
In Silico Identification of Potential Clovibactin-like Antibiotics Binding to Unique Cell Wall Precursors in Diverse Gram-Positive Bacterial Strains

 , , , , ,
, , , , ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Data Collection
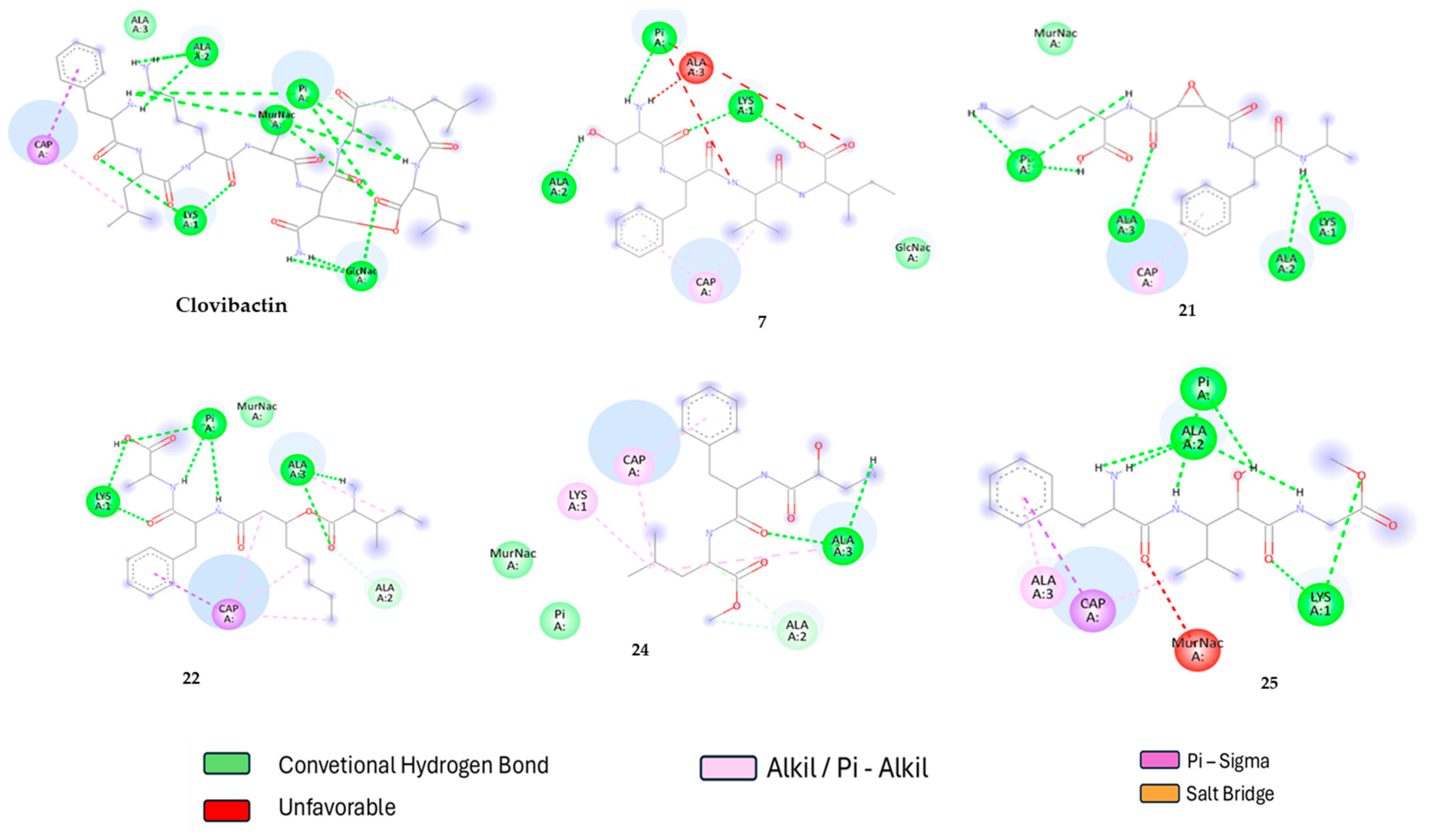
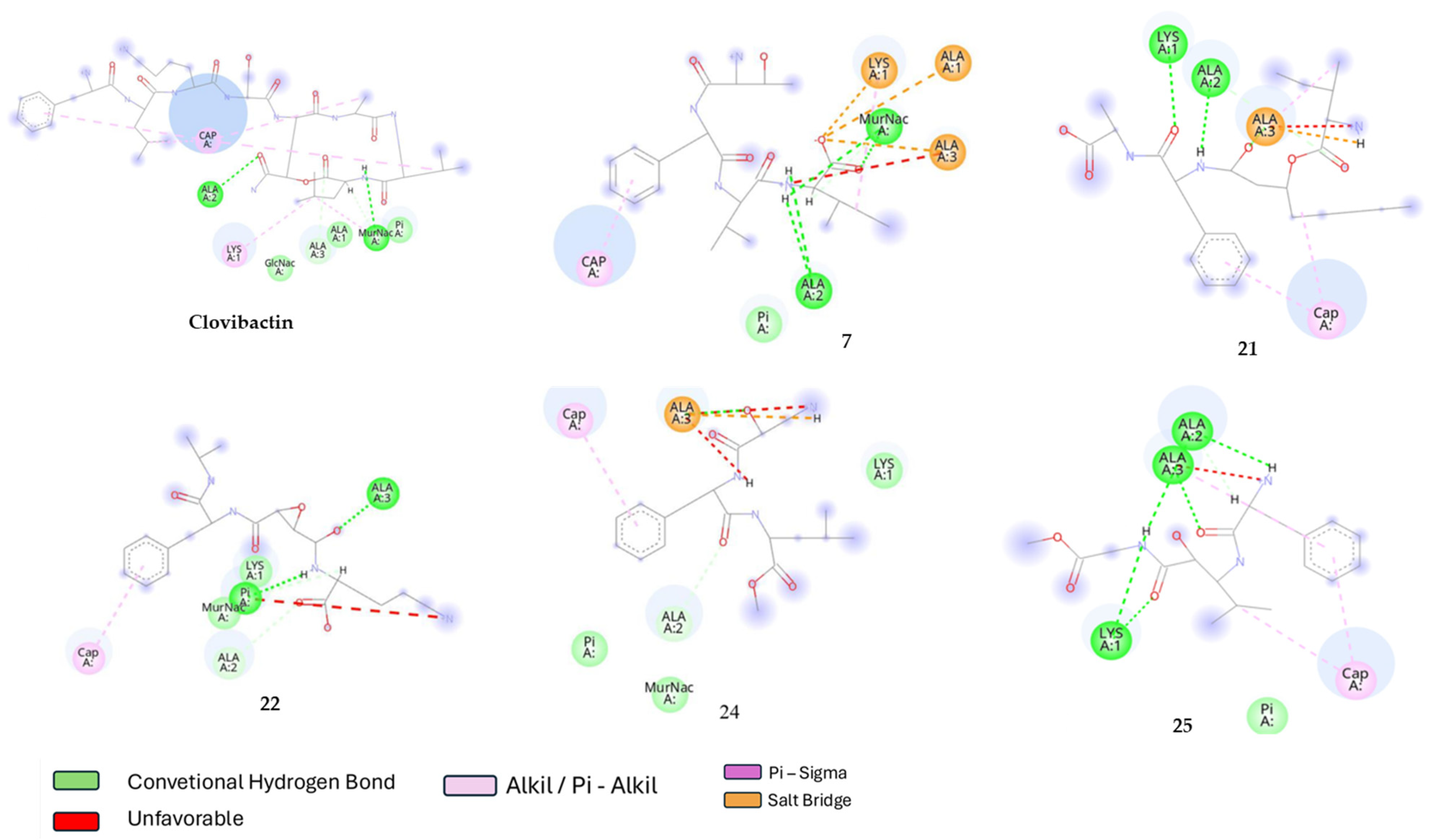
2.2. Molecular Docking
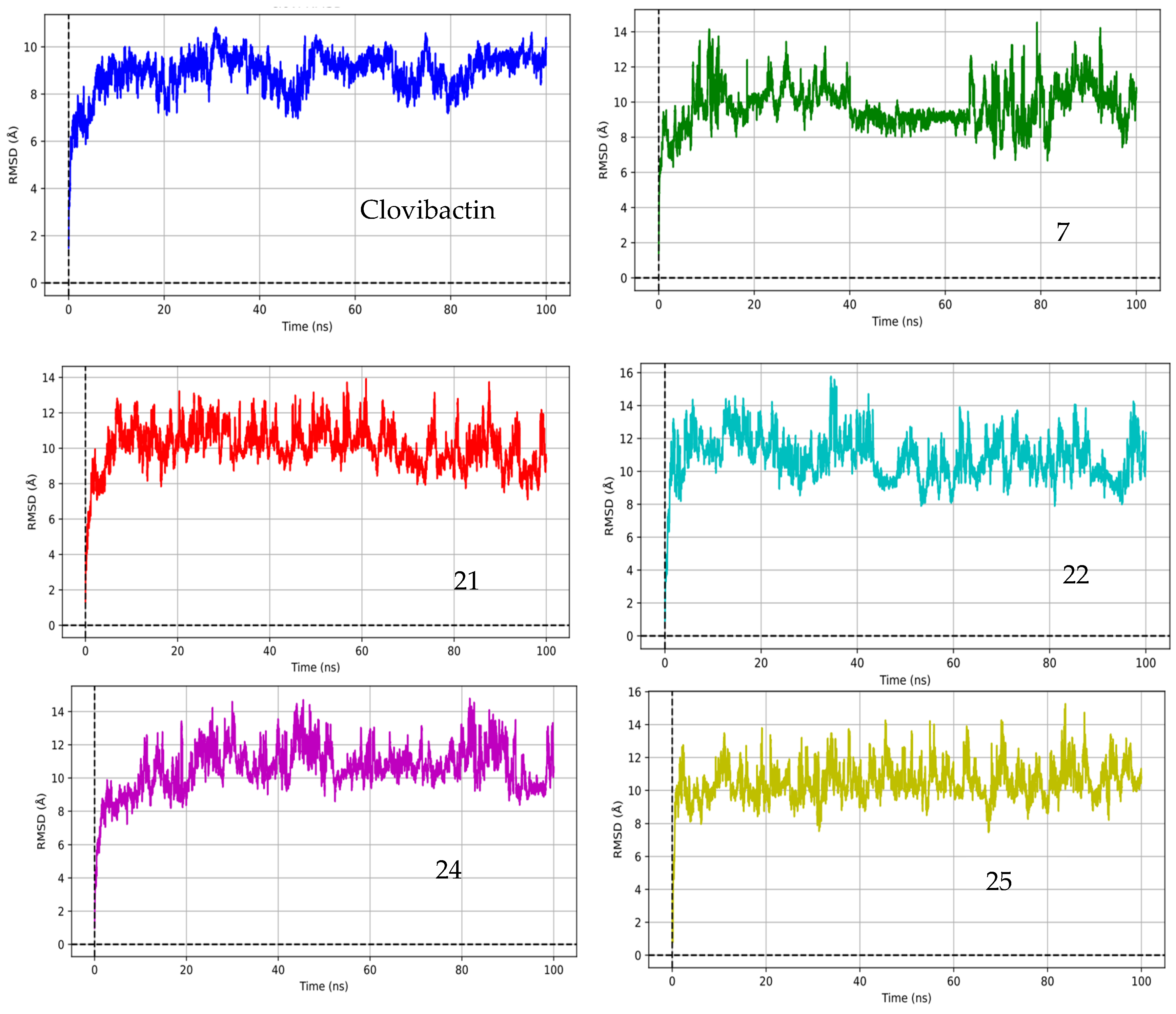
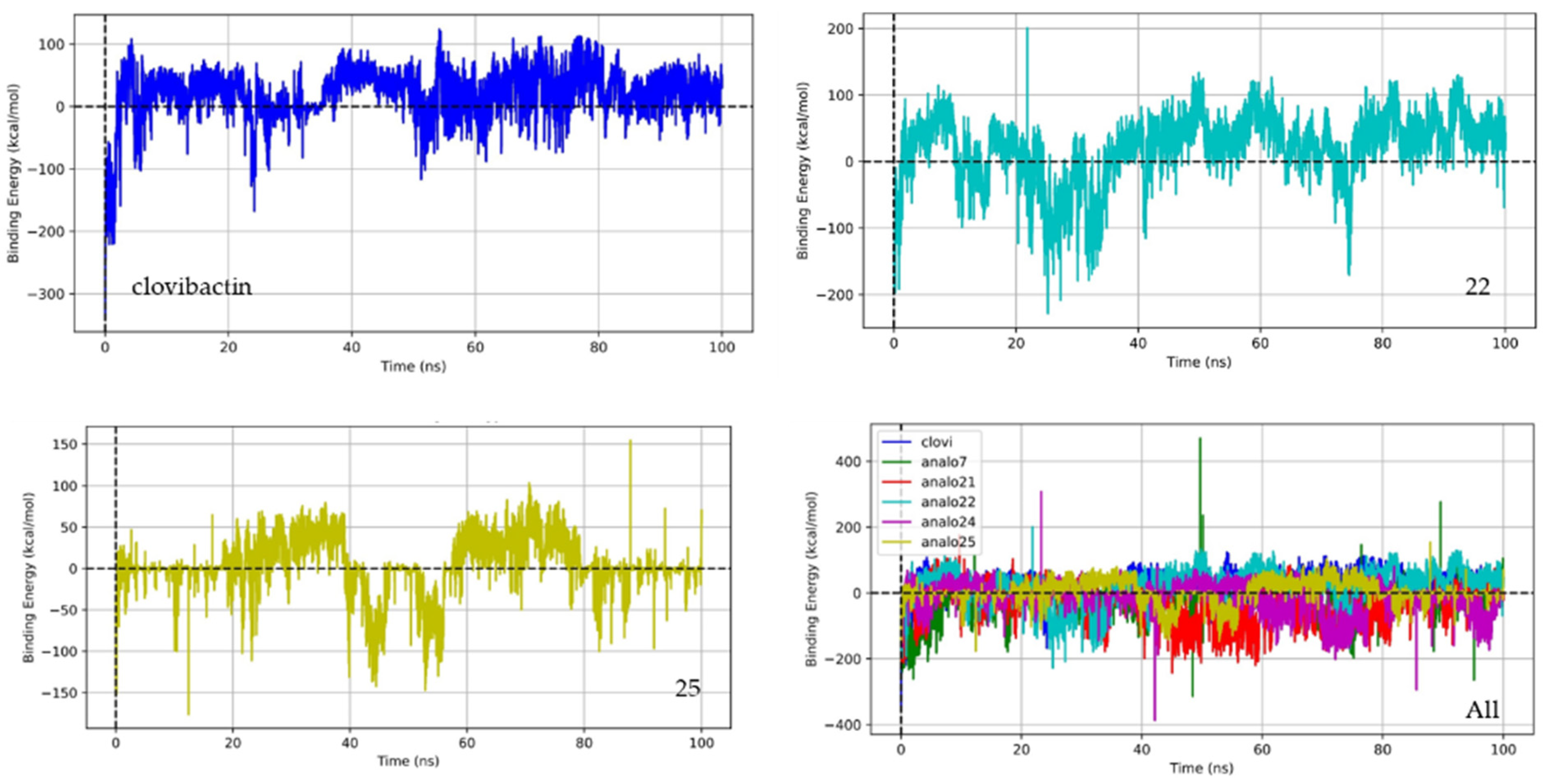
2.3. Molecular Dynamic
3. Materials and Methods
3.1. Data Collection
3.2. Minimum Energy Structures
3.3. ADME-Tox Properties
3.4. Molecular Docking
3.4.1. The Preparation of Ligands and Targets
3.4.2. Docking Protocol
3.5. Molecular Dynamics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilham, E.L.; Pearce-Smith, N.; Carter, V.; Ashiru-Oredope, D. Assessment of Global Antimicrobial Resistance Campaigns Conducted to Improve Public Awareness and Antimicrobial Use Behaviours: A Rapid Systematic Review. BMC Public Health 2024, 24, 396. [Google Scholar] [CrossRef] [PubMed]
- Sijbom, M.; Büchner, F.L.; Saadah, N.H.; Numans, M.E.; De Boer, M.G.J. Trends in Antibiotic Selection Pressure Generated in Primary Care and Their Association with Sentinel Antimicrobial Resistance Patterns in Europe. J. Antimicrob. Chemother. 2023, 78, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Hughes, D. Selection and Evolution of Resistance to Antimicrobial Drugs. IUBMB Life 2014, 66, 521–529. [Google Scholar] [CrossRef]
- Albrich, W.C.; Monnet, D.L.; Harbarth, S. Antibiotic Selection Pressure and Resistance in Streptococcus pneumoniae and Streptococcus pyogenes. Emerg. Infect. Dis. 2004, 10, 514–517. [Google Scholar] [CrossRef]
- Vestergaard, M.; Frees, D.; Ingmer, H. Antibiotic Resistance and the MRSA Problem. Microbiol. Spectr. 2019, 7, 10-1128. [Google Scholar] [CrossRef]
- Maraolo, A.E.; Giaccone, A.; Gentile, I.; Saracino, A.; Bavaro, D.F. Daptomycin versus Vancomycin for the Treatment of Methicillin-Resistant Staphylococcus Aureus Bloodstream Infection with or without Endocarditis: A Systematic Review and Meta-Analysis. Antibiotics 2021, 10, 1014. [Google Scholar] [CrossRef]
- Garbo, V.; Venuti, L.; Boncori, G.; Albano, C.; Condemi, A.; Natoli, G.; Frasca Polara, V.; Billone, S.; Canduscio, L.A.; Cascio, A.; et al. Severe Panton–Valentine-Leukocidin-Positive Staphylococcus Aureus Infections in Pediatric Age: A Case Report and a Literature Review. Antibiotics 2024, 13, 1192. [Google Scholar] [CrossRef]
- Li, G.; Walker, M.J.; De Oliveira, D.M.P. Vancomycin Resistance in Enterococcus and Staphylococcus Aureus. Microorganisms 2022, 11, 24. [Google Scholar] [CrossRef]
- Berbel, D.; González-Díaz, A.; López de Egea, G.; Càmara, J.; Ardanuy, C. An Overview of Macrolide Resistance in Streptococci: Prevalence, Mobile Elements and Dynamics. Microorganisms 2022, 10, 2316. [Google Scholar] [CrossRef]
