If not otherwise noted, all reactions were magnetically stirred and conducted in oven-dried glassware. Moisture sensitive reactions were performed in an argon atmosphere, using standard Schlenk techniques. Solvents for moisture sensitive reactions (diethyl ether, dichloromethane and tetrahydrofuran) were dried according to standard procedures and distilled prior to use or purchased as extra dry reagents (N,N-dimethylformamide) from Acros Organics (Geel, Belgium). Commercially available reagents were purchased from Sigma-Aldrich (via Merck KGaA, Darmstadt, Germany), TCI Deutschland GmbH (Eschborn, Germany) and were used without further purification.
Analytical thin-layer chromatography (TLC) was used for monitoring reactions. TLC was performed on pre-coated silica gel 60 F254 aluminum plates (Merck KGaA, Darmstadt, Germany) and visualized by exposure to ultraviolet light (UV, 254 nm) and/or staining with a 1:1 mixture of 1 M ethanolic H2SO4 and 4-methoxyphenol in EtOH (3%). Alternatively, staining was performed with Seebach’s reagent [Cerium phosphomolybdic acid (5.0 g), conc. H2SO4 (16 mL), water (200 mL) and Cerium (IV) sulfate (2.0 g)]. If not otherwise stated, purification of substances was achieved by standard flash column chromatography on silica (35–70 μm particle size) from Acros Organics (Geel, Belgium). Amberlite® IR120 and Celite® Hyflo Supercel were purchased from Merck KGaA (Darmstadt, Germany).
Analytical RP-HPLC was performed using a JASCO system (PU-2080 Plus, LG-2080-02-S, DG-2080-53 and MD-2010 Plus, JASCO Deutschland GmbH, Pfungstadt, Germany) on a Phenomenex luna column (C18, 5 μm, 250 mm × 4.6 mm). As eluent, a gradient of water (A) and acetonitrile (B) containing 0.1% TFA with a flow rate of 1 mL/min was applied.
