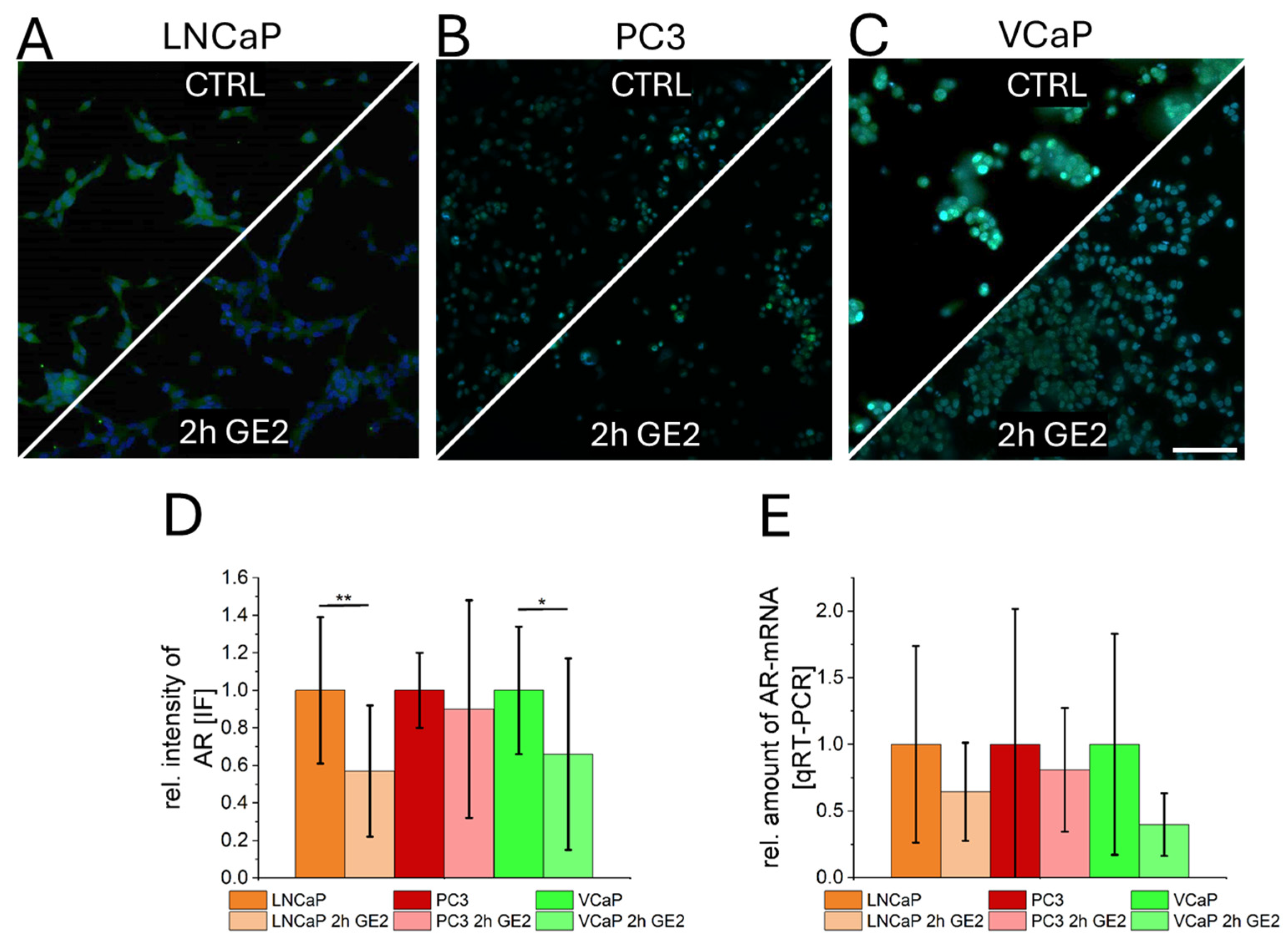
2.3. Interaction of GE with Androgen Receptors in Prostate Cancer Cells
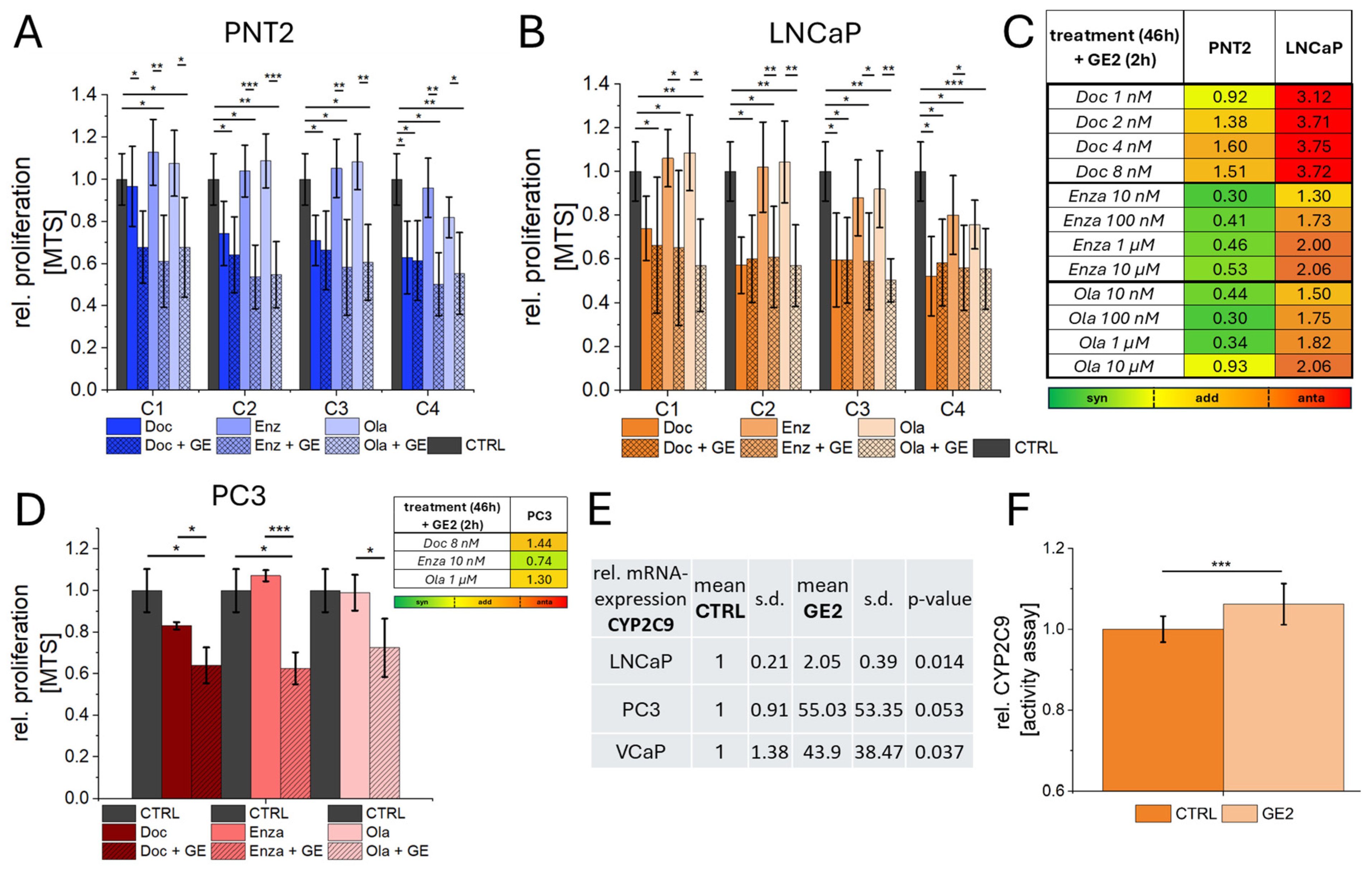
2.4. Combination of GE with Chemotherapy, ADT, and PARPi
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Preparation and Treatment of GE
4.3. Preparation and Treatment of PCa Therapeutic Agents
4.4. Proliferation Assay (MTS), Live Dead Staining, and Cell Cycle Analysis
4.5. Comet Assay (DNA Damage) Analysis
4.6. Immunefluorescence Staining
4.7. Microscopy and Image Analysis
4.8. Characterization of Protein Alterations
4.8.1. Protein Isolation
4.8.2. Determination of Protein Concentrations
4.8.3. SDS-PAGE
4.8.4. Western Blot and Antibody Stain
4.9. Characterization of mRNA Alterations
4.9.1. RNA Isolation
4.9.2. cDNA Synthesis
4.9.3. qRT-PCR
4.10. CYP2C9 Activity Assay
4.11. Calculation of Combination Indices (CIs)
4.12. Statistics and Graphical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Oliva, J.; Anastay, V.; Baboudjian, M.; Roumiguie, M.; Peltier, A.; Dariane, C.; Fiard, G.; Roumeguere, T.; Diamand, R.; Bakhri, M.; et al. Active surveillance for low-risk prostate cancer with high tumor burden at biopsy: Lessons learned from a contemporary radical prostatectomy cohort. World J. Urol. 2024, 42, 513. [Google Scholar] [CrossRef] [PubMed]
- Martini, A.; Fallara, G.; Ploussard, G.; Malavaud, B. Combination therapy with olaparib and abiraterone acetate for metastatic castration-resistant prostate cancer. Lancet Oncol. 2023, 24, 1056–1057. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Foller, S.; Leucht, K. Combination therapy with poly(adenosine diphosphate-ribose) polymerase (PARPi) and androgen receptor signaling pathway (ARPi) inhibitors for metastatic castration-resistant prostate cancer. Urologie 2023, 62, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Kato, D.; Tobisawa, Y.; Iinuma, K.; Nakane, K.; Koie, T. Efficacy and safety of combination neoadjuvant chemo-hormonal therapy and robot-assisted radical prostatectomy for oligometastatic prostate cancer. Int. J. Urol. 2024, 31, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Kendi, A.T.; Sartor, O. Status of PSMA-targeted radioligand therapy in prostate cancer: Current data and future trials. Ther. Adv. Med. Oncol. 2023, 15, 17588359231157632. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Van Asseldonk, B.; Black, P.; Elterman, D.S. Chemical vs Surgical ADT in Metastatic Prostate Cancer: A Comparison of Side Effects. Commentary on Comparison of Gonadotropin-releasing Hormone Agonists and Orchiectomy: Effects of Androgen Deprivation Therapy. Urology 2016, 93, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Pimenta, M.; Estevinho, L.M.; Szopa, A.; Basit, M.; Khan, K.; Armaghan, M.; Ibrayeva, M.; Sonmez Gurer, E.; Calina, D.; Hano, C.; et al. Chemotherapeutic properties and side-effects associated with the clinical practice of terpene alkaloids: Paclitaxel, docetaxel, and cabazitaxel. Front. Pharmacol. 2023, 14, 1157306. [Google Scholar] [CrossRef]
- Gou, R.; Horikawa, N.; Kosaka, K. Olaparib-induced cutaneous side effects in a patient with recurrent ovarian cancer. BMJ Case Rep. 2022, 15, e249177. [Google Scholar] [CrossRef]
- Stanislawiak-Rudowicz, J.; Szalek, E.; Adamiak, A.; Urjasz, H.; Wieckowska, B.; Grzeskowiak, E.; Madry, R. A retrospective analysis of haematological side effects of olaparib in excess-weight and normal BMI patients with ovarian cancer. Rep. Pract. Oncol. Radiother. 2024, 29, 113–121. [Google Scholar] [CrossRef]
- Tascilar, M.; de Jong, F.A.; Verweij, J.; Mathijssen, R.H. Complementary and alternative medicine during cancer treatment: Beyond innocence. Oncologist 2006, 11, 732–741. [Google Scholar] [CrossRef]
- Jeremic, J.N.; Jakovljevic, V.L.; Zivkovic, V.I.; Srejovic, I.M.; Bradic, J.V.; Milosavljevic, I.M.; Mitrovic, S.L.; Jovicic, N.U.; Bolevich, S.B.; Svistunov, A.A.; et al. Garlic Derived Diallyl Trisulfide in Experimental Metabolic Syndrome: Metabolic Effects and Cardioprotective Role. Int. J. Mol. Sci. 2020, 21, 9100. [Google Scholar] [CrossRef] [PubMed]
- Torres-Narvaez, J.C.; Perez-Torres, I.; Del Valle-Mondragon, L.; Castrejon-Tellez, V.; Guarner-Lans, V.; Sanchez-Aguilar, M.; Varela-Lopez, E.; Vargas-Gonzalez, A.; Pastelin-Hernandez, G.; Diaz-Juarez, J.A. Garlic prevents the oxidizing and inflammatory effects of sepsis induced by bacterial lipopolysaccharide at the systemic and aortic level in the rat. Role of trpv1. Heliyon 2023, 9, e21230. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Hay, R.I.; Mazroa, S.A.; El-Mohandes, E.; Am, M. The possible protective role of melatonin versus garlic on monosodium Glutamate-induced changes in rat cerebellar cortex: Histological, immunohistochemical and electron microscope study. Ultrastruct. Pathol. 2023, 47, 49–72. [Google Scholar] [CrossRef]
