
Proteomics Profiling Reveals Pharmaceutical Excipient PEG400 Induces Nuclear-Receptor-Activation-Affected Lipid Metabolism and Metabolic Enzyme Expression

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Histomorphology and Body Weight of Rat Liver
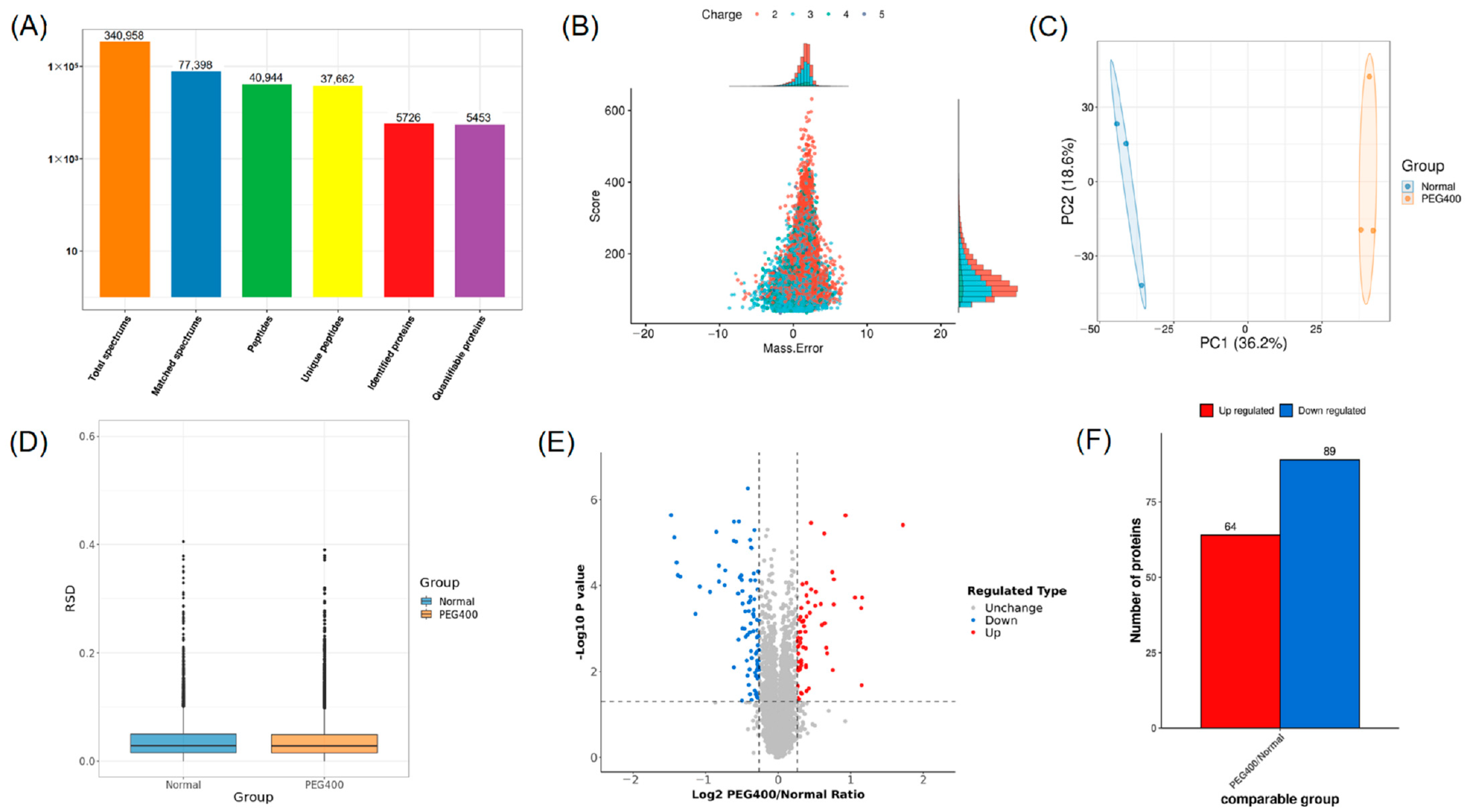
2.2. Proteomic Profiling Results
2.2.1. Differential Protein Screening and Analysis
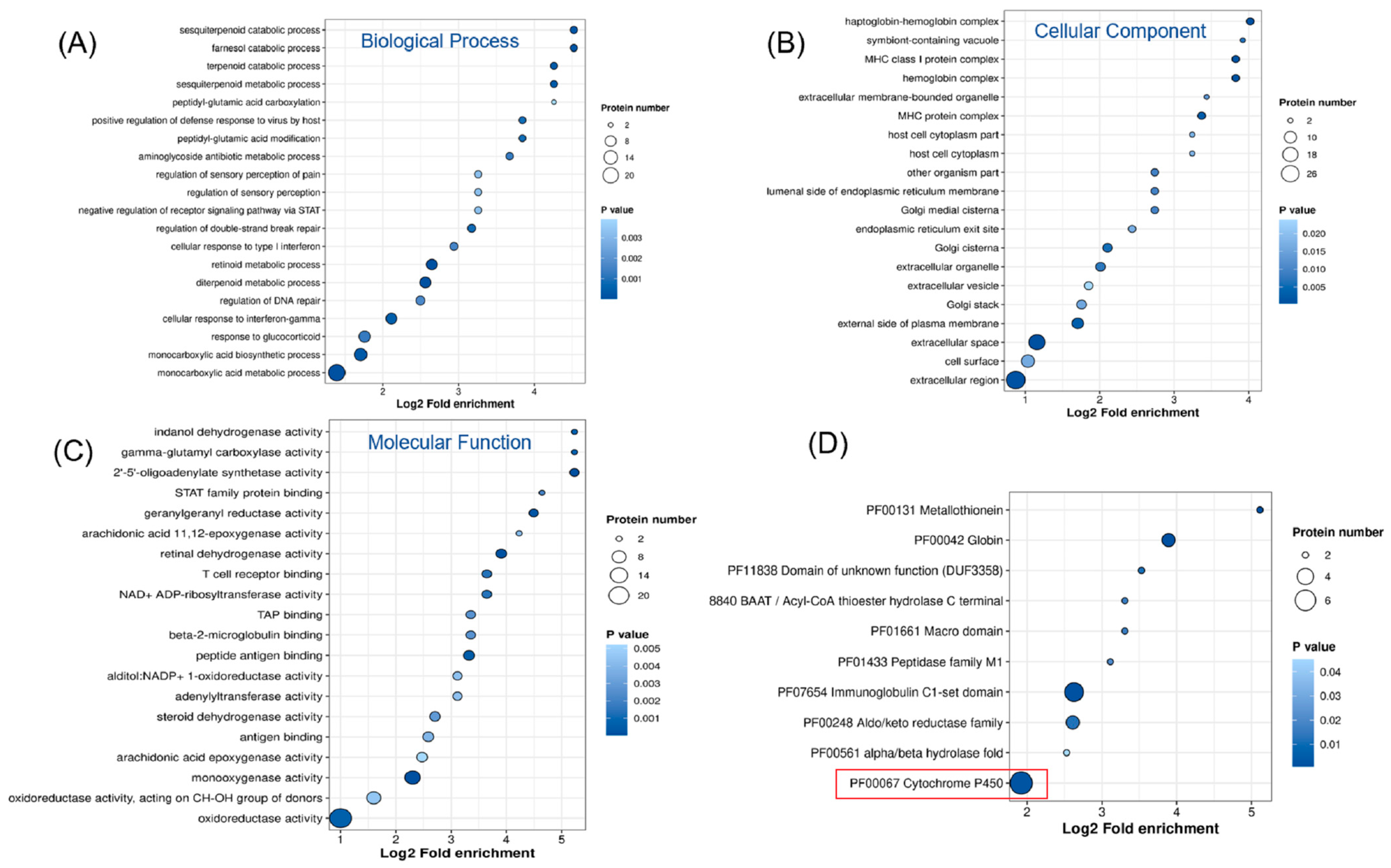
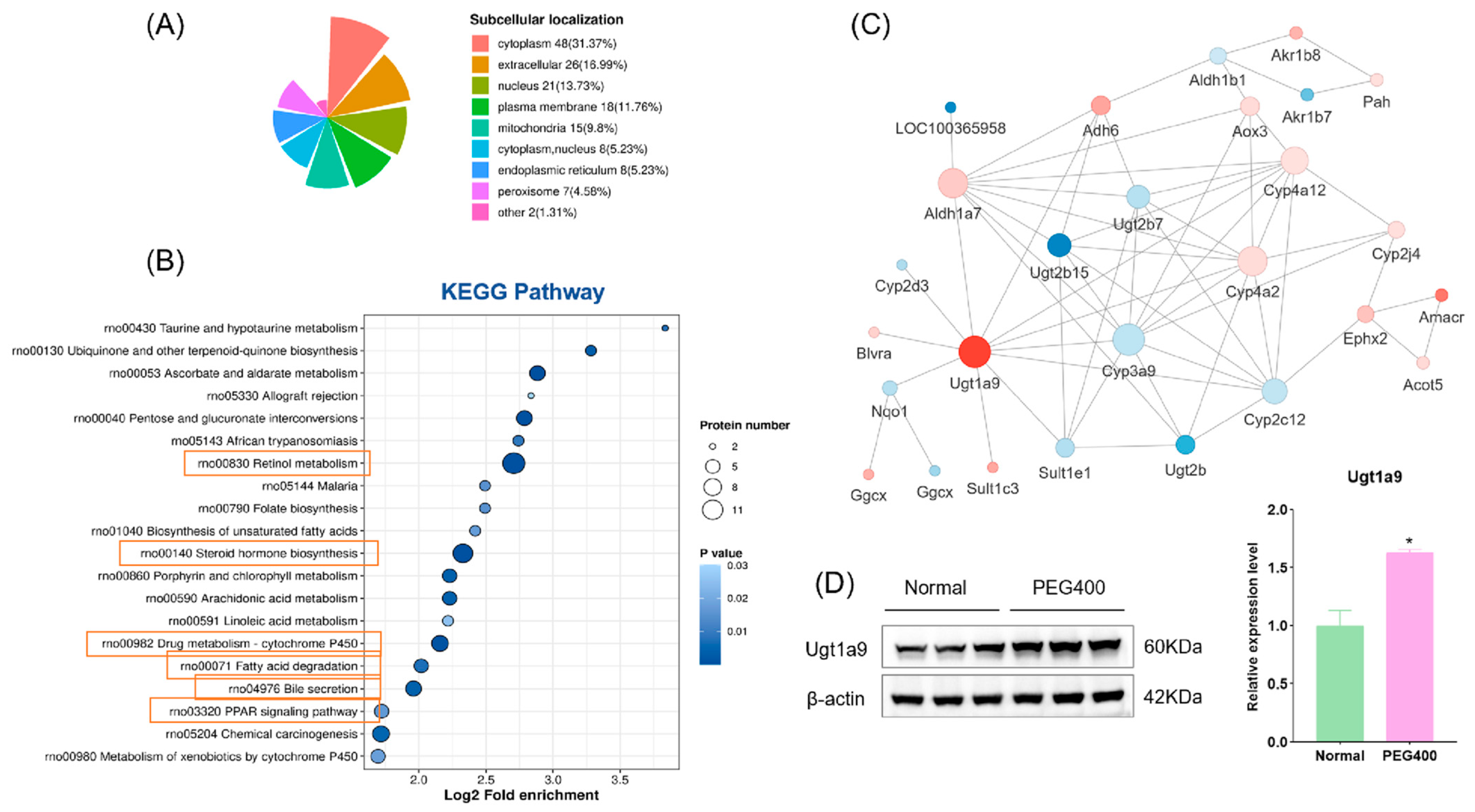
2.2.2. GO and KEGG Enrichment Analysis
2.2.3. PPI Network Analysis
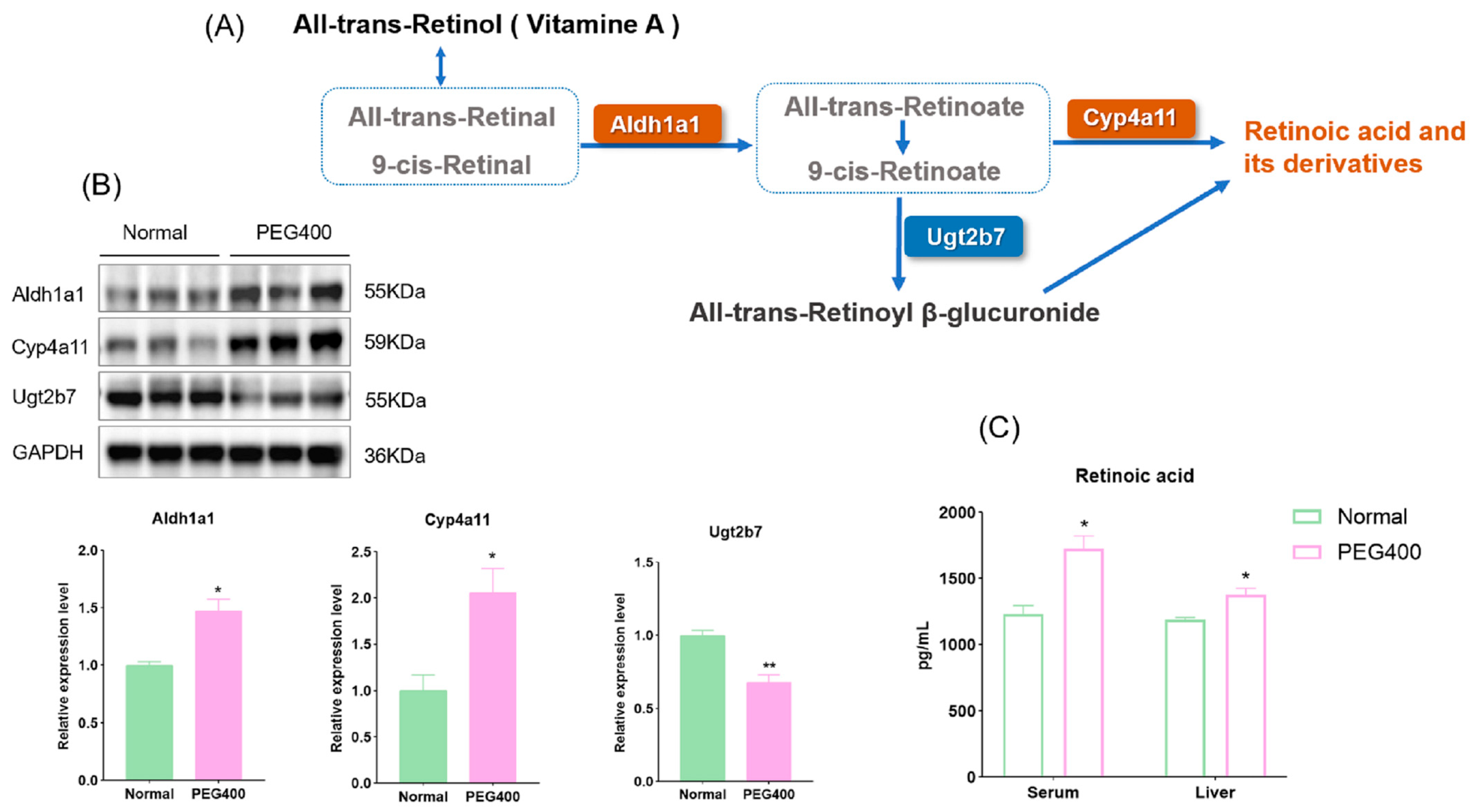
2.3. Effect of PEG400 on Retinol Metabolism
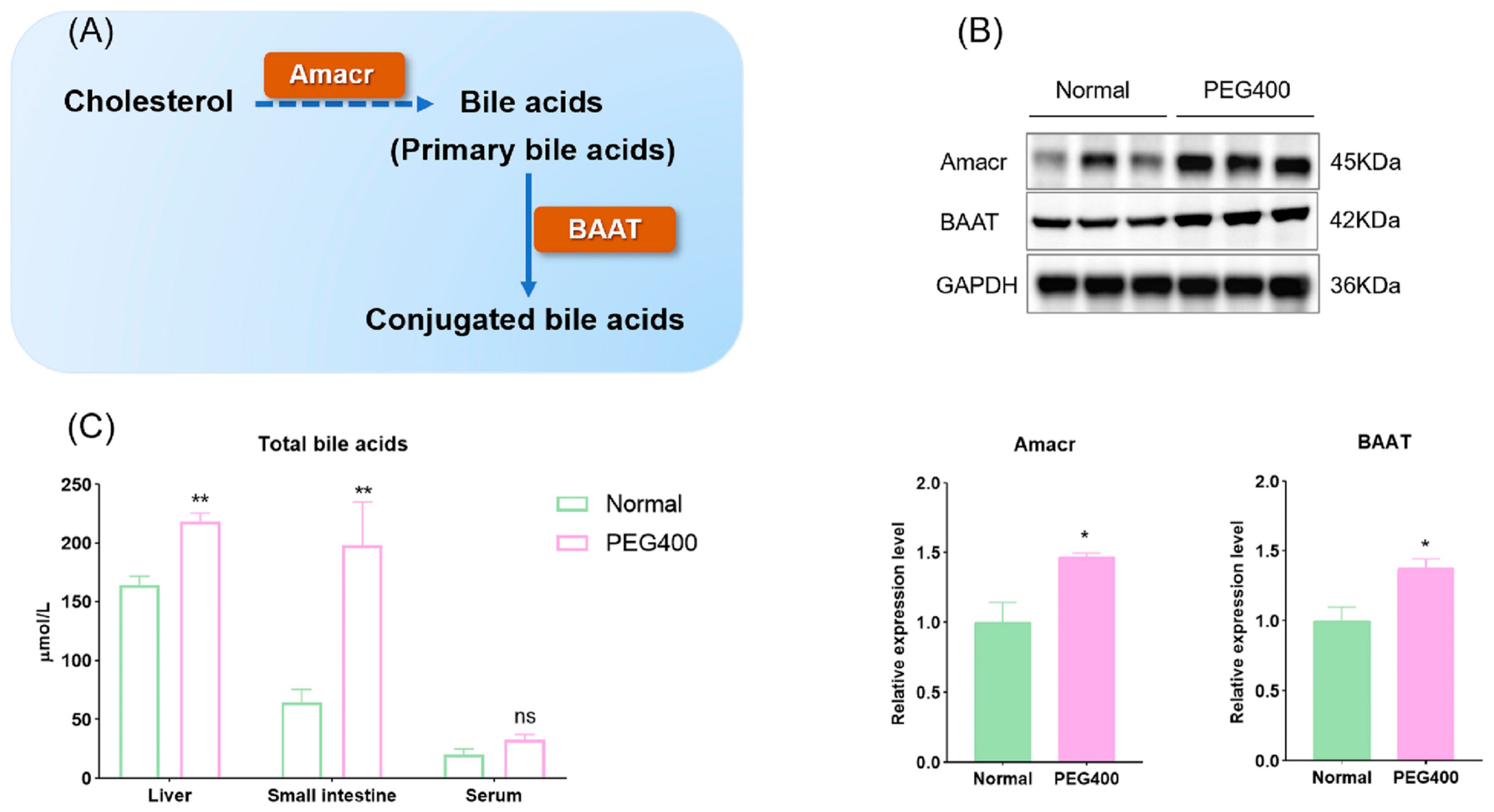
2.4. PEG400 Promotes Bile Secretion
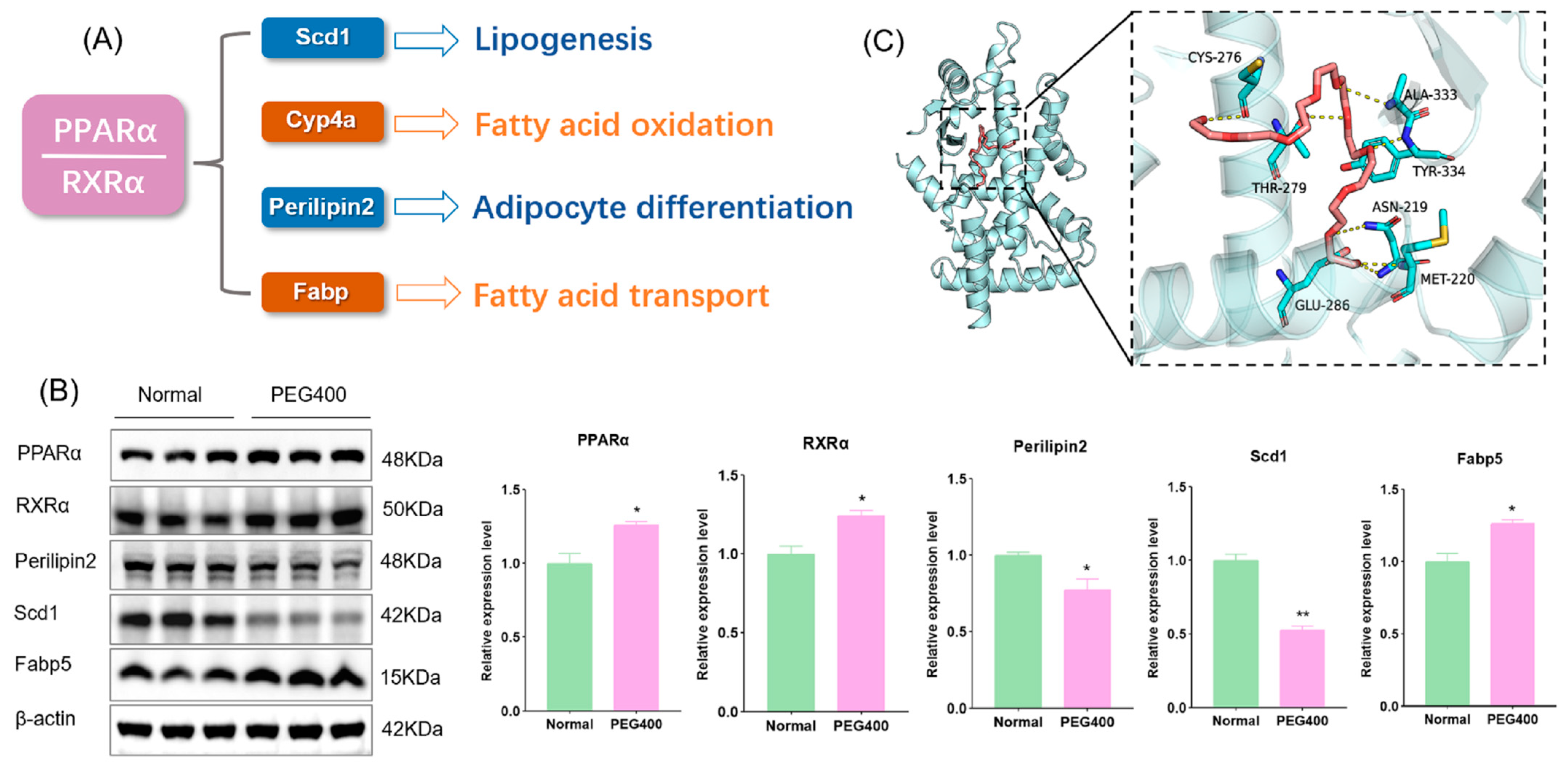
2.5. PEG400 Affects the Activation of the PPAR Signaling Pathway
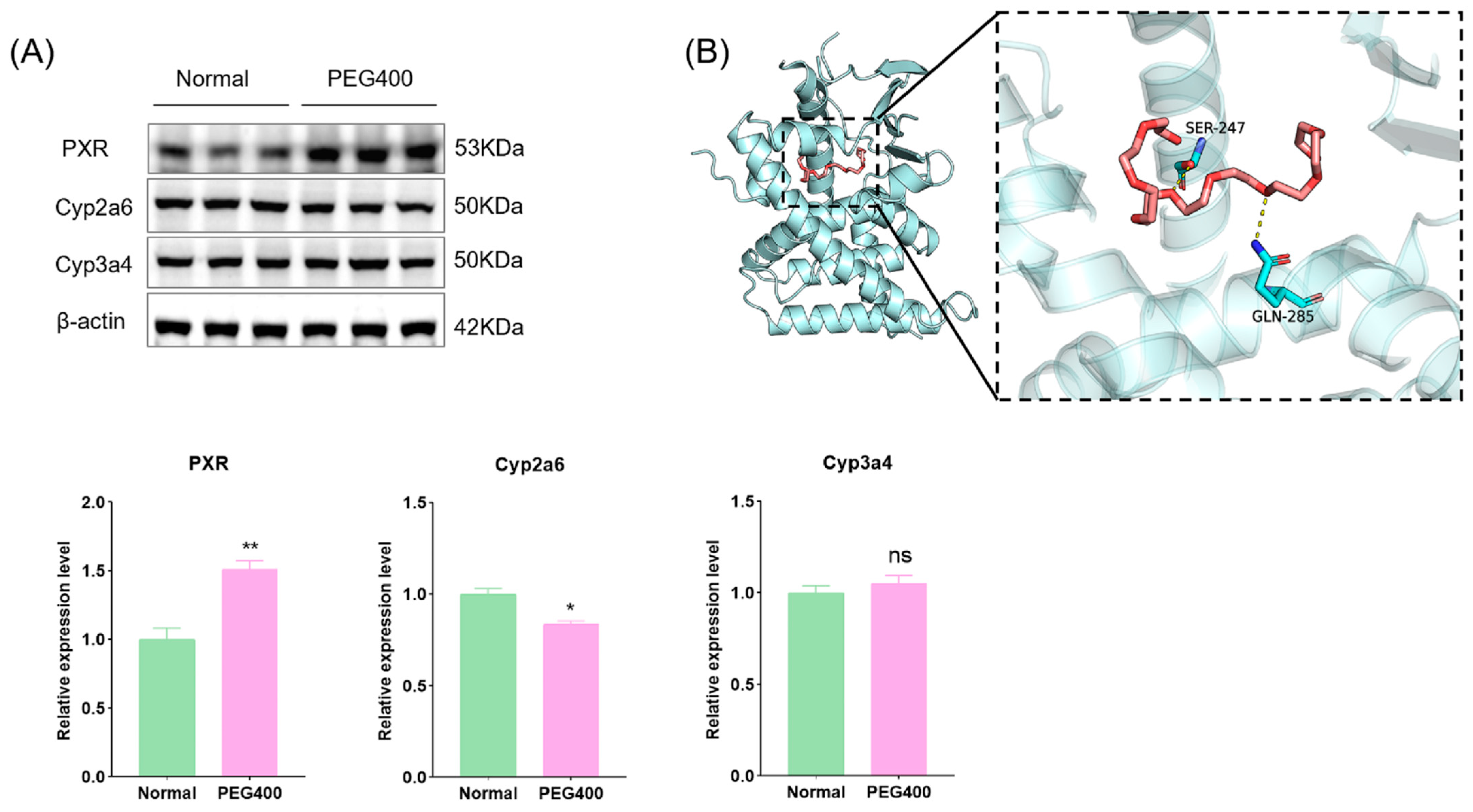
2.6. Effect of PEG400 on Metabolic Enzymes
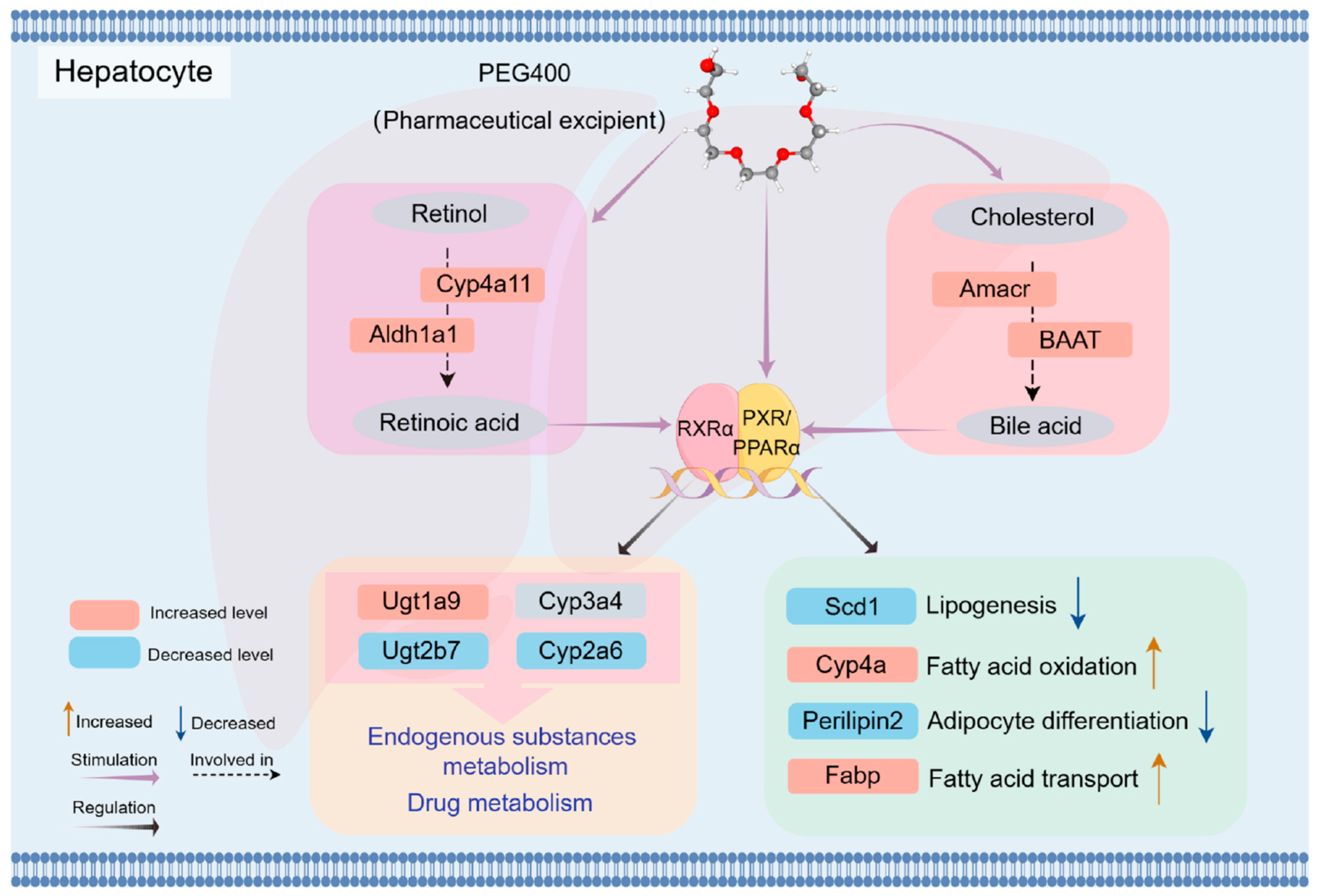
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Animal Experiments
4.3. Liver Histopathology
4.4. TMT Labeling Quantitative Proteomics Analysis
4.5. Determination of Retinoic Acid and Bile Acid
4.6. Molecular Docking
4.7. Western Blotting Verification
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalász, H.; Antal, I. Drug excipients. Curr. Med. Chem. 2006, 13, 2535–2563. [Google Scholar] [CrossRef]
- Flanagan, T. Potential for pharmaceutical excipients to impact absorption: A mechanistic review for BCS Class 1 and 3 drugs. Eur. J. Pharm. Biopharm. 2019, 141, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, Y.; He, Y.; Ren, T.; Yin, L.; Fawcett, J.P.; Gu, J.; Sun, H. Impact of molecular weight on the mechanism of cellular uptake of polyethylene glycols (PEGs) with particular reference to P-glycoprotein. Acta Pharm. Sin. B 2020, 10, 2002–2009. [Google Scholar] [CrossRef]