High resolution (HR-ESI) mass spectra were recorded on a Thermo Finnigan LTQ FT spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) either in positive or negative ionization mode. MALDI-TOF spectra were recorded on a Bruker Daltonics Autoflex II Time-of-flight spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with a N2-laser (λ = 337 nm).
Optical rotations were measured on a PerkinElmer polarimeter 241 (PerkinElmer, Inc., Waltham, MA, USA) at the Sodium-D-line (589 nm) and at the given temperature in °C. Concentrations, c, are given in g/100 mL, and the solvents used are stated in brackets (CHCl3).
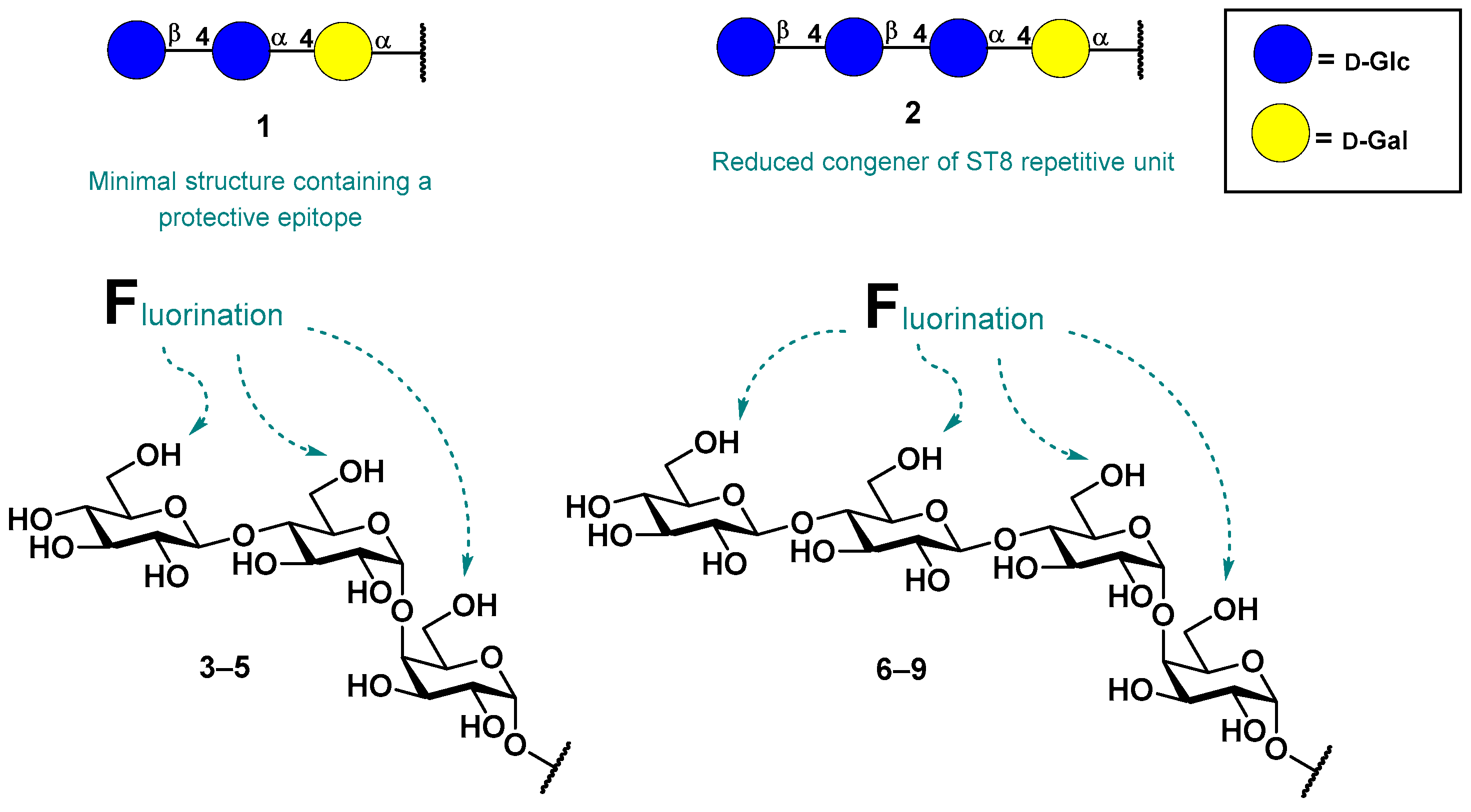
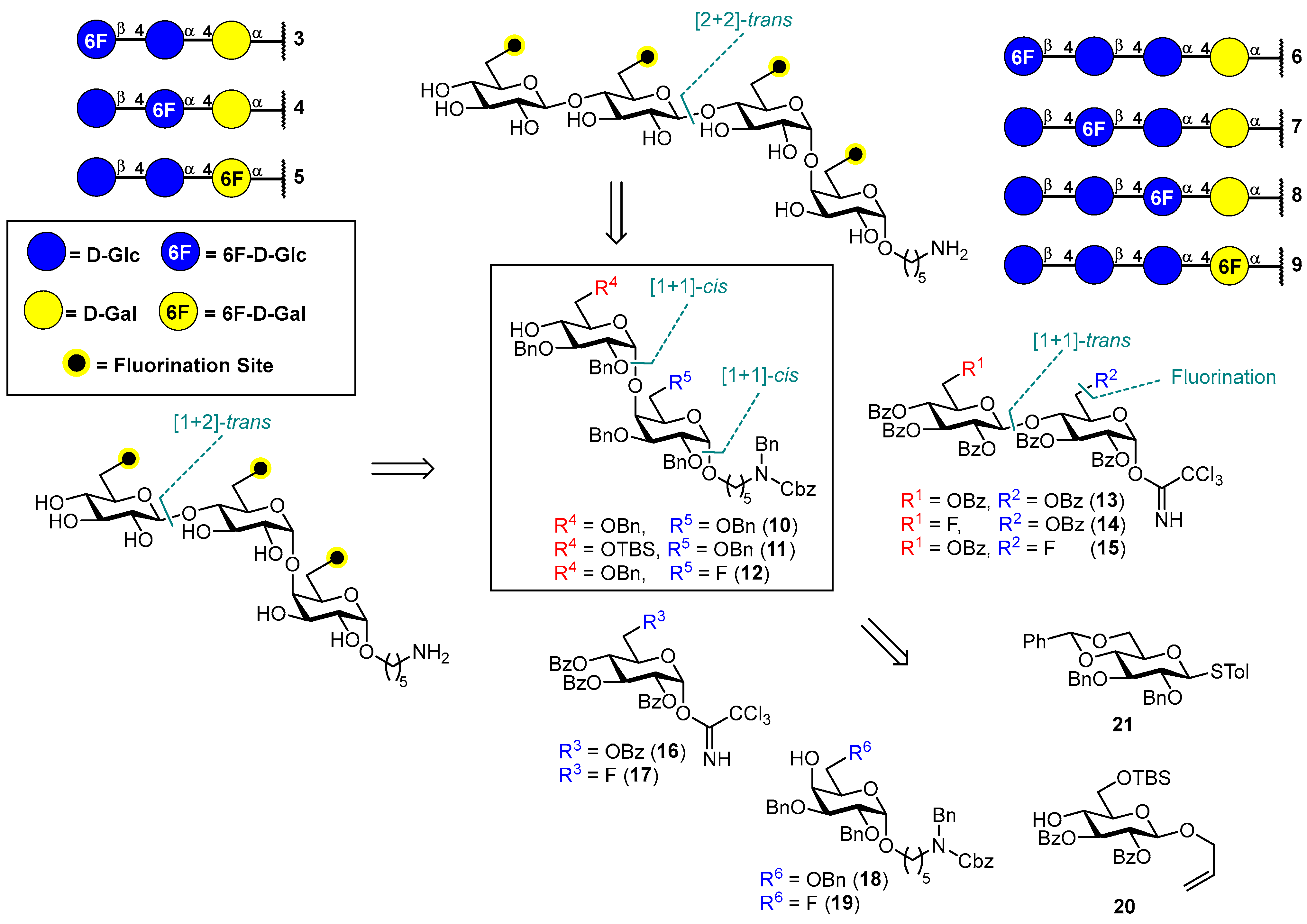
3.1. Synthesis of Fluorinated Trisaccharides 3–5
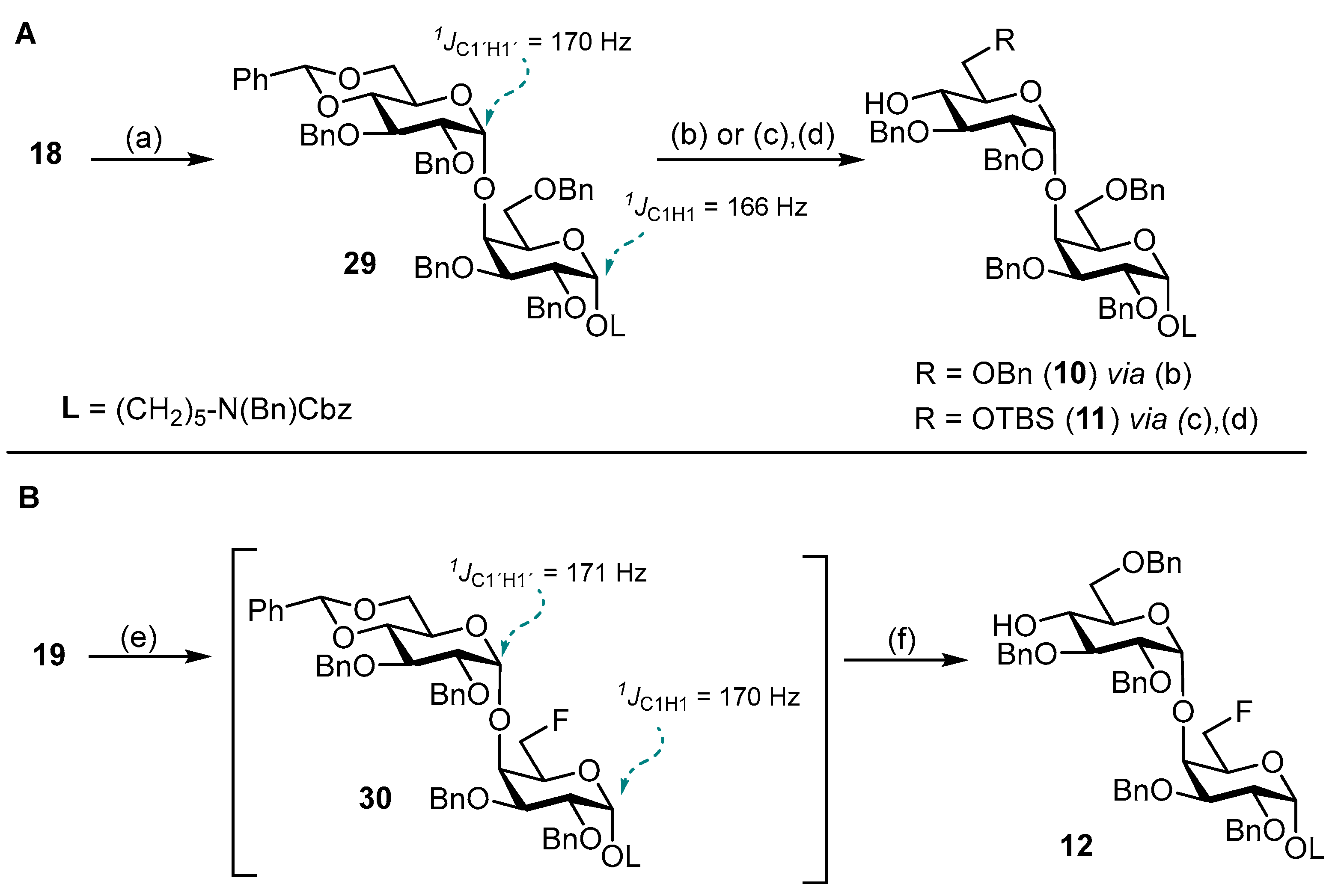
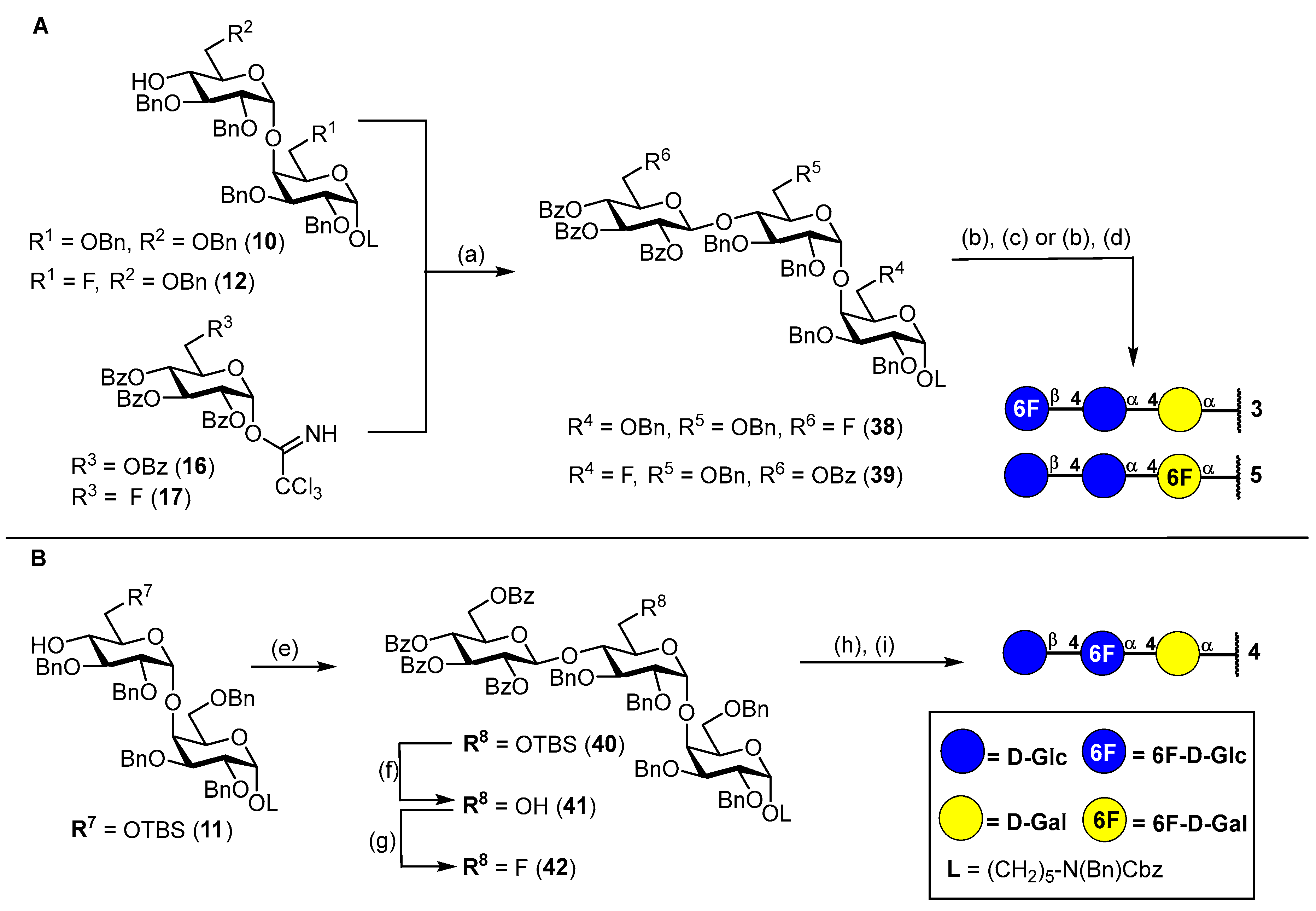
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (38). Acceptor 10 (280 mg, 235 μmol, 1.0 eq.) and fluorinated glucosyl donor 17 (210 mg, 329 μmol, 1.4 eq.) were combined and co-evaporated with toluene (2 × 10 mL). Educts were dried 1 h under high vacuo and subsequently dissolved in dry CH2Cl2 (15 mL). Freshly activated 4 Å molecular sieves were added, and the mixture was stirred for 1 h at ambient temperature. The reaction was chilled to 0 °C and TMSOTf (4.00 μL, 24.0 μmol, 0.1 eq.) was added. After the reaction was deemed complete (approximately 2.5 h) it was neutralized by the addition of NEt3 (300 μL) and filtered through a pad of Celite® Hyflo Supercel. The solvents were removed under reducd pressure and the crude product was subjected to column chromatography (cHex/EtOAc v/v = 5:1) to obtain 38 (300 mg, 0.18 mmol, 77%) as a colorless oil. Rf = 0.28 (cHex/EtOAc v/v = 3:1 + 1% NEt3). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min) tR = 22.19 min, λ = 230 nm. [α]D22 = −9.9° (c = 0.6; CHCl3). 