
Identification of the B7-H3 Interaction Partners Using a Proximity Labeling Strategy

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
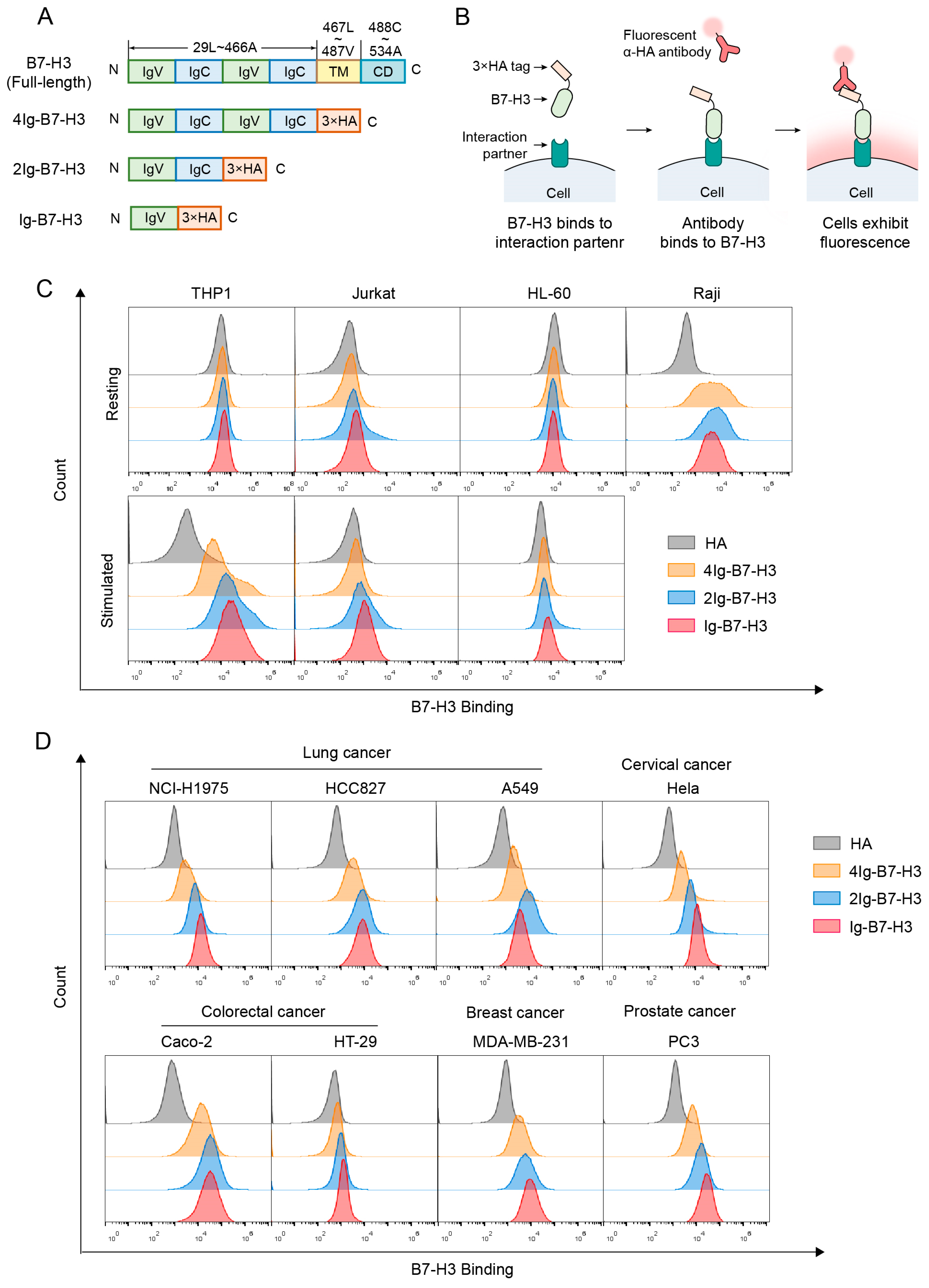
2.1. B7-H3 Strongly Binds to Resting Raji Cells and Stimulated THP-1 Cells
2.2. The Interaction Partner(s) of B7-H3 Are Widely Present on Cancer Cells
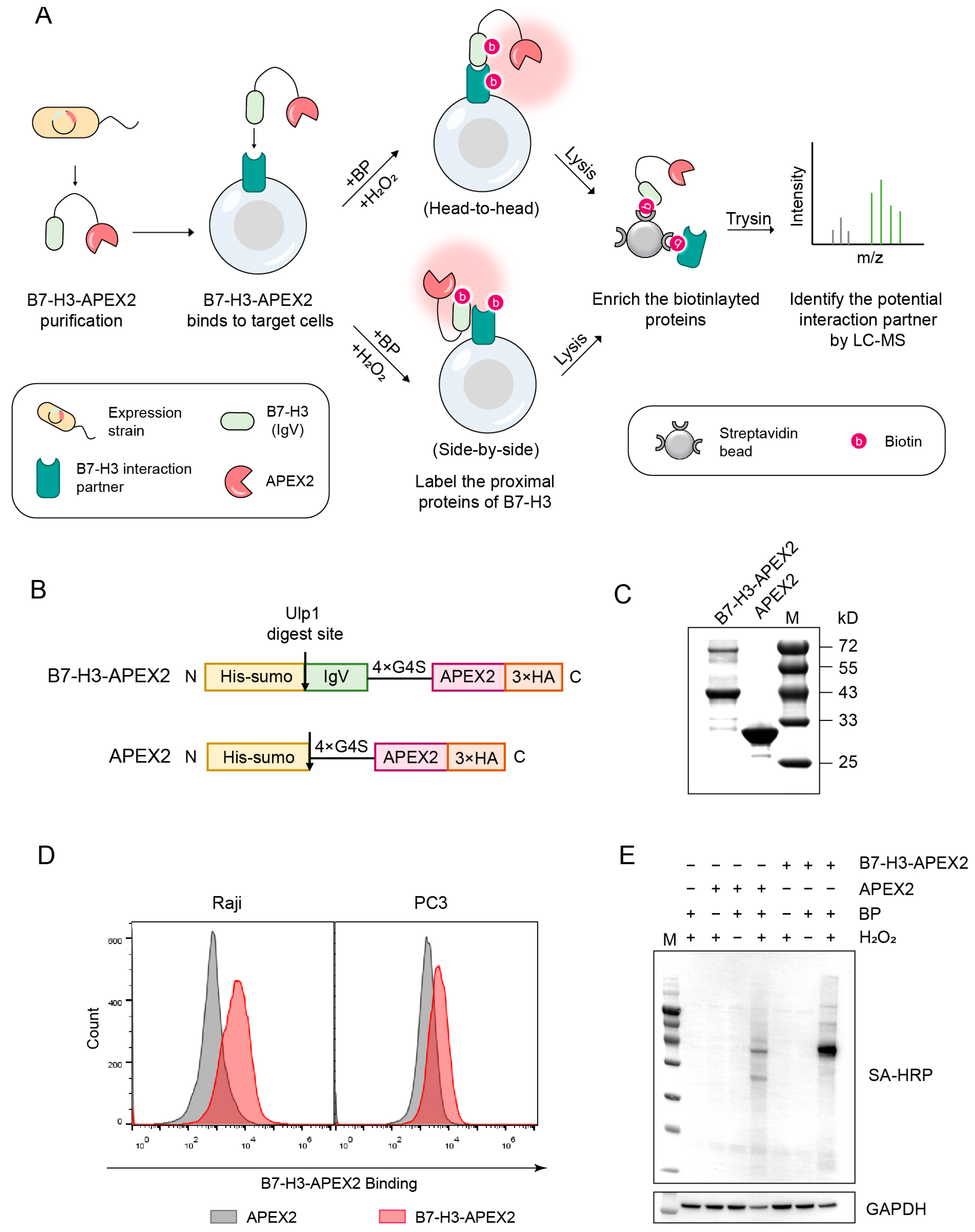
2.3. An APEX2-Based Proximity Labeling Approach for Identifying B7-H3 Interaction Partner(s)
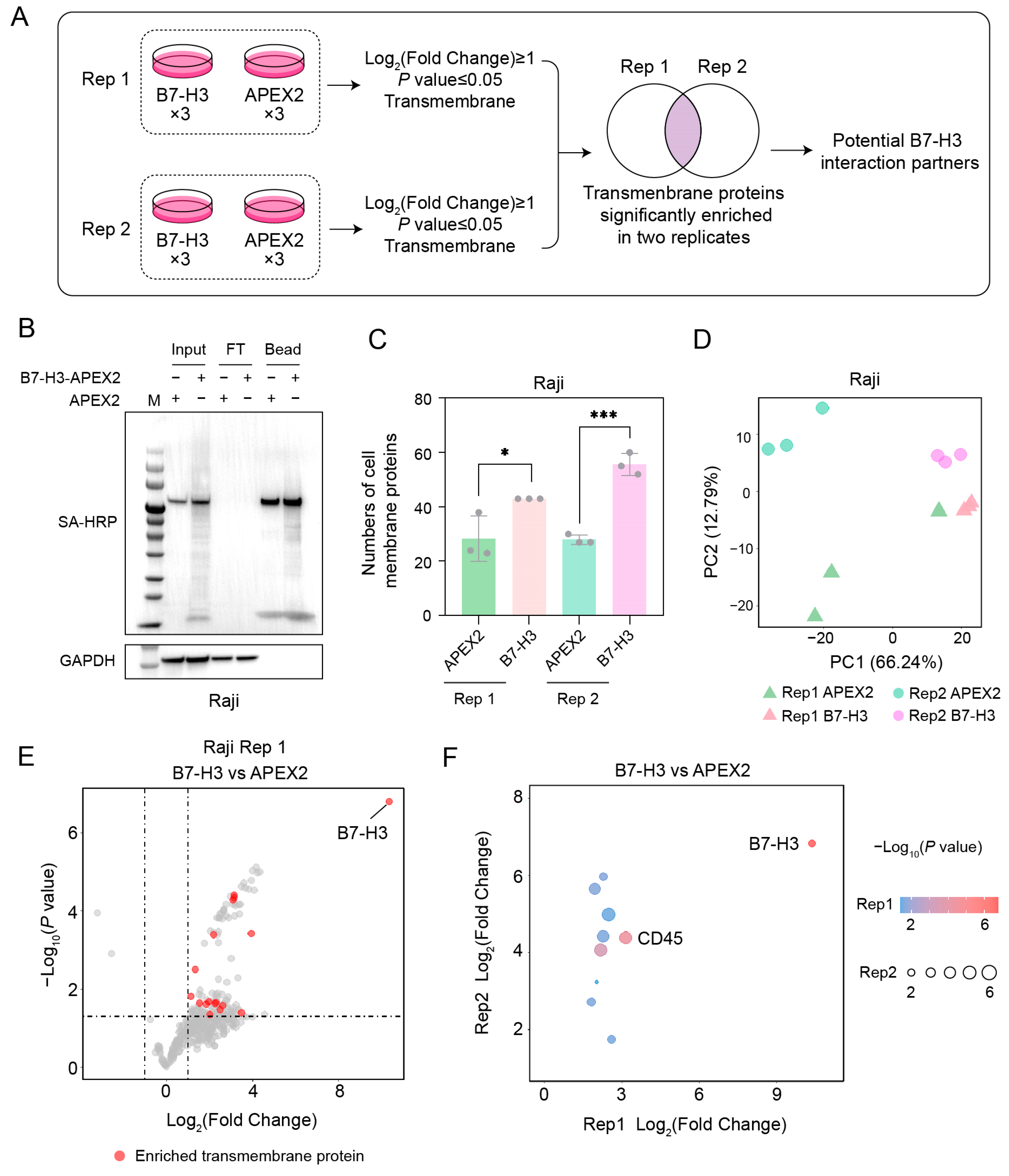
2.4. CD45 Is a Potential B7-H3 Receptor on Raji Cells
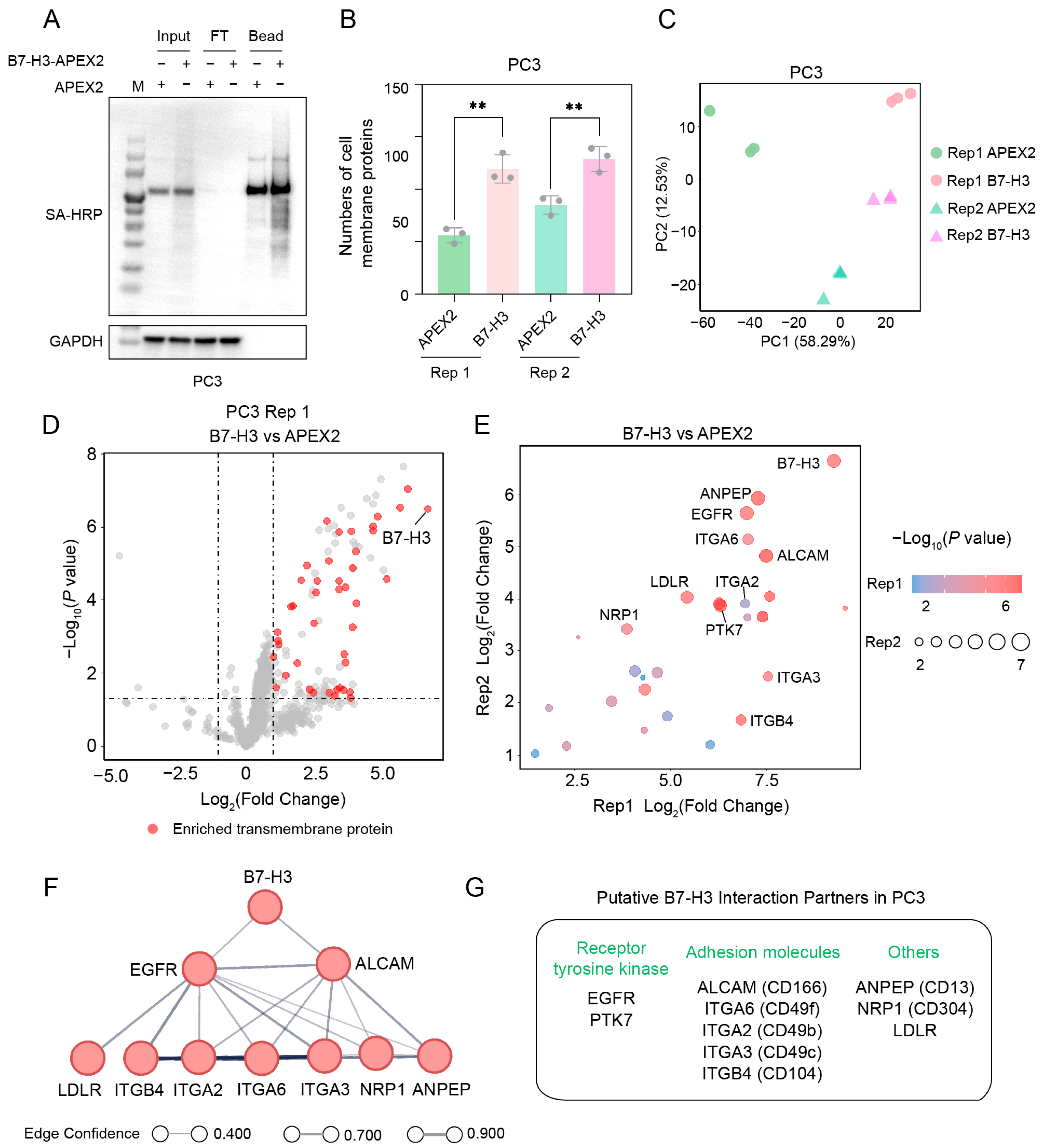
2.5. Identification of B7-H3 Interaction Partner(s) on PC3 Cells
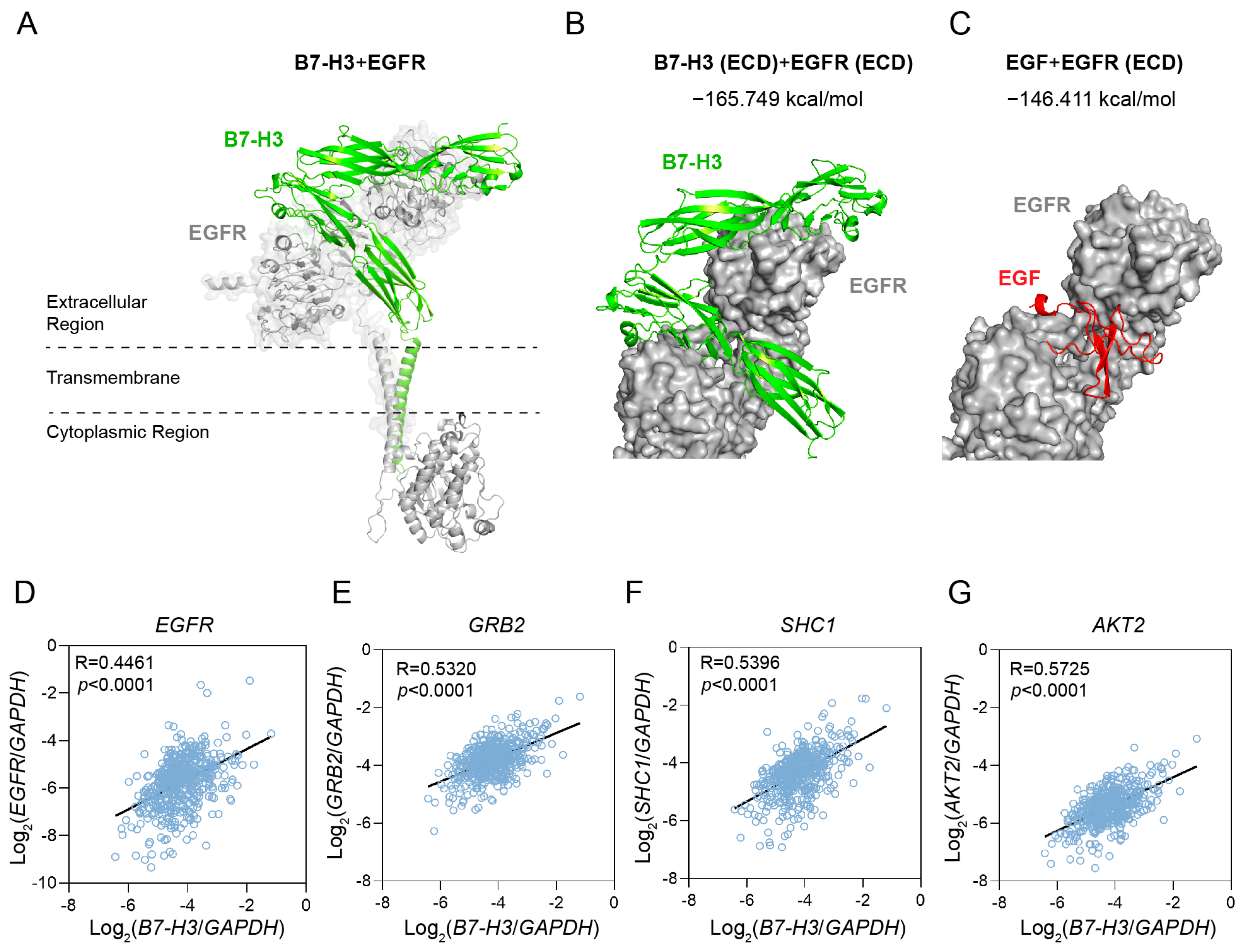
2.6. B7-H3 Is Predicted to Bind to the EGF Binding Pocket of EGFR
3. Discussion
4. Materials and Methods
4.1. Construct Design
4.2. Cell Lines and Culture Conditions
4.3. Immune Cell Stimulation
4.4. Protein Purification
4.5. Flow Cytometry
4.6. APEX2 Labeling In Vitro
4.7. Western Blotting
4.8. Proximity Labeling In Vivo
4.9. Pulldown and Sample Preparation for Mass Spectrometry
4.10. Mass Spectrometry
4.11. Data Analysis
4.12. Structure Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef]
- Suh, W.K.; Gajewska, B.U.; Okada, H.; Gronski, M.A.; Bertram, E.M.; Dawicki, W.; Duncan, G.S.; Bukczynski, J.; Plyte, S.; Elia, A.; et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 2003, 4, 899–906. [Google Scholar] [CrossRef]