- Mudra, D.R.; Borchardt, R.T. Absorption barriers in the rat intestinal mucosa. 3: Effects of polyethoxylated solubilizing agents on drug permeation and metabolism. J. Pharm. Sci. 2010, 99, 1016–1027. [Google Scholar] [CrossRef]
- Li, L.; Yi, T.; Lam, C.W. Interactions between human multidrug resistance related protein (MRP2; ABCC2) and excipients commonly used in self-emulsifying drug delivery systems (SEDDS). Int. J. Pharm. 2013, 447, 192–198. [Google Scholar] [CrossRef]
- Mai, Y.; Dou, L.; Murdan, S.; Basit, A.W. An animal’s sex influences the effects of the excipient PEG 400 on the intestinal P-gp protein and mRNA levels, which has implications for oral drug absorption. Eur. J. Pharm. Sci. 2018, 120, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, R.; Matsuhisa, R.; Nomoto, M.; Chudan, S.; Nishikawa, M.; Tabuchi, Y.; Ikushiro, S.; Nagai, Y.; Furusawa, Y. Effect of Oral Administration of Polyethylene Glycol 400 on Gut Microbiota Composition and Diet-Induced Obesity in Mice. Microorganisms 2023, 11, 1882. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, P.; Sun, X.; Yang, D.; Zhang, S.; Meng, X.; Zhang, M.; Gao, X. Detrimental Impacts of Pharmaceutical Excipient PEG400 on Gut Microbiota and Metabolome in Healthy Mice. Molecules 2023, 28, 7562. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, S.; Zhang, M.; Meng, X.; Wang, P.; Zhang, R.; Gao, X. Study of Pharmaceutical Excipient PEG400 Alteration of Pharmacokinetics and Tissue Distribution of Main Flavonoids Metabolites of Baicalin. Curr. Pharm. Anal. 2021, 17, 609–623. [Google Scholar] [CrossRef]