- Graham, D.Y. Crises in Antimicrobial Stewardship: Misuse of Clarithromycin for Helicobacter Pylori Therapy. Pharmacoepidemiology 2024, 3, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.A.; Shie, C.B.; Hsu, P.I. Update on the First-Line Treatment of Helicobacter Pylori Infection in Areas with High and Low Clarithromycin Resistances. Ther. Adv. Gastroenterol. 2022, 15, 17562848221138168. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Antunes, W.; Mota, S.; Madureira-Carvalho, Á.; Dinis-Oliveira, R.J.; Dias da Silva, D. An Overview of the Recent Advances in Antimicrobial Resistance. Microorganisms 2024, 12, 1920. [Google Scholar] [CrossRef] [PubMed]
- Chinemerem Nwobodo, D.; Ugwu, M.C.; Oliseloke Anie, C.; Al-Ouqaili, M.T.S.; Chinedu Ikem, J.; Victor Chigozie, U.; Saki, M. Antibiotic Resistance: The Challenges and Some Emerging Strategies for Tackling a Global Menace. J. Clin. Lab. Anal. 2022, 36, 24655. [Google Scholar] [CrossRef]
- Maeda, T.; Furusawa, C. Laboratory Evolution of Antimicrobial Resistance in Bacteria to Develop Rational Treatment Strategies. Antibiotics 2024, 13, 94. [Google Scholar] [CrossRef]
- Kealey, C.; Creaven, C.A.; Murphy, C.D.; Brady, C.B. New Approaches to Antibiotic Discovery. Biotechnol. Lett. 2017, 39, 805–817. [Google Scholar] [CrossRef]
- Jackson, N.; Czaplewski, L.; Piddock, L.J. V Discovery and Development of New Antibacterial Drugs: Learning from Experience? J. Antimicrob. Chemother. 2018, 73, 1452–1459. [Google Scholar] [CrossRef]
- Liu, F.; Rajabi, S.; Shi, C.; Afifirad, G.; Omidi, N.; Kouhsari, E.; Khoshnood, S.; Azizian, K. Antibacterial Activity of Recently Approved Antibiotics against Methicillin-Resistant Staphylococcus Aureus (MRSA) Strains: A Systematic Review and Meta-Analysis. Ann. Clin. Microbiol. Antimicrob. 2022, 21, 37. [Google Scholar] [CrossRef]
- Terreni, M.; Taccani, M.; Pregnolato, M. New Antibiotics for Multidrug-Resistant Bacterial Strains: Latest Research Developments and Future Perspectives. Molecules 2021, 26, 2671. [Google Scholar] [CrossRef]
- Dalbanjan, N.P.; Praveen Kumar, S.K. A Chronicle Review of In-Silico Approaches for Discovering Novel Antimicrobial Agents to Combat Antimicrobial Resistance. Indian J. Microbiol. 2024, 64, 879–893. [Google Scholar] [CrossRef]
- Atasever, S. In Silico Drug Discovery: A Machine Learning-Driven Systematic Review. Med. Chem. Res. 2024, 33, 1465–1490. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [PubMed]
- Calianu, A.; Tamaian, R. Computational Design of New Teixobactin Analogues as Inhibitors of Lipid II Flippase MurJ. In Proceedings of the ECMC 2022, Online, 1 November 2022; MDPI: Basel, Switzerland, 2022; p. 111. [Google Scholar]
- Zhang, X.; Wu, F.; Yang, N.; Zhan, X.; Liao, J.; Mai, S.; Huang, Z. In Silico Methods for Identification of Potential Therapeutic Targets. Interdiscip. Sci. 2022, 14, 285–310. [Google Scholar] [CrossRef] [PubMed]
- van Staden, A.D.P.; van Zyl, W.F.; Trindade, M.; Dicks, L.M.T.; Smith, C. Therapeutic Application of Lantibiotics and Other Lanthipeptides: Old and New Findings. Appl. Environ. Microbiol. 2021, 87, e00186-21. [Google Scholar] [CrossRef]