- Hassan, H.T. Prospective clinical role for anticancer garlic organosulfur compounds. Anticancer. Agents Med. Chem. 2011, 11, 247–248. [Google Scholar] [CrossRef]
- Perez-Rubio, K.G.; Mendez-Del Villar, M.; Cortez-Navarrete, M. The Role of Garlic in Metabolic Diseases: A Review. J. Med. Food 2022, 25, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gloria, J.L.; Rada, K.M.; Juarez-Rojas, J.G.; Sanchez-Lozada, L.G.; Rubio-Gayosso, I.; Sanchez-Munoz, F.; Osorio-Alonso, H. Role of Sulfur Compounds in Garlic as Potential Therapeutic Option for Inflammation and Oxidative Stress in Asthma. Int. J. Mol. Sci. 2022, 23, 15599. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Gunadharini, D.N.; Arunkumar, A.; Krishnamoorthy, G.; Muthuvel, R.; Vijayababu, M.R.; Kanagaraj, P.; Srinivasan, N.; Aruldhas, M.M.; Arunakaran, J. Antiproliferative effect of diallyl disulfide (DADS) on prostate cancer cell line LNCaP. Cell Biochem. Funct. 2006, 24, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, K.; Danilenko, M.; Giat, J.; Miron, T.; Rabinkov, A.; Wilchek, M.; Mirelman, D.; Levy, J.; Sharoni, Y. Effect of purified allicin, the major ingredient of freshly crushed garlic, on cancer cell proliferation. Nutr. Cancer 2000, 38, 245–254. [Google Scholar] [CrossRef]
- Arunkumar, A.; Vijayababu, M.R.; Srinivasan, N.; Aruldhas, M.M.; Arunakaran, J. Garlic compound, diallyl disulfide induces cell cycle arrest in prostate cancer cell line PC-3. Mol. Cell Biochem. 2006, 288, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Pinto, J.T. Cysteine S-conjugate beta-lyases. Amino Acids 2006, 30, 1–15. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Duan, W.; Feng, C.; He, X. Allicin induces apoptosis of the MGC-803 human gastric carcinoma cell line through the p38 mitogen-activated protein kinase/caspase-3 signaling pathway. Mol. Med. Rep. 2015, 11, 2755–2760. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Cho, S.J.; Kwon, H.C.; Lee, K.R.; Rhee, D.K.; Pyo, S. Caspase-independent cell death by allicin in human epithelial carcinoma cells: Involvement of PKA. Cancer Lett. 2005, 224, 123–132. [Google Scholar] [CrossRef]