- Shang, L.; Zhou, M.; Cao, S.; Zhang, M.; Wang, P.; Zhang, S.; Meng, X.; Yang, Q.; Gao, X. Effect of polyethylene glycol 400 on the pharmacokinetics and tissue distribution of baicalin by intravenous injection based on the enzyme activity of UGT1A8/1A9. Eur. J. Pharm. Sci. 2023, 180, 106328. [Google Scholar] [CrossRef]
- Cao, S.; Zhang, M.; Yuan, M.; Yang, D.; Zhao, M.; Zhang, S.; Wang, P.; Zhang, R.; Gao, X. The pharmaceutical excipient PEG400 affect the absorption of baicalein in Caco-2 monolayer model by interacting with UDP-glucuronosyltransferases and efflux transport proteins. Pharmacol. Res. Perspect. 2022, 10, e00928. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, M.; Zhao, M.; Li, C.; Shang, L.; Zhang, S.; Wang, P.; Gao, X. Study on the Effect of Pharmaceutical Excipient PEG400 on the Pharmacokinetics of Baicalin in Cells Based on MRP2, MRP3, and BCRP Efflux Transporters. Pharmaceutics 2024, 16, 731. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, F.; Gu, Y.; He, X.; Fan, N.; Zheng, Q.; Qin, S.; He, Z.; Wei, Y.; Song, X. Peg400-mediated nanocarriers improve the delivery and therapeutic efficiency of mRNA tumor vaccines. Chin. Chem. Lett. 2024, 35, 314–319. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Mcardle, A.J.; Menikou, S. What is proteomics? Arch. Dis. Child. Educ. Pract. Ed. 2021, 106, 178–181. [Google Scholar] [CrossRef]