- Field, D.; Cotter, P.D.; Hill, C.; Ross, R.P. Bioengineering Lantibiotics for Therapeutic Success. Front. Microbiol. 2015, 6, 1363. [Google Scholar] [CrossRef]
- Hasper, H.E.; Kramer, N.E.; Smith, J.L.; Hillman, J.D.; Zachariah, C.; Kuipers, O.P.; de Kruijff, B.; Breukink, E. An Alternative Bactericidal Mechanism of Action for Lantibiotic Peptides That Target Lipid II. Science 2006, 313, 1636–1637. [Google Scholar] [CrossRef]
- Dickman, R.; Mitchell, S.A.; Figueiredo, A.M.; Hansen, D.F.; Tabor, A.B. Molecular Recognition of Lipid II by Lantibiotics: Synthesis and Conformational Studies of Analogues of Nisin and Mutacin Rings A and B. J. Org. Chem. 2019, 84, 11493–11512. [Google Scholar] [CrossRef]
- Zhao, X.; Yin, Z.; Breukink, E.; Moll, G.N.; Kuipers, O.P. An Engineered Double Lipid II Binding Motifs-Containing Lantibiotic Displays Potent and Selective Antimicrobial Activity against Enterococcus Faecium. Antimicrob. Agents Chemother. 2020, 64, 10-1128. [Google Scholar] [CrossRef]
- Kingwell, K. Microbial ‘Dark Matter’ Yields New Antibiotic. Nat. Rev. Drug Discov. 2023, 22, 872. [Google Scholar] [CrossRef]
- Shukla, R.; Peoples, A.J.; Ludwig, K.C.; Maity, S.; Derks, M.G.N.; de Benedetti, S.; Krueger, A.M.; Vermeulen, B.J.A.; Lavore, F.; Honorato, R.V.; et al. A New Antibiotic from an Uncultured Bacterium Binds to an Immutable Target. Cell 2023, 186, 4059–4073. [Google Scholar] [CrossRef]
- Brunicardi, J.E.H.; Griffin, J.H.; Ferracane, M.J.; Kreutzer, A.G.; Small, J.; Mendoza, A.-T.; Ziller, J.W.; Nowick, J.S. Structure–Activity Relationship Studies of the Peptide Antibiotic Clovibactin. J. Org. Chem. 2024, 89, 12479–12484. [Google Scholar] [CrossRef] [PubMed]
- Varney, K.M.; Bonvin, A.M.J.J.; Pazgier, M.; Malin, J.; Yu, W.; Ateh, E.; Oashi, T.; Lu, W.; Huang, J.; Diepeveen-de Buin, M.; et al. Turning Defense into Offense: Defensin Mimetics as Novel Antibiotics Targeting Lipid II. PLoS Pathog. 2013, 9, e1003732. [Google Scholar] [CrossRef] [PubMed]
- MacDermott-Opeskin, H.I.; Gupta, V.; O’Mara, M.L. Lipid-Mediated Antimicrobial Resistance: A Phantom Menace or a New Hope? Biophys. Rev. 2022, 14, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Mingeot-Leclercq, M.-P.; Décout, J.-L. Bacterial Lipid Membranes as Promising Targets to Fight Antimicrobial Resistance, Molecular Foundations and Illustration through the Renewal of Aminoglycoside Antibiotics and Emergence of Amphiphilic Aminoglycosides. Medchemcomm 2016, 7, 586–611. [Google Scholar] [CrossRef]
- Snaebjarnarson, A.S.; Helgadottir, A.; Arnadottir, G.A.; Ivarsdottir, E.V.; Thorleifsson, G.; Ferkingstad, E.; Einarsson, G.; Sveinbjornsson, G.; Thorgeirsson, T.E.; Ulfarsson, M.O.; et al. Complex Effects of Sequence Variants on Lipid Levels and Coronary Artery Disease. Cell 2023, 186, 4085–4099.e15. [Google Scholar] [CrossRef]
- Garde, S.; Chodisetti, P.K.; Reddy, M. Peptidoglycan: Structure, Synthesis, and Regulation. EcoSal Plus 2021, 9, 1. [Google Scholar] [CrossRef]