1H NMR (600 MHz, CDCl3) δ = 7.97–7.92 (m, 2H, Ar-H), 7.78–7.76 (m, 2H, Ar-H), 7.68–7.64 (m, 2H, Ar-H), 7.58–7.11 (m, 49H, Ar-H), 5.53 (t, JH3″,H2″ = JH3″,H4″ = 9.5 Hz, 1H, H-3″), 5.47–5.41 (m, 2H, H-2″, H-4″), 5.20–5.11 (m, 3H, 2 × CHCbz, CHBn), 4.98 (d, JH1′,H2′ = 3.5 Hz, 1H, H-1′), 4.82 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.78 (d, JCH,CH = 10.6 Hz, 1H, CHBn), 4.71–4.64 (m, 3H, H-1″, 2 × CHBn), 4.59 (d, JCH,CH = 12.5 Hz, 1H, CHBn), 4.56–4.50 (m, 2H, H-1, CHBn), 4.50–4.38 (m, 3H, H-6a’’, 2 × NCHBn), 4.37–4.25 (m, 2H, H-6b’’, CHBn), 4.20–4.15 (m, 3H, 3 × CHBn), 4.12–4.00 (m, 5H, H-4′, H-5′,H-4, 2 × CHBn), 3.93–3.87 (m, 2H, H-3′, H-6a), 3.78–3.69 (m, 3H, H-2, H-3, H-5), 3.59–3.51 (m, 3H, H-5″, H-6a’,H-2′), 3.51–3.40 (m, 1H, CHLinker), 3.41–3.37 (m, 1H, H-6b), 3.35–3.24 (m, 1H, CHLinker), 3.23–3.11 (m, 2H, 2 × CHLinker), 2.98 (dd, JH6b,H6a = 11.0 Hz, JH6b,H5 = 1.6 Hz, 1H, H-6b’), 1.56–1.41 (m, 4H, 4 × CHLinker), 1.33–1.13 (m, 2H, 2 × CHLinker). 13C NMR (150 MHz, CDCl3) δ = 165.8 (C=O), 165.2 (C=O), 164.7 (C=O), 156.8/156.3 (C=OCbz), 139.5, 139.1, 138.7, 138.4, 138.2, 138.1, 137.0/136.9 (7 × Cq), 133.6, 133.3, 133.2, 129.9, 129.8 (2C), 129.1 (2C), 129.0 (2C), 128.7 (3C), 128.6 (2C), 128.4 (5C), 128.2, 128.0 (2C), 127.7, 127.6, 127.5, 127.4, 127.3 (28 × C-Ar), 100.2 (C-1″), 100.0 (C-1′), 97.9 (C-1), 81.2 (d, JC6″,F = 175.8 Hz, C-6″), 80.2 (C-3′), 79.7 (C-2′), 77.7 (C-3/C-2/C-5), 77.3 (2C, C-4, C-4′)*, 75.5 (CHBn), 75.0 (C-3/C-2/C-5), 74.3 (CHBn), 73.7 (CHBn), 73.6 (CHBn), 73.3 (C-3″), 73.0 (CHBn), 72.9 (d, JC5″,F = 19.9 Hz, C-5″), 72.5 (CHBn), 72.2 (C-2″), 70.5 (C-5′), 69.5 (C-3/C-2/C-5), 69.1 (d, JC4″,F = 6.5 Hz, C-4″), 68.0 (2C, C-6, CHLinker), 67.3 (CHCbz), 67.2 (C-6′), 50.6/50.3 (NCHBn), 47.3/46.3 (CHLinker), 29.2 (CHLinker) 28.1/27.7 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 70 out of 101 carbon atoms were assigned. *Assigned from HSQC due to signal superimposition with solvent peak. 19F NMR (377 MHz, CDCl3) δ = -230.5 (td, JF,H6a = JF,H6b = 46.9 Hz, JF,H5 = 22.1 Hz). 1H-13C-coupled HSQC (CDCl3) JC1,H1 = 171 Hz, JC1′H1′ = 171 Hz, JC1″,H1″ = 161 Hz. HRMS (ESI+) calculated for C101H106O20FN2F+ [M + NH4]+: 1686.7352; found: 1686.7282.