- ALJohani, A.M.; Syed, D.N.; Ntambi, J.M. Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E.; Kaczocha, M. FABP5 as a novel molecular target in prostate cancer. Drug Discov. Today 2020, 20, 2056–2061. [Google Scholar] [CrossRef]
- Sun, X.; Yang, S.; Feng, X.; Zheng, Y.; Zhou, J.; Wang, H.; Zhang, Y.; Sun, H.; He, C. The modification of ferroptosis and abnormal lipometabolism through overexpression and knockdown of potential prognostic biomarker perilipin2 in gastric carcinoma. Gastric Cancer 2020, 23, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Kamata, S.; Honda, A.; Ishii, I. Current Clinical Trial Status and Future Prospects of PPAR-Targeted Drugs for Treating Nonalcoholic Fatty Liver Disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Esplugues, E.; Bort, A.; Cardelo, M.P.; Ruz-Maldonado, I.; Fernández-Tussy, P.; Wong, C.; Wang, H.; Ojima, I.; Kaczocha, M.; et al. Fatty acid binding protein 5 suppression attenuates obesity-induced hepatocellular carcinoma by promoting ferroptosis and intratumoral immune rewiring. Nat. Metab. 2024, 6, 741–763. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Luo, Y.; Yan, N.; Hamada, K.; Zhao, N.; Xia, Y.; Wang, P.; Zhao, C.; Qi, D.; Yang, S.; et al. Intestinal peroxisome proliferator-activated receptor α-fatty acid-binding protein 1 axis modulates nonalcoholic steatohepatitis. Hepatology 2023, 77, 239–255. [Google Scholar] [CrossRef]
- Ning, M.; Tao, Y.; Hu, X.; Guo, L.; Ni, J.; Hu, J.; Shen, H.; Chen, Y. Roles of UGT2B7 C802T gene polymorphism on the efficacy of morphine treatment on cancer pain among the Chinese han population. Niger. J. Clin. Pract. 2019, 22, 1319–1323. [Google Scholar] [CrossRef]