- Reeves, P. Chapter 13 Biosynthesis and Assembly of Lipopolysaccharide. In New Comprehensive Biochemistry; Elsevier: Amsterdam, The Netherlands, 1994; pp. 281–317. [Google Scholar]
- Müller, A.; Klöckner, A.; Schneider, T. Targeting a Cell Wall Biosynthesis Hot Spot. Nat. Prod. Rep. 2017, 34, 909–932. [Google Scholar] [CrossRef]
- Scheffers, D.-J.; Tol, M.B. LipidII: Just Another Brick in the Wall? PLoS Pathog. 2015, 11, e1005213. [Google Scholar] [CrossRef]
- United Nations. Globally Harmonized System of Classification and Labelling of Chemicals (GHS); United Nations: New York, NY, USA, 2017; ISBN 9789210604574. [Google Scholar]
- Jaeger, T.; Mayer, C. N-Acetylmuramic Acid 6-Phosphate Lyases (MurNAc Etherases): Role in Cell Wall Metabolism, Distribution, Structure, and Mechanism. Cell. Mol. Life Sci. 2008, 65, 928–939. [Google Scholar] [CrossRef]
- Friedman, R.; Khalid, S.; Aponte-Santamaría, C.; Arutyunova, E.; Becker, M.; Boyd, K.J.; Christensen, M.; Coimbra, J.T.S.; Concilio, S.; Daday, C.; et al. Understanding Conformational Dynamics of Complex Lipid Mixtures Relevant to Biology. J. Membr. Biol. 2018, 251, 609–631. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Khandelia, H.; Marsh, D. Lipid Configurations from Molecular Dynamics Simulations. Biophys. J. 2018, 114, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- McTiernan, T.J.; Diaz, D.B.; Saunders, G.J.; Sprang, F.; Yudin, A.K. Navigating Complex Peptide Structures Using Macrocycle Conformational Maps. RSC Chem. Biol. 2022, 3, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Alekseenko, A.; Kozakov, D.; Miao, Y. Improved Modeling of Peptide-Protein Binding Through Global Docking and Accelerated Molecular Dynamics Simulations. Front. Mol. Biosci. 2019, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, S.; Shukla, R.; Tripathi, T.; Saudagar, P. PH-Based Molecular Dynamics Simulation for Analysing Protein Structure and Folding. In Protein Folding Dynamics and Stability; Springer Nature: Singapore, 2023; pp. 203–219. [Google Scholar]
- Luthfiana, D.; Utomo, D.H. Network Pharmacology Reveals the Potential of Dolastatin 16 as a Diabetic Wound Healing Agent. In Silico Pharmacol. 2023, 11, 23. [Google Scholar] [CrossRef]
- Fath, T.; Theodorea, C.F.; Idrus, E.; Mashima, I.; Suniarti, D.F.; Soekanto, S.A. Binding Modes of the Metabolites Docosahexaenoic Acid, Eicosapentaenoic Acid, and Eicosapentaenoic Acid Ethyl Ester from Caulerpa Racemosa as COX-2 Inhibitors Revealed via Metabolomics and Molecular Dynamics. Inform. Med. Unlocked 2024, 49, 101539. [Google Scholar] [CrossRef]
- Chan, L.-C.; Mat Yassim, A.S.; Ahmad Fuaad, A.A.H.; Leow, T.C.; Sabri, S.; Radin Yahaya, R.S.; Abu Bakar, A.M.S. Inhibition of SARS-CoV-2 3CL Protease by the Anti-Viral Chimeric Protein RetroMAD1. Sci. Rep. 2023, 13, 20178. [Google Scholar] [CrossRef]
- Wen, P.-C.; Vanegas, J.M.; Rempe, S.B.; Tajkhorshid, E. Probing Key Elements of Teixobactin–Lipid II Interactions in Membranes. Chem. Sci. 2018, 9, 6997–7008. [Google Scholar] [CrossRef]