5-Aminopentyl-(6-deoxy-6-fluoro-β-D-glucopyranosyl)-(1→4)-(α-D-glucopyranosyl)-(1→4)-α-D-galactopyranoside (3). To a stirred solution of the fluorinated trisaccharide 38 (95 mg, 56.9 μmol, 1.0 eq.) in a mixture of MeOH/THF (v/v = 1:1, 7 mL), NaOMe (1.00 mL, 1.5 M in MeOH) was added. The reaction solution was stirred 6 h at ambient temperature before being neutralized by addition of Amberlite® IR120 The reaction mixture was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo. Subsequently, the crude residue was dissolved in a mixture of CH2Cl2/t-BuOH/H2O (v/v = 1:6:2 25 mL), and Pd(OH)2/C (100 mg) was added under Ar atmosphere. The reaction was then purged three times with H2 and stirred for 36 h at ambient temperature. The catalyst was removed by filtration through Celite® Hyflo Supercel, and the solvents were removed under reduced pressure. The crude product was dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 3 (30 mg, 50.7 μmol, 89% over 2 steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6) δ = 8.13–8.05 (m, 2H, NH2), 4.82 (d, JH1′,H2′ = 3.7 Hz, 1H, H-1′), 4.65 (d, JH1,H2 = 3.6 Hz, 1H, H-1), 4.63– 4.45 (m, 2H, H-6a″, H-6b″), 4.35 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 4.15 (dt, JH5′,H4′ = 10.1 Hz, JH5′,H6a′ = JH5′,H6b′ = 3.0 Hz, 1H, H-5′), 3.86 (bs, 1H, H-4), 3.74–3.67 (m, 2H, H-6a, H-6a′), 3.67–3.64 (m, 2H, H-3, H-5), 3.61–3.51 (m, 4H, H-2, H-3′, H-6b/H-6b′, CHLinker), 3.49–3.41 (m, 2H, H- 5″, H-6b/H-6b′), 3.38 (t, JH4′,H3′ = JH4′,H5′ = 9.5 Hz, 1H, H-4′), 3.31 (dt, JCH,CH = 9.6 Hz, JCH,CH = 6.1 Hz, 1H, CHLinker), 3.26 (dd, JH2′,H3′ = 9.7 Hz, JH2′,H1′ = 3.7 Hz, 1H, H-2′), 3.22 (t, JH3″,H2″ = JH3″,H4″ = 8.9 Hz, 1H, H-3″), 3.11 (t, JH4″,H3″ = JH4″,H5″ = 9.4 Hz, 1H, H-4″), 2.99 (t, JH2″,H1″ = JH2″,H3″ = 8.5 Hz, 1H, H-2″), 2.73 (q, JCH,CH = 6.7 Hz, 2H, CHLinker), 1.60–1.46 (m, 4H, CHLinker), 1.41–1.32 (m, 2H, CHLinker). 13C NMR (200 MHz, DMSO-d6) δ = 102.9 (C-1″), 99.4 (C-1′), 99.0 (C-1), 82.7 (d, JC6″,F = 169.1 Hz, C-6″), 79.6 (C-4′), 77.1 (C- 4), 76.3 (C-3″), 74.6 (d, JC5″,F = 17.1 Hz, C-5″), 73.2 (C-2″), 72.2 (C-2′), 71.2 (C-3′), 70.9 (C-3/C-5), 70.1 (C-5′), 68.8/68.7 (2C, C-4″, C-3/C-5), 68.4 (C-2), 66.8 (CHLinker), 59.4 (C-6/C-6′), 58.9 (C-6/C-6′), 38.7 (CHLinker), 28.6 (CHLinker), 26.7 (CHLinker), 22.8 (CHLinker). 19F NMR (377 MHz, DMSO-d6) δ = -232.1 (td, JF,H6a = JF,H6b = 47.8 Hz, JF,H5 = 23.8 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 167 Hz, JC1′H1′ = 170 Hz, JC1″,H1″ = 161 Hz. HRMS (ESI+) calculated for C23H43FNO15+ [M + H]+: 592.2611; found: 592.2610.
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3-di-O-benzyl-6-deoxy-6-fluoro-α-D-galactopyranoside (39). Disaccharide acceptor 12 (100 mg, 90.6 μmol, 1.0 eq.) and donor 16 (101 mg, 136 μmol, 1.5 eq.) were combined and co-evaporated with toluene (2 × 10 mL). Starting materials were dried for 1 h under high vacuo and subsequently dissolved in dry CH2Cl2 (10 mL). Freshly activated 4 Å molecular sieves were added, and the mixture was stirred for 1 h at ambient temperature. The reaction was cooled to 0 °C and TMSOTf (1.60 μL, 9.00 μmol, 0.1 eq.) was added in one portion. After the TLC indicated complete conversion of acceptor 12, the reaction was neutralized by addition of NEt3 (100 μL) and filtered through a pad of Celite® Hyflo Supercel. The filtrate was washed with 1 M HCl (10 mL), sat. aq. NaHCO3 (10 mL) and brine (10 mL) and dried with MgSO4. The solvents were removed under reduced pressure, and the crude product was subjected to column chromatography (cHex/EtOAc v/v = 4:1) to obtain 39 (133 mg, 79.0 μmol, 87%) as a colorless oil. Rf= 0.23 (cHex/EtOAc v/v = 3:1 + 1% NEt3). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min) tR = 25.48 min, λ = 230 nm. [α]D22 = +8.4° (c = 0.3; CHCl3). 