- Suddek, G.M. Allicin enhances chemotherapeutic response and ameliorates tamoxifen-induced liver injury in experimental animals. Pharm. Biol. 2014, 52, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.C.t.; Haar, C.P.; Vandergrift, W.A., 3rd; Giglio, P.; Dixon-Mah, Y.N.; Varma, A.K.; Ray, S.K.; Patel, S.J.; Banik, N.L.; Das, A. Multi-targeted DATS prevents tumor progression and promotes apoptosis in ectopic glioblastoma xenografts in SCID mice via HDAC inhibition. J. Neurooncol 2013, 114, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Puccinelli, M.T.; Stan, S.D. Dietary Bioactive Diallyl Trisulfide in Cancer Prevention and Treatment. Int. J. Mol. Sci. 2017, 18, 1645. [Google Scholar] [CrossRef]
- Bhadra, R.; Ravakhah, K.; Ghosh, R.K. Herb-drug interaction: The importance of communicating with primary care physicians. Australas. Med. J. 2015, 8, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Isvoran, A.; Louet, M.; Vladoiu, D.L.; Craciun, D.; Loriot, M.A.; Villoutreix, B.O.; Miteva, M.A. Pharmacogenomics of the cytochrome P450 2C family: Impacts of amino acid variations on drug metabolism. Drug Discov. Today 2017, 22, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, Y.; Jing, X.; Qian, D.; Tang, Y.; Duan, J.A. In vitro and in vivo assessment of CYP2C9-mediated herb-herb interaction of Euphorbiae Pekinensis Radix and Glycyrrhizae Radix. Front. Pharmacol. 2014, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gao, Q.; Zhang, T.; Rao, J.; Ding, L.; Qiu, F. Inhibition of CYP2C9 by natural products: Insight into the potential risk of herb-drug interactions. Drug Metab. Rev. 2020, 52, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Paltiel, O.; Avitzour, M.; Peretz, T.; Cherny, N.; Kaduri, L.; Pfeffer, R.M.; Wagner, N.; Soskolne, V. Determinants of the use of complementary therapies by patients with cancer. J. Clin. Oncol. 2001, 19, 2439–2448. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.S.; Halabi, S.; Kaplan, E.B.; Baron, J.A.; Paskett, E.; Petrelli, N.J. Use of vitamins, minerals, and nutritional supplements by participants in a chemoprevention trial. Cancer 2001, 91, 1040–1045. [Google Scholar] [CrossRef]
- Meijerman, I.; Beijnen, J.H.; Schellens, J.H. Herb-drug interactions in oncology: Focus on mechanisms of induction. Oncologist 2006, 11, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Koon, H.K.; Ma, C.T.; Au, K.Y.; Zuo, Z.; Chung, V.C.; Cheung, Y.T. Real-world data on herb-drug interactions in oncology: A scoping review of pharmacoepidemiological studies. Phytomedicine 2022, 103, 154247. [Google Scholar] [CrossRef]