- Chugunov, A.; Pyrkova, D.; Nolde, D.; Polyansky, A.; Pentkovsky, V.; Efremov, R. Lipid-II Forms Potential “Landing Terrain” for Lantibiotics in Simulated Bacterial Membrane. Sci. Rep. 2013, 3, 1678. [Google Scholar] [CrossRef]
- Mitra, R.P.; Malhotra, H.C.; Jain, D.V.S. Dissociation Equilibria in Pyrophosphates and Kinetics of Degradation. Part 1.—Dissociation Constants of Pyrophosphoric Acid. Trans. Faraday Soc. 1966, 62, 167–172. [Google Scholar] [CrossRef]
- Choi, H.; Kang, H.; Park, H. New Solvation Free Energy Function Comprising Intermolecular Solvation and Intramolecular Self-Solvation Terms. J. Cheminform. 2013, 5, 8. [Google Scholar] [CrossRef]
- Gilson, M.K.; Kurtzman, T. Free Energy Density of a Fluid and Its Role in Solvation and Binding. J. Chem. Theory Comput. 2024, 20, 2871–2887. [Google Scholar] [CrossRef] [PubMed]
- Leahy, D.E. Intrinsic Molecular Volume as a Measure of the Cavity Term in Linear Solvation Energy Relationships: Octanol-Water Partition Coefficients and Aqueous Solubilities. J. Pharm. Sci. 1986, 75, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.S.G.; Williams, M.A.; Pitt, W.R.; Ladbury, J.E. The Thermodynamics of Protein–Ligand Interaction and Solvation: Insights for Ligand Design. J. Mol. Biol. 2008, 384, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Prince, A.; Sandhu, P.; Ror, P.; Dash, E.; Sharma, S.; Arakha, M.; Jha, S.; Akhter, Y.; Saleem, M. Lipid-II Independent Antimicrobial Mechanism of Nisin Depends On Its Crowding And Degree Of Oligomerization. Sci. Rep. 2016, 6, 37908. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Chan-Park, M.B.; Mu, Y. Binding Modes of Teixobactin to Lipid II: Molecular Dynamics Study. Sci. Rep. 2017, 7, 17197. [Google Scholar] [CrossRef]
- de Leeuw, E.; Fletcher, S.; Yu, W.; Huang, J.; Kwasny, S.; Chauhan, J.; Opperman, T.; MacKerrel, A. Structure–Activity Exploration of a Small-Molecule Lipid II Inhibitor. Drug Des. Devel Ther. 2015, 9, 2383. [Google Scholar] [CrossRef]
- Kim, S.; Bolton, E.E.; Bryant, S.H. Similar Compounds versus Similar Conformers: Complementarity between PubChem 2-D and 3-D Neighboring Sets. J. Cheminform. 2016, 8, 62. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Zhong, H.A. ADMET Properties: Overview and Current Topics. In Drug Design: Principles and Applications; Springer: Singapore, 2017; pp. 113–133. [Google Scholar]
- Dong, J.; Wang, N.-N.; Yao, Z.-J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A Platform for Systematic ADMET Evaluation Based on a Comprehensively Collected ADMET Database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef]
- Westhouse, R.A. Safety Assessment Considerations and Strategies for Targeted Small Molecule Cancer Therapeutics in Drug Discovery. Toxicol. Pathol. 2010, 38, 165–168. [Google Scholar] [CrossRef]
- Lazic, S.E.; Williams, D.P. Improving Drug Safety Predictions by Reducing Poor Analytical Practices. Toxicol. Res. Appl. 2020, 4, 2397847320978633. [Google Scholar] [CrossRef]