1H NMR (800 MHz, CD2Cl2) δ = 7.97–7.95 (m, 2H, Ar-H), 7.90–7.87 (m, 2H, Ar-H), 7.77–7.74 (m, 2H, Ar-H), 7.71–7.69 (m, 2H, Ar-H), 7.55–7.52 (m, 2H, Ar-H), 7.50–7.47 (m, 2H, Ar-H), 7.47–7.09 (m, 43H, Ar-H), 5.61 (t, JH3″,H4″ = JH3″,H2″ = 9.5 Hz, 1H, H-3″), 5.57 (t, JH4″,H3″ = JH4″,H5″ = 9.6 Hz, 1H, H-4″), 5.51 (dd, JH2″,H3″ = 9.6 Hz, JH2″,H1″ = 8.1 Hz, 1H, H-2″), 5.22 (d, JCH,CH = 11.0 Hz, 1H, CHBn), 5.13 (d, JCH,CH = 20.4 Hz, 2H, CHCbz), 4.86 (d, JH1″,H2″ = 8.1 Hz, 1H, H-1″), 4.83 (d, JH1′,H2′ = 3.5 Hz, 1H, H-1′), 4.76 (d, JCH,CH = 11.1 Hz, 1H, CHBn), 4.70 (d, JCH,CH = 11.3 Hz, 1H, CHBn), 4.68–4.53 (m, 5H, H-1, H-6a, 3 × CHBn), 4.49–4.45 (m, 3H, CHBn, 2 × NCHBn), 4.39 (dd, JH6a″,H6b″ = 11.9 Hz, JH6a″,H5″ = 3.3 Hz, 1H, H-6a″), 4.38–4.35 (m, 1H, CHBn), 4.34–4.25 (m, 2H, H-6b, H-6b″), 4.19 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.15 (d, JCH,CH = 12.0 Hz, 1H, CHBn), 4.06 (dd, JH4′,H5′ = 10.3 Hz, JH4′,H3′ = 8.8 Hz, 1H, H-4′), 4.00 (dt, JH5′,H3′ = 10.3 Hz, JH5′,H6a′ = JH5′,H6b′ = 2.0 Hz, 1H, H-5′), 3.99–3.95 (m,1H, H-4), 3.89–3.79 (m, 3H, H-5″, H-3′, H-5), 3.73–3.67 (m, 2H, H-2, H-3), 3.56 (dd, JH6a′,H6b′ = 11.0 Hz, JH6a′,H5′ = 2.3 Hz, 1H, H-6a′), 3.52–3.42 (m, 2H, H-2′, CHLinker), 3.33–3.24 (m, 1H, CHLinker), 3.23–3.15 (m, 2H, 2 × CHLinker), 3.01 (dd, JH6b′,H6a′ = 11.0 Hz, JH6b′,H5′ = 1.6 Hz, 1H, H-6b′), 1.58–1.44 (m, 4H, 4 × CHLinker), 1.33–1.16 (m, 2H, 2× CHLinker). 13C-NMR (200 MHz, CD2Cl2) δ = 166.4 (C=O), 166.0 (C=O), 165.6 (C=O), 165.2 (C=O), 157.1/156.5 (C=OCbz), 140.2, 139.4, 139.0, 138.9, 138.7, 137.8/137.7 (6 × Cq), 134.0, 133.8, 133.7, 133.5, 130.3, 130.2, 130.1, 130.0, 129.6 (2C), 129.5, 129.3, 129.0 (2C), 128.9 (2C), 128.8 (3C), 128.5, 128.3, 128.2 (2C), 128.1, 128.0, 127.9, 127.7, 127.5 (28 × C-Ar), 100.9 (C-1″), 100.4 (C-1′), 98.4 (C-1), 81.6 (d, JC6,F = 164.4 Hz, C-6), 80.6 (C-3′), 80.2 (C-2′), 77.9 (C-4′), 77.5 (C-2/C-3), 77.2 (C-4), 75.6 (2C, CHBn, C-2/C-3), 75.0 (CHBn), 74.1 (CHBn), 73.8 (2C, CHBn, C-3″), 72.8 (2C, CHBn, C-2″), 72.4 (C-5″), 71.1 (C-5′), 70.4 (C-4″), 69.3 (d, JC5,F = 24.9 Hz, C-5), 68.6 (CHLinker), 67.8 (C-6′), 67.5 (CHCbz), 63.6 (C-6″), 51.0/50.6 (NCHBn), 47.7/46.9 (CHLinker), 29.7 (CHLinker), 28.5/28.0 (CHLinker), 23.9 (CHLinker). Due to signal overlap, 69 out of 101 carbon atoms were assigned. 19F NMR (377 MHz, CD2Cl2) δ = −230.8 – −231.3 (m). 1H-13C-coupled HSQC (CD2Cl2) JC1,H1 = 171 Hz, JC1′H1′ = 171 Hz, JC1″,H1″ = 166 Hz. HRMS (ESI+) calculated for C101H104FN2O21+ [M + NH4]+: 1700.7144; found: 1700.7185.
5-Aminopentyl-(β-D-glucopyranosyl)-(1→4)-(α-D-glucopyranosyl)-(1→4)-6-deoxy-6-fluoro-α-D-galacto-pyranoside (5). To a stirred solution of the fluorinated trisaccharide 39 (100 mg, 59.4 μmol, 1.0 eq.) in a mixture of MeOH/THF (v/v = 1:1, 6 mL), NaOMe (1.78 mL, 1.5 M in MeOH) was added. The reaction solution was stirred for 4 h at ambient temperature before being neutralized by the addition of Amberlite® IR120. The reaction mixture was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo. Subsequently, the crude residue was dissolved in a mixture of MeOH/THF/H2O/AcOH (v/v = 10:5:4:1, 10 mL), and Pd/C (50 mg) was added under Ar atmosphere. The reaction was purged three times with H2 and stirred for 27 h at ambient temperature. The catalyst was filtered off by Celite® Hyflo Supercel, and the solvents were removed under reduced pressure. The crude product was dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 5 (32 mg, 54.5 μmol, 92% over two steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6) δ = 4.77–4.57 (m, 4H, H-1, H-1′, H-6a, H-6b), 4.23 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 4.10–4.03 (m, 1H, H-5′/H-5″), 3.96–3.90 (m, 1H, H-5), 3.83 (s, 1H, H-4), 3.71–3.64 (m, 3H, H-3, H-6a′, H-6a″), 3.61–3.53 (m, 3H, H-2, CHLinker, H-6b′/H-6b″), 3.50 (t, JH3′,H2′ = JH3′,H4′ = 9.2 Hz, 1H, H-3′), 3.42 (dd, JH6b′/H-6b″,H6a′/H-6a″ = 11.7 Hz, JH6b′/H-6b″,H5′/H5″ = 6.4 Hz, 1H, H-6b′/H-6b″), 3.38–3.31 (m, 2H, H-4′, CHLinker), 3.29–3.23 (m, 1H, H-2′), 3.16 (s, 2H, H-3″, H-5′/H-5″), 3.06 (t, JH4″, H3″ = JH4″, H5″ = 9.3 Hz, 1H, H-4″), 2.98 (t, JH2″, H1″ = JH2″, H3″ = 8.6 Hz, 1H, H-2″), 2.68 (s, 2H, 2 × CHLinker), 1.56–1.47 (m, 4H, 4 × CHLinker), 1.38–1.32 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, DMSO-d6): δ [ppm] =103.1 (C-1″), 100.1 (C-1′), 99.0 (C-1), 82.8 (d, JC6,F = 163.2 Hz, C-6), 79.9 (C-4′), 78.4 (d, JC4,F = 6.4 Hz, C-4), 76.8 (C-3″, C-5′/C-5″), 76.5 (C-3″, C-5′/C-5″), 73.3 (C-2″), 72.1 (C-2′), 71.4 (C-3′), 70.4 (C-5′/C-5″), 70.0 (C-4″) 69.7 (d, JC5,F = 21.1 Hz, C-5), 68.3 (2C, C-3, C-2), 67.2 (CHLinker), 61.0 (C-6′/C-6″), 59.7 (C-6′/C-6″), 39.5 (CHLinker)*, 28.6 (CHLinker), 28.5 (CHLinker), 22.9 (CHLinker). *Assigned from HSQC due to signal superimposition with solvent peak. 19F NMR (377 MHz, DMSO-d6) δ = −227.4 (td, JF,H6 = 47.4 Hz, JF,H5 = 14.5 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 167 Hz, JC1′H1′ = 168 Hz, JC1″,H1″ = 159 Hz. HRMS (ESI+) calculated for C23H43FNO15+ [M + H]+: 592.2611; found: 592.2606.