- Li, C.; Zhang, M.; Zhao, Y.; Yang, D.; Zhao, M.; Shang, L.; Sun, X.; Zhang, S.; Wang, P.; Gao, X. Exploring the Impact of Pharmaceutical Excipient PEG400 on the Pharmacokinetics of Mycophenolic Acid Through In Vitro and In Vivo Experiments. Int. J. Mol. Sci. 2024, 26, 72. [Google Scholar] [CrossRef]
- Gutierrez-Mazariegos, J.; Schubert, M.; Laudet, V. Evolution of retinoic acid receptors and retinoic acid signaling. Subcell. Biochem. 2014, 70, 55–73. [Google Scholar] [CrossRef]
- Kumar, S.; Duester, G. SnapShot: Retinoic acid signaling. Cell 2011, 147, 1422.e1. [Google Scholar] [CrossRef]
- Bian, Y.; Shan, G.; Bi, G.; Liang, J.; Hu, Z.; Sui, Q.; Shi, H.; Zheng, Z.; Yao, G.; Wang, Q.; et al. Targeting ALDH1A1 to enhance the efficacy of KRAS-targeted therapy through ferroptosis. Redox Biol. 2024, 77, 103361. [Google Scholar] [CrossRef]
- Petkovich, M. Regulation of gene expression by vitamin A: The role of nuclear retinoic acid receptors. Annu. Rev. Nutr. 1992, 12, 443–471. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signaling and neuronal differentiation. Cell Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef]
- Menéndez-Gutiérrez, M.P.; Rőszer, T.; Fuentes, L.; Núñez, V.; Escolano, A.; Redondo, J.M.; De Clerck, N.; Metzger, D.; Valledor, A.F.; Ricote, M. Retinoid X receptors orchestrate osteoclast differentiation and postnatal bone remodeling. J. Clin. Investig. 2015, 125, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Suga, T.; Yamaguchi, H.; Ogura, J.; Shoji, S.; Maekawa, M.; Mano, N. Altered bile acid composition and disposition in a mouse model of non-alcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 2019, 379, 114664. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.J.; Schmitz, W.; Nair, R.R.; Selkälä, E.M.; Sormunen, R.T.; Miinalainen, I.J.; Crick, P.J.; Wang, Y.; Griffiths, W.J.; Reddy, J.K.; et al. Role of AMACR (α-methylacyl-CoA racemase) and MFE-1 (peroxisomal multifunctional enzyme-1) in bile acid synthesis in mice. Biochem. J. 2014, 461, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Schoonjans, K. Metabolic Messengers: Bile acids. Nat. Metab. 2022, 4, 416–423. [Google Scholar] [CrossRef]
- Zhang, Y.; Edwards, P.A. FXR signaling in metabolic disease. FEBS Lett. 2008, 582, 10–18. [Google Scholar] [CrossRef]
- Chen, L.; Jiao, T.; Liu, W.; Luo, Y.; Wang, J.; Guo, X.; Tong, X.; Lin, Z.; Sun, C.; Wang, K.; et al. Hepatic cytochrome P450 8B1 and cholic acid potentiate intestinal epithelial injury in colitis by suppressing intestinal stem cell renewal. Cell Stem Cell 2022, 29, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.N.; Chen, C. The role of CYP3A4 in the biotransformation of bile acids and therapeutic implication for cholestasis. Ann. Transl. Med. 2014, 2, 7. [Google Scholar] [CrossRef]
- Yuan, X.; Lu, H.; Zhao, A.; Ding, Y.; Min, Q.; Wang, R. Transcriptional regulation of CYP3A4 by nuclear receptors in human hepatocytes under hypoxia. Drug Metab. Rev. 2020, 52, 225–234. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Woody, S.; Sun, M.; Cui, W. Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver. Drug Metab. Rev. 2013, 45, 48–59. [Google Scholar] [CrossRef]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Korashy, H.M.; Shayeganpour, A.; Brocks, D.R.; El-Kadi, A.O. Induction of cytochrome P450 1A1 by ketoconazole and itraconazole but not fluconazole in murine and human hepatoma cell lines. Toxicol. Sci. 2007, 97, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, P.I.; Hu, D.G.; Gardner-Stephen, D.A. The regulation of UDP-glucuronosyltransferase genes by tissue-specific and ligand-activated transcription factors. Drug Metab. Rev. 2010, 42, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.T.; Chen, T. PXR variants: The impact on drug metabolism and therapeutic responses. Acta Pharm. Sin. B 2016, 6, 441–449. [Google Scholar] [CrossRef]
- Yang, J.; Luan, X.; Gui, H.; Yan, P.; Yang, D.; Song, X.; Liu, W.; Hu, G.; Yan, B. Byakangelicin induces cytochrome P450 3A4 expression via transactivation of pregnane X receptors in human hepatocytes. Br. J. Pharmacol. 2011, 162, 441–451. [Google Scholar] [CrossRef]
- Song, P.; Shen, X. Proteomic analysis of liver in diet-induced Hyperlipidemic mice under Fructus Rosa roxburghii action. J. Proteom. 2020, 230, 103982. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Western Blot Dilution | Item No. |
|---|---|---|---|
| Aldh1a1 | Rabbit | 1:2000 | IPB0967 |
| Cyp4a11 | Rabbit | 1:1000 | HA722348 |
| Ugt2b7 | Rabbit | 1:2000 | 16661-1-AP |
| Amacr | Mouse | 1:1000 | IMB0226 |
| BAAT | Rabbit | 1:2000 | IPB6862 |
| ARαPP | Rabbit | 1:2000 | IPB0660 |
| RXRα | Rabbit | 1:2000 | IPB8970 |
| Perilipin2 | Rabbit | 1:1000 | ET1704-17 |
| Scd1 | Mouse | 1:1000 | HA601180 |
| Fabp5 | Rabbit | 1:2000 | IPB7383 |
| Ugt1a9 | Rabbit | 1:1000 | TD6537 |
| Cyp3a4 | Rabbit | 1:500 | IPB2432 |