- Gundert-Remy, U. Health-Based Threshold Values Versus MOS in Toxicology. In Regulatory Toxicology; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1–6. [Google Scholar]
- Guth, B.D. Preclinical Cardiovascular Risk Assessment in Modern Drug Development. Toxicol. Sci. 2007, 97, 4–20. [Google Scholar] [CrossRef]
- Shamim, S.; Munawar, R.; Rashid, Y.; Muhammad Zesshan Qadar, S.; Bushra, R.; Begum, I.; Imran, M.; Quds, T. Molecular Docking: An Insight from Drug Discovery to Drug Repurposing Approach. In Unravelling Molecular Docking—From Theory to Practice [Working Title]; IntechOpen: London, UK, 2024. [Google Scholar]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock 4 with GPUs and Gradient-Based Local Search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef]
- Mulholland, S.; Turpin, E.R.; Bonev, B.B.; Hirst, J.D. Docking and Molecular Dynamics Simulations of the Ternary Complex Nisin2:Lipid II. Sci. Rep. 2016, 6, 21185. [Google Scholar] [CrossRef]
- Cochrane, S.A.; Findlay, B.; Bakhtiary, A.; Acedo, J.Z.; Rodriguez-Lopez, E.M.; Mercier, P.; Vederas, J.C. Antimicrobial Lipopeptide Tridecaptin A 1 Selectively Binds to Gram-Negative Lipid II. Proc. Natl. Acad. Sci. USA 2016, 113, 11561–11566. [Google Scholar] [CrossRef]
- Wood, T.M.; Zeronian, M.R.; Buijs, N.; Bertheussen, K.; Abedian, H.K.; Johnson, A.V.; Pearce, N.M.; Lutz, M.; Kemmink, J.; Seirsma, T.; et al. Mechanistic Insights into the C55-P Targeting Lipopeptide Antibiotics Revealed by Structure–Activity Studies and High-Resolution Crystal Structures. Chem. Sci. 2022, 13, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Yuan, S. Genetic Optimization Method of Pantograph and Catenary Comprehensive Monitor Status Prediction Model Based on Adadelta Deep Neural Network. IEEE Access 2019, 7, 12345–12356. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Iso-SMILES | Tox-Level | CID Pubchem |
|---|---|---|---|
| Clovibactin | C[C@H]1C(=O)N[C@H](C(=O)N[C@H](C(=O)O[C@H]([C@H](C(=O)N1)NC(=O)[C@H](CO)NC(=O)[C@@H](CCCCN)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC2=CC=CC=C2)N)C(=O)N)CC(C)C)CC(C)C | 4 | 146342518 |
| Teixobactin | CC[C@H](C)[C@H]1C(=O)O[C@H]([C@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N1)C[C@H]2CN=C(N2)N)C)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]([C@@H](C)CC)NC(=O)[C@@H](CCC(=O)N)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H](CC3=CC=CC=C3)NC)C | 4 | 86341926 |
| 1 | CC(C(C(=O)NC(CC1=CC=CC=C1)C(=O)O)NC(=O)C(CC(=O)N)NC(=O)CN)O | 4 | 18485869 |
| 2 | CCCC[C@H](C)[C@@H]1CC(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](C(=O)O1)CC(=O)O)C)CC2=CC=CC=C2 | 3 | 70881873 |
| 3 | C1CCC(CC1)NC(=O)[C@H](CC2=CC=CC=C2)NC(=O)C3C(O3)C(=O)N[C@@H](CCCCN)C(=O)O | 3 | 88197590 |
| 4 | CC(C(C(=O)NC(CC(=O)N)C(=O)NC(CC1=CC=CC=C1)C(=O)NC(C(C)O)C(=O)O)N)O | 4 | 18746171 |
| 5 | CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)[C@H](CC(=O)O)N | 4 | 16122630 |