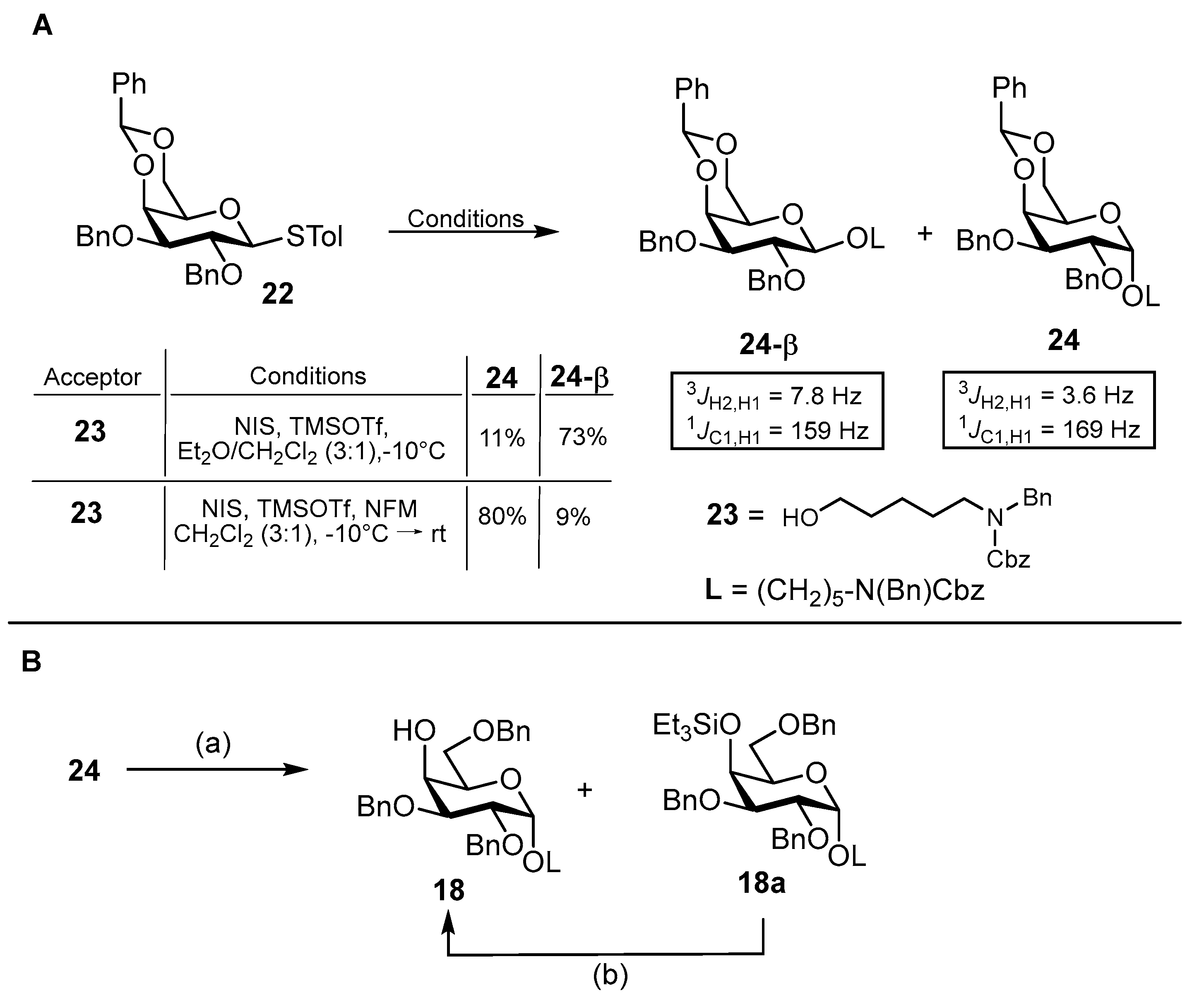
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-6-O-tert-butyldimethylsilyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (40). Acceptor 11 (170 mg, 0.14 mmol, 1.0 eq.) and donor 16 (156 mg, 0.21 mmol, 1.5 eq.) were combined and co-evaporated with toluene (2 × 10 mL). Educts were dried for 1 h under high vacuo and subsequently dissolved in dry CH2Cl2 (10 mL). Freshly activated 4 Å molecular sieves were added, and the mixture was stirred for 1 h at ambient temperature. The reaction was cooled to 0 °C, and TMSOTf (3.60 μL, 20.0 μmol, 0.1 eq.) was added in one portion. After the reaction was deemed complete by TLC, it was neutralized by the addition of NEt3 (100 μL) and filtered through a pad of Celite® Hyflo Supercel. The solvents were removed under reduced pressure, and the crude product was subjected to column chromatography (cHex/EtOAc v/v = 6:1) to obtain 40 (240 mg, 0.13 mmol, 93%) as a colorless oil. Rf = 0.43 (cHex/EtOAc v/v = 3:1). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min) tR = 39.67 min, λ = 230 nm. [α]D22 = −10.0° (c = 1.1, CHCl3). 1H NMR (600 MHz, CD2Cl2) δ = 8.02–7.98 (m, 2H, Ar-H), 7.92–7.88 (m, 2H, Ar-H), 7.85–7.83 (m, 2H, Ar-H), 7.80–7.75 (m, 2H, Ar- H), 7.53 (dtd, JCH,CH = 8.3 Hz, JCH,CH = 7.3 Hz, JCH,CH = 1.4 Hz, 2H, Ar-H), 7.47–7.40 (m, 4H, Ar-H), 7.39–7.15 (m, 41H, Ar-H), 5.83 (t, JH3″,H4″ = JH3″,H2″ = 9.6 Hz, 1H, H-3″), 5.67–5.60 (m, 2H, H-2″, H-4″), 5.31–5.26 (m, 2H, H-1″, CHBn), 5.13 (d, JCH,CH = 12.1 Hz, 2H, CHCbz), 4.91 (d, JH1′,H2′ = 3.6 Hz, 1H, H-1′), 4.75 (d, JCH,CH = 11.1 Hz, 1H, CHBn), 4.72 (d, JCH,CH = 11.4 Hz, 1H, CHBn), 4.63 (s, 1H, H-1), 4.58–4.52 (m, 3H, 3× CHBn), 4.49 (dd, JH6a″,H6b″ = 12.0 Hz, JH6a″,H5″ = 3.0 Hz, 1H, H-6a″), 4.45 (s, 2H, NCHBn), 4.35–4.30 (m, 2H, H-6b″, CHBn), 4.29–4.22 (m, 3H, 3 × CHBn), 4.13–4.07 (m, 3H, H-5″, H-4′, H-5′), 4.00 (s, 1H, H-4), 3.91 (dd, JH3′,H2′/H-4′ = 9.8 Hz, JH3′,H2′/H-4′ = 8.1 Hz, 1H, H-3′), 3.87–3.70 (m, 4H, H-6a′, H-6a, H-2, H-3), 3.68–3.64 (m, 1H, H-5), 3.51–3.45 (m, 1H, CHLinker), 3.42–3.34 (m, 3H, H-2′, H-6b′, H-6b), 3.32–3.23 (m, 1H, CHLinker) 3.22–3.11 (m, 2H, 2× CHLinker), 1.60–1.41 (m, 4H, 4× CHLinker), 1.33–1.14 (m, 2H, 2× CHLinker), 0.95 (s, 9H, Si-t-Bu), 0.09 (s, 3H, Si-CH3), 0.02 (s, 3H, Si-CH3). 13C NMR (150 MHz, CD2Cl2) δ = 166.4 (C=O), 166.1 (C=O), 165.7 (C=O), 165.3 (C=O), 157.1/156.5 (C=OCbz), 140.3, 139.4, 139.2, 139.0, 138.9, 137.8/137.7 (6 × Cq), 134.0, 133.9, 133.8, 133.5, 130.4, 130.3, 130.1 (2C), 129.7, 129.6, 129.5, 129.0 (3C), 128.9 (2C), 128.8 (3C), 128.7, 128.5, 128.2, 128.1 (2C), 128.0 (2C), 127.5, 127.4 (28 × C-Ar), 101.0 (C-1″), 99.7 (C-1′), 98.3 (C-1), 80.6 (2C, C-2′, C-3′), 77.7 (C-4′), 77.6 (C-5), 76.5 (C-4), 75.7 (CHBn), 75.6 (C-2), 75.1 (CHBn), 74.2 (C-3″), 73.6 (CHBn), 73.4 (CHBn), 73.0 (C-2″/C-4″), 72.8 (C-5″), 72.3 (CHBn), 71.9 (C-5′), 70.5 (C-2″/C-4″), 69.8 (C-3), 68.4 (2C, C-6, CHLinker), 67.4 (CHCbz), 63.8 (C-6″), 61.5 (C-6′), 51.0/50.6 (NCHBn), 47.7/46.9 (CHLinker), 29.7 (CHLinker), 28.6/28.0 (CHLinker), 26.4 (3C, Si-t-Bu), 23.9 (CHLinker), 18.7 (Cq, Si-t-Bu), −4.60 (Si-CH3), −4.80 (Si-CH3). Due to signal overlap, 75 out of 107 carbon atoms were assigned. HRMS (ESI+) calculated for C107H119N2O22Si+ [M + NH4]+: 1812.8052; found: 1812.8062.