- Bolam, K.A.; Bojsen-Moller, E.; Wallin, P.; Paulsson, S.; Lindwall, M.; Rundqvist, H.; Ekblom-Bak, E. Association between change in cardiorespiratory fitness and prostate cancer incidence and mortality in 57 652 Swedish men. Br. J. Sports Med. 2024, 58, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Byeon, J.Y.; Park, D.H.; Oh, C.G.; Lee, J.; Choi, Y.D.; Kang, D.W.; An, K.Y.; Courneya, K.S.; Lee, D.H.; et al. Effects of exercise during active surveillance for prostate cancer: A systematic review and meta-analysis. Support. Care Cancer 2024, 32, 406. [Google Scholar] [CrossRef] [PubMed]
- Richman, E.L.; Kenfield, S.A.; Stampfer, M.J.; Paciorek, A.; Carroll, P.R.; Chan, J.M. Physical activity after diagnosis and risk of prostate cancer progression: Data from the cancer of the prostate strategic urologic research endeavor. Cancer Res. 2011, 71, 3889–3895. [Google Scholar] [CrossRef]
- Veras, A.S.C.; Batista, V.R.G.; Correia, R.R.; de Almeida Tavares, M.E.; Rubira, R.J.G.; Tavares, E.R.; Giometti, I.C.; Maranhao, R.C.; Teixeira, G.R. Integrated aerobic exercise with LDE-docetaxel treatment: A novel approach to combat prostate cancer progression. Sci. Rep. 2024, 14, 9626. [Google Scholar] [CrossRef]
- Santos Pimentel, L.; Sommerfeld, S.; Fernanda de Sousa Braga, P.; Flores Coleto, A.; Beatriz Fonseca, B.; Machado Bastos, L.; Ricardo Goulart, L.; Nunes de Morais Ribeiro, L. Antitumor activity of essential oils-based nanostructured lipid carriers on prostate cancer cells. Int. J. Pharm. 2024, 657, 124149. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Ravicz, J.R.; Liu, S.; Chawla, S.P.; Hall, F.L. Cell cycle checkpoint control: The cyclin G1/Mdm2/p53 axis emerges as a strategic target for broad-spectrum cancer gene therapy—A review of molecular mechanisms for oncologists. Mol. Clin. Oncol. 2018, 9, 115–134. [Google Scholar] [CrossRef]
- Yam, C.Q.X.; Lim, H.H.; Surana, U. DNA damage checkpoint execution and the rules of its disengagement. Front. Cell Dev. Biol. 2022, 10, 1020643. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Sharma, S.; Sharma, N.R.; Kabra, D. Phytochemicals enriched in spices: A source of natural epigenetic therapy. Arch. Pharm. Res. 2020, 43, 171–186. [Google Scholar] [CrossRef]