| 6 | CC(C)C[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)NC(=O)[C@H](CO)N | 4 | 16741160 |
| 7 | CCC(C)C(C(=O)O)NC(=O)C(C(C)C)NC(=O)C(CC1=CC=CC=C1)NC(=O)C(C(C)O)N | 4 | 18750676 |
| 8 | CCCC[C@H](C)[C@@H]1CC(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](C(=O)O1)[C@H](C)O)C)CC2=CC=CC=C2 | 3 | 101371349 |
| 9 | CC(C(C(=O)O)NC(=O)C(CC(=O)N)NC(=O)C(CC1=CC=CC=C1)NC(=O)C(CO)N)O | 4 | 18742358 |
| 10 | CCC(C)C(C(=O)O)NC(=O)C(C(C)O)NC(=O)C(CC1=CC=CC=C1)NC(=O)C(CO)N | 4 | 18742630 |
| 11 | C1=CC=C(C=C1)C[C@@H](C(=O)NCC(=O)OCC=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(=O)O)N | 3 | 11081110 |
| 12 | CC(C)C(C(=O)O)NC(=O)C(CC(=O)N)NC(=O)C(CC1=CC=CC=C1)NC(=O)C(CO)N | 4 | 18742361 |
| 13 | C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)OC)NC(=O)CNC(=O)[C@H](CC(=O)O)N)O | 5 | 91975530 |
| 14 | CCNC(=O)[C@H](CC1=CC=CC=C1)NC(=O)C2C(O2)C(=O)N[C@@H](CCCCN)C(=O)O | 3 | 88197746 |
| 15 | CC(C(C(=O)NC(CC1=CC=CC=C1)C(=O)O)NC(=O)C(CC(=O)N)NC(=O)C(C)N)O | 4 | 18233707 |
| 16 | CC(C)C[C@@H]([C@@H](C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)OC)O)N | 5 | 44370882 |
| 17 | CNC(=O)[C@H](CC1=CC=CC=C1)NC(=O)C2C(O2)C(=O)N[C@@H](CCCCN)C(=O)O | 3 | 88197044 |
| 18 | CC(C)CC(C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)OC(C)C)[C@](C(=O)N)(O)OC | 5 | 69037115 |
| 19 | C1=CC=C(C=C1)C[C@@H](C(=O)N)NC(=O)C2C(O2)C(=O)N[C@@H](CCCCN)C(=O)O | 3 | 88197052 |
| 20 | C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)NC(=O)[C@H](CC(C)C)NC(=O)CCN)O | 4 | 10225861 |
| 21 | CC(C)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)C2C(O2)C(=O)N[C@@H](CCCCN)C(=O)O | 4 | 88197600 |
| 22 | CCCCC[C@H](CC(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](C)C(=O)O)OC(=O)[C@@H]([C@@H](C)CC)N | 3 | 11663205 |
| 23 | CC(C(C(=O)NC(CC1=CC=CC=C1)C(=O)O)NC(=O)C(CC(=O)N)N)O | 4 | 18219295 |
| 24 | CC(C)C[C@@H](C(=O)OC)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)[C@H](CN)O | 5 | 129012247 |
| 25 | CC(C)[C@@H](C(C(=O)NCC(=O)OC)O)NC(=O)[C@H](CC1=CC=CC=C1)N | 5 | 101177680 |
| Compound | C55P | C55PP | Lipid I | Lipid II | Lipid III |
|---|---|---|---|---|---|
| Clovibactin | −1.89 | −2.57 | −9.91 | −7.61 | −7.07 |
| Teixobactin | 0.94 | 0.31 | −7.17 | −4.25 | −4.95 |
| 1 | −2.29 | −0.82 | −5.39 | −5.76 | −3.99 |
| 2 | −2.58 | −1.66 | −4.99 | −5.69 | −3.18 |
| 3 | −2.79 | −2.2 | −6.44 | −5.83 | −5.30 |
| 4 | −1.54 | −1.14 | −5.14 | −4.58 | −4.12 |
| 5 | −0.18 | 0.11 | −2.65 | −4.79 | −3.20 |
| 6 | −1.29 | −1.11 | −5.91 | −5.13 | −5.04 |
| 7 | −1.4 | −0.93 | −4.99 | −6.10 | −4.73 |
| 8 | −2.95 | −2.43 | −6.74 | −5.20 | −3.88 |
| 9 | −1.04 | −1.22 | −5.38 | −4.09 | −4.66 |
| 10 | −1.15 | −0.93 | −3.26 | −5.71 | −4.05 |
| 11 | −1.01 | −0.18 | −4.19 | −4.41 | −2.97 |
| 12 | −1.21 | −0.59 | −4.82 | −4.24 | −4.90 |
| 13 | −0.95 | 0.35 | −4.06 | −3.90 | −4.63 |
| 14 | −2.02 | −2.49 | −4.81 | −4.87 | −3.94 |
| 15 | −1.71 | −2.44 | −5.11 | −5.48 | −4.4 |
| 16 | −2.41 | −3.11 | −7.14 | −5.68 | −5.66 |
| 17 | −2.65 | −2.36 | −5.00 | −4.99 | −4.98 |