- Vigdorovich, V.; Ramagopal, U.A.; Lázár-Molnár, E.; Sylvestre, E.; Lee, J.S.; Hofmeyer, K.A.; Zang, X.; Nathenson, S.G.; Almo, S.C. Structure and T cell inhibition properties of B7 family member, B7-H3. Structure 2013, 21, 707–717. [Google Scholar] [CrossRef]
- Dong, P.X.; Xiong, Y.; Yue, J.M.; Hanley, S.J.B.; Watari, H. B7H3 as a promoter of metastasis and promising therapeutic target. Front. Oncol. 2018, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Getu, A.A.; Tigabu, A.; Zhou, M.; Lu, J.; Fodstad, Ø.; Tan, M. New frontiers in immune checkpoint B7-H3 (CD276) research and drug development. Mol. Cancer 2023, 22, 43. [Google Scholar] [CrossRef]
- Loos, M.; Hedderich, D.M.; Ottenhausen, M.; Giese, N.A.; Laschinger, M.; Esposito, I.; Kleeff, J.; Friess, H. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer 2009, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An attractive target for antibody-based immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.D.; Sica, G.L.; Zhu, G.F.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-γ production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Leitner, J.; Klauser, C.; Pickl, W.F.; Stöckl, J.; Majdic, O.; Bardet, A.F.; Kreil, D.P.; Dong, C.; Yamazaki, T.; Zlabinger, G.; et al. B7-H3 is a potent inhibitor of human T-cell activation: No evidence for B7-H3 and TREML2 interaction. Eur. J. Immunol. 