- Kallenbach, J.; Atri Roozbahani, G.; Heidari Horestani, M.; Baniahmad, A. Distinct mechanisms mediating therapy-induced cellular senescence in prostate cancer. Cell Biosci. 2022, 12, 200. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Gonzalez, V.C.; Tellez-Banuelos, M.C.; Hernandez-Flores, G.; Bravo-Cuellar, A.; Aguilar-Lemarroy, A.; Jave-Suarez, L.F.; Haramati, J.; Solorzano-Ibarra, F.; Ortiz-Lazareno, P.C. Differential effects of alliin and allicin on apoptosis and senescence in luminal A and triple-negative breast cancer: Caspase, DeltaPsim, and pro-apoptotic gene involvement. Fundam. Clin. Pharmacol. 2020, 34, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef]
- Daly, A.K.; Rettie, A.E.; Fowler, D.M.; Miners, J.O. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. J. Pers. Med. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Maksymchuk, O.V.; Kashuba, V.I. Altered expression of cytochrome P450 enzymes involved in metabolism of androgens and vitamin D in the prostate as a risk factor for prostate cancer. Pharmacol. Rep. 2020, 72, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Maksymchuk, O.; Gerashchenko, G.; Rosohatska, I.; Kononenko, O.; Tymoshenko, A.; Stakhovsky, E.; Kashuba, V. Cytochrome P450 genes expression in human prostate cancer. Mol. Genet. Metab. Rep. 2024, 38, 101049. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Wang, Y.; Liu, X.; Wu, S.; Wang, M.; Turowski, S.G.; Spernyak, J.A.; Tracz, A.; Abdelaal, A.M.; Sudarshan, K.; et al. Developing Folate-Conjugated miR-34a Therapeutic for Prostate Cancer: Challenges and Promises. Int. J. Mol. Sci. 2024, 25, 2123. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, A.M.; Sohal, I.S.; Iyer, S.G.; Sudarshan, K.; Orellana, E.A.; Ozcan, K.E.; Dos Santos, A.P.; Low, P.S.; Kasinski, A.L. Selective targeting of chemically modified miR-34a to prostate cancer using a small molecule ligand and an endosomal escape agent. Mol. Ther. Nucleic Acids 2024, 35, 102193. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Fishman, J.; Vapiwala, N.; Li, S.Q.; Desai, K.; Xie, S.X.; Mao, J.J. Patient-physician communication about complementary and alternative medicine in a radiation oncology setting. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, e1–e6. [Google Scholar] [CrossRef]