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (41). To a stirred solution of 40 (180 mg, 100 μmol, 1.0 eq.) in a mixture of CH2Cl2/MeOH (v/v = 1:1, 20 mL), p-TsOH·H2O (19.0 mg, 100 μmol, 1.0 eq.) was added. The reaction solution was stirred 7 h at 50 °C and 12 h at ambient temperature. After the complete conversion of the starting material was observed by TLC, the reaction was neutralized by the addition of NEt3 (200 μL). The organic solvents were removed under reduced pressure, and the crude residue was dissolved in CH2Cl2 (75 mL), washed with 1 M HCl (30 mL), sat. aq. NaHCO3 (30 mL) and brine (20 mL) and dried with MgSO4. The solvent was removed under reduced pressure, and the crude product was subjected to column chromatography (cHex/EtOAc v/v = 2:1) to obtain 41 (150 mg, 89.2 μmol, 89%) as a colorless oil. Rf = 0.22 (cHex/EtOAc v/v = 2:1). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min) tR = 20.19 min, λ = 230 nm. [α]D22 = +15.0° (c = 0.3, CHCl3). 1H NMR (800 MHz, CDCl3) δ = 7.95 (d, JCH,CH = 7.7 Hz, 2H, Ar-H), 7.87 (d, JCH,CH = 7.7 Hz, 2H, Ar-H), 7.85–7.83 (m, 2H, Ar-H), 7.80–7.77 (m, 2H, Ar-H), 7.50 (td, JCH,CH = 7.4 Hz, JCH,CH = 1.4 Hz, 1H, Ar-H), 7.48–7.46 (m, 1H, Ar-H), 7.42–7.13 (m, 45H, Ar-H), 5.87 (t, JH3″,H4″ = JH3″,H2″ = 9.7 Hz, 1H, H-3″), 5.68 (t, JH4″,H3″ = JH4″,H5″ = 9.8 Hz, 1H, H-4″), 5.65–5.60 (m, 1H, H-2″), 5.21–5.13 (m, 4H, H-1″, 2 × CHCbz, CHBn), 4.87 (d, JH1′,H2′ = 3.5 Hz, 1H, H-1′), 4.85 (d, JCH,CH = 11.3 Hz, 1H, CHBn), 4.72 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.62–4.57 (m, 3H, H-1, 2 × CHBn), 4.53 (d, JCH,CH = 12.4 Hz, 1H, CHBn), 4.46 (d, JCH,CH = 26.7 Hz, 2H, NCHBn), 4.44–4.38 (m, 2H, H-6a″, CHBn), 4.32–4.28 (m, 2H, H-6b″, CHBn), 4.26–4.19 (m, 2H, 2 × CHBn), 4.05 (dd, JH5′,H4′ = 10.2 Hz, JH5′,H6′ = 2.4 Hz, 1H, H-5′), 4.03 (dt, JH5″,H4″ = 8.9 Hz, JH5″,H6a″ = JH5″,H6b″ = 4.1 Hz, 1H, H-5″), 3.97 (t, JH3′,H2′ = JH3′,H2′ = 9.2 Hz, 1H, H-3′), 3.95–3.89 (m, 2H, H-4, H-4′), 3.86–3.81 (m, 1H, H-6a), 3.80–3.74 (m, 2H, H-2, H-3), 3.74–3.70 (m, 1H, H-5), 3.60–3.55 (m, 1H, H-6a′), 3.53–3.41 (m, 3H, H-2′, H-6b, CHLinker), 3.39–3.28 (m, 2H, H-6b′, CHLinker), 3.22 (t, JCH,CH = 7.7 Hz, 1H, CHLinker), 3.14 (t, JCH,CH = 7.5 Hz, 1H, CHLinker), 1.59–1.41 (m, 4H, 4 × CHLinker), 1.34–1.11 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, CDCl3) δ = 166.1 (C=O), 165.9 (C=O), 165.2 (C=O), 165.0 (C=O), 156.8/156.3 (C=OCbz), 139.4, 138.7, 138.4 (2C), 138.1 (2C,), 137.0/136.9 (7 × Cq), 133.4 (2C), 133.3, 133.0, 129.9 (2C), 129.8 (2C), 129.7, 129.0, 128.9 (2C), 128.6 (2C), 128.5 (3C), 128.4 (4C), 128.3, 128.2, 128.0, 127.9 (2C), 127.7, 127.6 (2C), 127.5, 127.4, 127.3, 127.2, 127.0 (34 × C-Ar), 101.5 (C-1″), 99.6 (C-1′), 97.7 (C-1), 80.2 (C-3′), 80.0 (C-2′), 78.2 (C-4′), 77.8 (C-4), 77.4 (C-5)*, 75.3 (C-2), 75.1 (CHBn), 74.1 (CHBn), 73.5 (CHBn), 73.4 (C-3″), 73.0 (CHBn), 72.6 (CHBn), 72.5 (C-2″), 72.3 (C-5″), 71.1 (C-5′), 69.9 (C-4″), 69.5 (C-3), 68.4 (C-6), 68.1/68.0 (CHLinker), 67.3/67.2 (CHCbz), 63.2 (C-6″), 60.5 (C-6′), 50.6/50.3 (NCHBn), 47.2/46.3 (CHLinker), 29.2/29.1 (CHLinker), 28.0/27.6 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 76 out of 101 carbon atoms were assigned. *Assigned from HSQC due to signal superimposition with solvent peak. HRMS (ESI+) calculated for C101H102NO22+ [M + H]+: 1697.7153; found: 1697.7183.