2009, 39, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Ho, K.-H.; Huang, T.-W.; Shih, C.-M.; Hsu, S.-Y.; Liu, A.-J.; Chen, K.-C. A regulatory loop among CD276, miR-29c-3p, and Myc exists in cancer cells against natural killer cell cytotoxicity. Life Sci. 2021, 277, 119438. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Chen, L.J.; Wang, F.M.; Zhu, D.W.; Ge, X.S.; Hua, D.; Sun, J. Cancer cell-expressed B7-H3 regulates the differentiation of tumor-associated macrophages in human colorectal carcinoma. Oncol. Lett. 2017, 14, 6177–6183. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Liu, H.; da Silva, L.M.; Arora, R.; Liu, Z.X.; Phillips, J.B.; Schmitt, D.C.; Vu, T.; McClellan, S.; Lin, Y.F.; et al. Immunoregulatory protein B7-H3 reprograms glucose metabolism in cancer cells by ROS-mediated stabilization of HIF1α. Cancer Res. 2016, 76, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, Y.; Zhan, S.; Zhang, G.; Cao, L.; Zhang, X.; Shi, T.; Chen, W. B7-H3 promotes colorectal cancer angiogenesis through activating the NF-κB pathway to induce VEGFA expression. Cell Death Dis. 2020, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Ding, M.; Zhou, N.N.; Yang, Y.; Chen, L.W. B7-H3 promotes the epithelial-mesenchymal transition of NSCLC by targeting SIRT1 through the PI3K/AKT pathway. Mol. Med. Rep. 2022, 25, 79. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, W.; Phillips, J.B.; Arora, R.; McClellan, S.; Li, J.; Kim, J.-H.; Sobol, R.W.; Tan, M. Immunoregulatory protein B7-H3 regulates cancer stem cell enrichment and drug resistance through MVP-mediated MEK activation. Oncogene 2019, 38, 88–102. [Google Scholar] [CrossRef]
- Hashiguchi, M.; Kobori, H.; Ritprajak, P.; Kamimura, Y.; Kozono, H.; Azuma, M. Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses. Proc. Natl. Acad. Sci. USA 2008, 105, 10495–10500. [Google Scholar] [CrossRef] [PubMed]
- Husain, B.; Ramani, S.R.; Chiang, E.; Lehoux, I.; Paduchuri, S.; Arena, T.A.; Patel, A.; Wilson, B.; Chan, P.; Franke, Y.; et al. A platform for extracellular interactome discovery identifies novel functional binding partners for the immune receptors B7-H3/CD276 and PVR/CD155. Mol. Cell Proteom. 2019, 18, 2310–2323. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Peterson, S.M.; Müller, S.; Reichelt, M.; McRoberts Amador, C.; Martinez-Martin, N. A membrane protein display platform for receptor interactome discovery. Proc. Natl. Acad. Sci. USA 2021, 118, e2025451118. [Google Scholar] [CrossRef] [PubMed]
- Oslund, R.C.; Reyes-Robles, T.; White, C.H.; Tomlinson, J.H.; Crotty, K.A.; Bowman, E.P.; Chang, D.; Peterson, V.M.; Li, L.X.; Frutos, S.; et al. Detection of cell-cell interactions via photocatalytic cell tagging. Nat. Chem. Biol. 2022, 18, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, J.; Fijalkowska, D.; Eyckerman, S.; Gevaert, K. Mass spectrometry and the cellular surfaceome. Mass Spectrom. Rev. 2022, 41, 804–841. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Meth. 2021, 18, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Meth. 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Li, X.; Zhou, J.; Zhao, W.; Wen, Q.; Wang, W.; Peng, H.; Gao, Y.; Bouchonville, K.J.; Offer, S.M.; Chan, K.; et al. Defining proximity proteomics of histone modifications by antibody-mediated protein A-APEX2 labeling. Genom. Proteom. Bioinform. 2021, 20, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Yu, J.; Cao, Z.; Zhang, B.; Li, S.; Xin, Y.; Yang, L.; Lou, R.; Zhuang, M.; Shui, W. Endogenous cell membrane interactome mapping for the GLP-1 receptor in different cell types. Nat. Chem. Biol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, E.; Qian, D.; Enica, A.; Thakkar, K.; Augustine, M.; Gamble, S.; Reading, J.L.; Litchfield, K. A pan-cancer single-cell RNA-seq atlas of intratumoral B cells. Cancer Cell 2024, 42, 1784–1797. [Google Scholar] [CrossRef]
- Volkov, D.V.; Stepanova, V.M.; Rubtsov, Y.P.; Gabibov, A.G. Protein tyrosine phosphatase CD45 as an immunity regulator and a potential effector of CAR-T therapy. Acta Naturae 2023, 15, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.K.; Mine, J.A.; Biswas, S.; Chaurio, R.A.; Perales-Puchalt, A.; Anadon, C.M.; Costich, T.L.; Harro, C.M.; Walrath, J.; Ming, Q.; et al. BTN3A1 governs antitumor responses by coordinating αβ and γδ T cells. Science 2020, 369, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhang, D.; Wang, F.; Wang, Q.; Wu, Y.; Gou, M.; Hu, Y.; Zhang, W.; Huang, J.; Gong, Y.; et al. ALCAM-EGFR interaction regulates myelomagenesis. Blood Adv. 2021, 5, 5269–5282. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Geri, J.B.; Oakley, J.V.; Reyes-Robles, T.; Wang, T.; McCarver, S.J.; White, C.H.; Rodriguez-Rivera, F.P.; Parker, D.L.; Hett, E.C.; Fadeyi, O.O.; et al. Microenvironment mapping via Dexter energy transfer on immune cells. Science 2020, 367, 1091–1097. [Google Scholar] [CrossRef]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Ma, X.; Yang, Y.; Qu, Y.; Li, X.; Zhu, X.; Ma, W.; Duan, J.; Xue, J.; Yang, H.; et al. Phosphoantigens glue butyrophilin 3A1 and 2A1 to activate Vγ9Vδ2 T cells. Nature 2023, 621, 840–848. [Google Scholar] [CrossRef]
- Chen, L.-C.; Yang, P.-C.; Chen, C.-Y.; Chiang, S.-F.; Chen, T.-W.; Chen, W.T.-L.; Ke, T.-W.; Liang, J.-A.; Shiau, A.C.; Chao, K.S.C.; et al. Dual inhibition of B7-H3 and EGFR overcomes acquired chemoresistance in colon adenocarcinoma. J. Cancer 2024, 15, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Liao, H.; Zhou, N.; Yang, Y.; Guan, S.; Chen, L. B7-H3-induced signaling in lung adenocarcinoma cell lines with divergent epidermal growth factor receptor mutation patterns. BioMed Res. Int. 2020, 2020, 8824805. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.; Xiao, N.; Lu, Y.; Fu, Y.; Yang, C.; Li, M.; Wu, S.; et al. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2022, 50, D1522–D1527. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, S.; Huang, J.; Lupala, C.S.; Li, X.; Li, X.; Li, N. Identification of the B7-H3 Interaction Partners Using a Proximity Labeling Strategy. Int. J. Mol. Sci. 2025, 26, 1731. https://doi.org/10.3390/ijms26041731
Liao S, Huang J, Lupala CS, Li X, Li X, Li N. Identification of the B7-H3 Interaction Partners Using a Proximity Labeling Strategy. International Journal of Molecular Sciences. 2025; 26(4):1731. https://doi.org/10.3390/ijms26041731
Chicago/Turabian StyleLiao, Shujie, Jiamin Huang, Cecylia S. Lupala, Xiangcheng Li, Xuefei Li, and Nan Li. 2025. "Identification of the B7-H3 Interaction Partners Using a Proximity Labeling Strategy" International Journal of Molecular Sciences 26, no. 4: 1731. https://doi.org/10.3390/ijms26041731
APA StyleLiao, S., Huang, J., Lupala, C. S., Li, X., Li, X., & Li, N. (2025). Identification of the B7-H3 Interaction Partners Using a Proximity Labeling Strategy. International Journal of Molecular Sciences, 26(4), 1731. https://doi.org/10.3390/ijms26041731