- Petrovic, V.; Nepal, A.; Olaisen, C.; Bachke, S.; Hira, J.; Sogaard, C.K.; Rost, L.M.; Misund, K.; Andreassen, T.; Melo, T.M.; et al. Anti-Cancer Potential of Homemade Fresh Garlic Extract Is Related to Increased Endoplasmic Reticulum Stress. Nutrients 2018, 10, 450. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Ermler, T.F.; Hoffmann, F.; Alexa, R.; Kranz, J.; Steinke, N.; Leypold, S.; Gaisa, N.T.; Saar, M. Therapeutic and Diagnostic Potential of Folic Acid Receptors and Glycosylphosphatidylinositol (GPI) Transamidase in Prostate Cancer. Cancers 2024, 16, 2008. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Media | Supplemented with |
|---|---|---|
| LNCaP | RPMI 1640 (1) | 1% of 10,000 U/mL penicillin-streptomycin (PS) (2) 10% of fetal bovine serum (FBS) (3) |
| PC3 | DMEM (1) | |
| VCaP | DMEM F12 (1) | |
| PNT2 | RPMI 1640 (1) | 1% PS 10% FBS 1% of 200 mM L-glutamine (2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, M.; Sauer, J.; Book, M.; Ermler, T.F.; Fischer, P.; Gerlach, S.; Beltagi, K.; Morgenroth, A.; Alexa, R.; Kranz, J.; et al. Mechanism of Action and Interaction of Garlic Extract and Established Therapeutics in Prostate Cancer. Int. J. Mol. Sci. 2025, 26, 1777. https://doi.org/10.3390/ijms26041777
Hoffmann M, Sauer J, Book M, Ermler TF, Fischer P, Gerlach S, Beltagi K, Morgenroth A, Alexa R, Kranz J, et al. Mechanism of Action and Interaction of Garlic Extract and Established Therapeutics in Prostate Cancer. International Journal of Molecular Sciences. 2025; 26(4):1777. https://doi.org/10.3390/ijms26041777
Chicago/Turabian StyleHoffmann, Marco, Jana Sauer, Marie Book, Thomas Frank Ermler, Petra Fischer, Sven Gerlach, Kareem Beltagi, Agnieszka Morgenroth, Radu Alexa, Jennifer Kranz, and et al. 2025. "Mechanism of Action and Interaction of Garlic Extract and Established Therapeutics in Prostate Cancer" International Journal of Molecular Sciences 26, no. 4: 1777. https://doi.org/10.3390/ijms26041777
APA StyleHoffmann, M., Sauer, J., Book, M., Ermler, T. F., Fischer, P., Gerlach, S., Beltagi, K., Morgenroth, A., Alexa, R., Kranz, J., & Saar, M. (2025). Mechanism of Action and Interaction of Garlic Extract and Established Therapeutics in Prostate Cancer. International Journal of Molecular Sciences, 26(4), 1777. https://doi.org/10.3390/ijms26041777