| 18 | −2.34 | −2.17 | −5.59 | −5.36 | −3.25 |
| 19 | −2.40 | −2.87 | −5.40 | −5.40 | −4.36 |
| 20 | −1.55 | −0.70 | −4.88 | −4.82 | −5.45 |
| 21 | −2.61 | −2.61 | −5.28 | −6.08 | −4.6 |
| 22 | −1.37 | −0.99 | −4.78 | −7.41 | −4.91 |
| 23 | −1.76 | −1.74 | −5.58 | −4.82 | −4.61 |
| 24 | −3.55 | −3.28 | −7.84 | −5.02 | −6.45 |
| 25 | −2.58 | −2.63 | −7.06 | −5.75 | −6.06 |
| Complex Compound-Lipid II | Binding Energy (kcal/mol) | ΔE Potential (kcal/mol) | ΔE Solvation (kcal/mol) | Molecular Volume (Bohr3/mol) |
|---|---|---|---|---|
| Clovibactin | −16.73 | −40.82 | 24.09 | 4000.23 |
| 7 | 19.77 | −46.29 | 66.06 | 3752.27 |
| 21 | 25.45 | −100.15 | 125.61 | 4259.55 |
| 22 | −25.50 | −244.50 | 218.99 | 4392.21 |
| 24 | 4.85 | −41.84 | 46.69 | 3466.97 |
| 25 | −5.96 | −22.38 | 16.43 | 3088.73 |
| Lipid | X Center | X Size | Y Center | Y Size | Z Center | Z Size |
|---|---|---|---|---|---|---|
| C55PP | −11.761 | 50 | 1.152 | 50 | 1.231 | 48 |
| Lipid I | −5.019 | 50 | 1.152 | 66 | 9.083 | 78 |
| Lipid II | −11.577 | 60 | −8.455 | 68 | −4.373 | 62 |
| Lipid III | −14.81 | 50 | −13.122 | 66 | −7.562 | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sierra-Hernandez, O.; Saurith-Coronell, O.; Rodríguez-Macías, J.; Márquez, E.; Mora, J.R.; Paz, J.L.; Flores-Sumoza, M.; Mendoza-Mendoza, A.; Flores-Morales, V.; Marrero-Ponce, Y.; et al. In Silico Identification of Potential Clovibactin-like Antibiotics Binding to Unique Cell Wall Precursors in Diverse Gram-Positive Bacterial Strains. Int. J. Mol. Sci. 2025, 26, 1724. https://doi.org/10.3390/ijms26041724
Sierra-Hernandez O, Saurith-Coronell O, Rodríguez-Macías J, Márquez E, Mora JR, Paz JL, Flores-Sumoza M, Mendoza-Mendoza A, Flores-Morales V, Marrero-Ponce Y, et al. In Silico Identification of Potential Clovibactin-like Antibiotics Binding to Unique Cell Wall Precursors in Diverse Gram-Positive Bacterial Strains. International Journal of Molecular Sciences. 2025; 26(4):1724. https://doi.org/10.3390/ijms26041724
Chicago/Turabian StyleSierra-Hernandez, Olimpo, Oscar Saurith-Coronell, Juan Rodríguez-Macías, Edgar Márquez, José Ramón Mora, José L. Paz, Maryury Flores-Sumoza, Adel Mendoza-Mendoza, Virginia Flores-Morales, Yovani Marrero-Ponce, and et al. 2025. "In Silico Identification of Potential Clovibactin-like Antibiotics Binding to Unique Cell Wall Precursors in Diverse Gram-Positive Bacterial Strains" International Journal of Molecular Sciences 26, no. 4: 1724. https://doi.org/10.3390/ijms26041724
APA StyleSierra-Hernandez, O., Saurith-Coronell, O., Rodríguez-Macías, J., Márquez, E., Mora, J. R., Paz, J. L., Flores-Sumoza, M., Mendoza-Mendoza, A., Flores-Morales, V., Marrero-Ponce, Y., Barigye, S. J., & Martinez-Rios, F. (2025). In Silico Identification of Potential Clovibactin-like Antibiotics Binding to Unique Cell Wall Precursors in Diverse Gram-Positive Bacterial Strains. International Journal of Molecular Sciences, 26(4), 1724. https://doi.org/10.3390/ijms26041724