| Cyp2a6 | Rabbit | 1:500 | IPB0396 |
| PXR | Rabbit | 1:2000 | IPB0584 |
| GAPDH | Rabbit | 1:8000 | ET1601-4 |
| β-actin | Rabbit | 1:5000 | GB15003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Cao, S.; Yang, D.; Shang, L.; Hang, Y.; Wang, P.; Zhang, S.; Li, C.; Zhang, M.; Gao, X. Proteomics Profiling Reveals Pharmaceutical Excipient PEG400 Induces Nuclear-Receptor-Activation-Affected Lipid Metabolism and Metabolic Enzyme Expression. Int. J. Mol. Sci. 2025, 26, 1732. https://doi.org/10.3390/ijms26041732
Zhao M, Cao S, Yang D, Shang L, Hang Y, Wang P, Zhang S, Li C, Zhang M, Gao X. Proteomics Profiling Reveals Pharmaceutical Excipient PEG400 Induces Nuclear-Receptor-Activation-Affected Lipid Metabolism and Metabolic Enzyme Expression. International Journal of Molecular Sciences. 2025; 26(4):1732. https://doi.org/10.3390/ijms26041732
Chicago/Turabian StyleZhao, Mei, Siyuan Cao, Dan Yang, Leyuan Shang, Ye Hang, Pengjiao Wang, Shuo Zhang, Chaoji Li, Min Zhang, and Xiuli Gao. 2025. "Proteomics Profiling Reveals Pharmaceutical Excipient PEG400 Induces Nuclear-Receptor-Activation-Affected Lipid Metabolism and Metabolic Enzyme Expression" International Journal of Molecular Sciences 26, no. 4: 1732. https://doi.org/10.3390/ijms26041732
APA StyleZhao, M., Cao, S., Yang, D., Shang, L., Hang, Y., Wang, P., Zhang, S., Li, C., Zhang, M., & Gao, X. (2025). Proteomics Profiling Reveals Pharmaceutical Excipient PEG400 Induces Nuclear-Receptor-Activation-Affected Lipid Metabolism and Metabolic Enzyme Expression. International Journal of Molecular Sciences, 26(4), 1732. https://doi.org/10.3390/ijms26041732