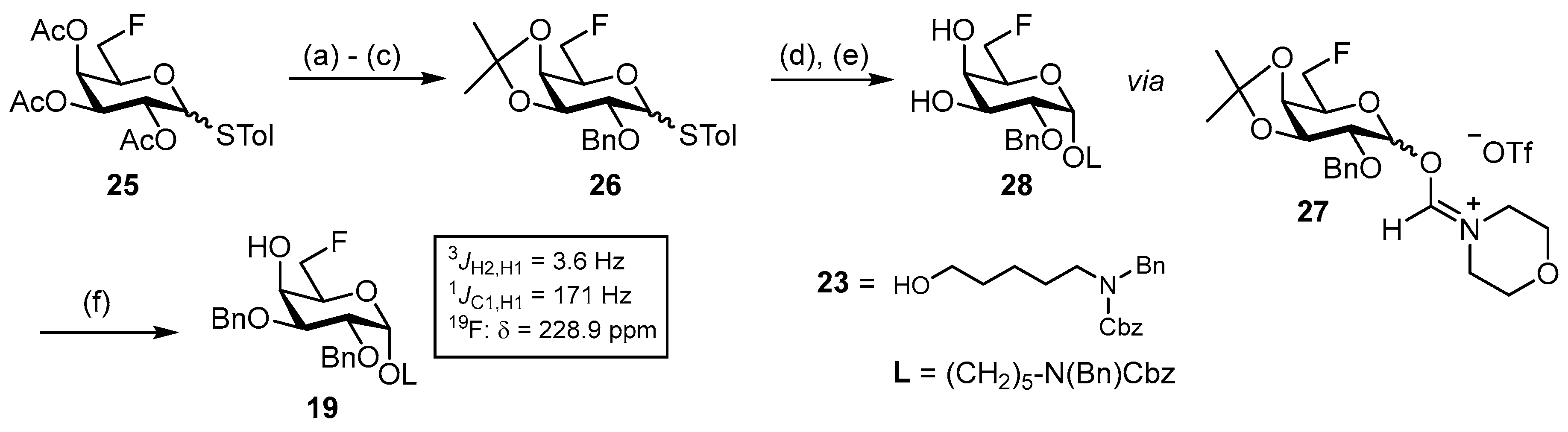
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-6-deoxy-6-fluoro-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (42). To a stirred solution of 41 (150 mg, 89.2 μmol, 1.0 eq.) in dry CH2Cl2 (4 mL), 2,4,6-collidine (48.0 μL, 357 μmol, 4.0 eq.) and DAST (24.0 μL, 179 μmol, 2.0 eq.) were added. The reaction was subjected to microwave irradiation (80 °C, 100 W) for 1 h and subsequently poured into MeOH (50 mL). The solvents were removed under reduced pressure, obtaining a brown oil, which was dissolved in CH2Cl2 (50 mL). The organic layer was washed with sat. aq. NaHCO3 (20 mL), 1 M HCl (20 mL) and brine (15 mL) and dried with MgSO4. The solvent was removed under reduced pressure, and the crude product was subjected to column chromatography (cHex/EtOAc v/v = 4:1) to obtain 42 (113 mg, 67.2 μmol, 75%) as a colorless oil. Rf = 0.46 (cHex/EtOAc v/v = 2:1). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min) tR = 21.80 min, λ = 230 nm. [α]D22 = +21.4° (c = 0.3, CHCl3). 1H NMR (600 MHz, CDCl3) δ = 7.95 (dd, JCH,CH = 8.3 Hz, JCH,CH = 1.4 Hz, 2H, Ar-H), 7.84 (td, JCH,CH = 7.8 Hz, JCH,CH = 7.3 Hz JCH,CH = 1.4 Hz, 4H, Ar-H), 7.81–7.76 (m, 2H, Ar-H), 7.54–7.11 (m, 47H, Ar-H), 5.85 (t, JH3″,H4″ = JH3″,H2″ = 9.7 Hz, 1H, H-3″), 5.67 (t, JH4″,H3″ = JH4″,H5″ = 9.7 Hz, 1H, H-4″), 5.62 (dd, JH2″,H3″ = 9.8 Hz, JH2″,H1″ = 8.0 Hz, 1H, H-2″), 5.21–5.14 (m, 3H, CHCbz, CHBn), 5.11 (d, JH1″,H2″ = 8.0 Hz, 1H, H-1″), 4.95 (d, JH1′,H2′ = 3.5 Hz, 1H, H-1′), 4.90 (d, JCH,CH = 11.5 Hz, 1H, CHBn), 4.71 (d, JCH,CH = 12.0 Hz, 1H, CHBn), 4.67 (d, JCH,CH = 12.2 Hz, 1H, CHBn), 4.64 (bs, 1H, H-1), 4.60 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.55 (d, JCH,CH = 12.3 Hz, 1H, CHBn), 4.47 (d, JCH,CH = 17.2 Hz, 2H, NCHBn), 4.44–4.27 (m, 4H, H6a/b′, H6a″, 2 × CHBn), 4.26–4.20 (m, 3H, H-6b″, 2 × CHBn), 4.19–4.09 (m, 1H, H-5′), 4.02–3.96 (m, 3H, H-5″, H-3′, H-4), 3.96–3.91 (m, 1H, H-4′), 3.91–3.70 (m, 5H, H-6a, H-6a/b′, H-3, H-2, H-5), 3.54–3.49 (m, 1H, CHLinker), 3.48 (dd, JH2′,H3′ = 9.6 Hz, JH2′,H1′ = 3.5 Hz, 1H, H-2′), 3.41 (dd, JH6b,H6a = 9.5 Hz, JH6b,H5 = 5.9 Hz, 1H, H-6b), 3.38–3.29 (m, 1H, CHLinker), 3.27–3.10 (m, 2H, 2 × CHLinker), 1.61–1.43 (m, 4H, 4 × CHLinker), 1.33–1.15 (m, 2H, 2 × CHLinker). 13C NMR (150 MHz, CDCl3) δ = 166.1 (C=O), 165.9 (C=O), 165.2 (C=O), 165.1 (C=O), 156.8/156.3 (C=OCbz), 139.4, 138.6, 138.4, 138.3, 138.2, 138.1 137.0/136.9 (7 × Cq), 133.4 (2C), 133.3, 133.0, 129.9 (2C), 129.8 (2C), 129.7, 129.1, 129.0, 128.9, 128.6 (2C), 128.5 (2C), 128.4 (2C), 128.3, 128.0 (3C), 127.7 (2C), 127.6, 127.5, 127.4, 127.2, 126.8 (29 × C-Ar), 101.6 (C-1″), 99.5 (C-1′), 97.7 (C-1), 81.2 (d, JC6′,F = 170.7 Hz, C-6′), 80.2 (C-5″/C-3′/C-4), 79.8 (C-2′), 77.7 (d, JC4′,F = 5.2 Hz C-4′), 77.3 (2C, C-5″/C-3′/C-4, C-3/C-2/C-5), 75.2 (C-3/C-2/C-5), 74.9 (CHBn), 74.2 (CHBn), 73.4 (2C, C-3″, CHBn), 73.1 (CHBn), 72.8 (CHBn), 72.6 (C-2″), 72.4 (C-5″/C-3′/C-4), 70.0 (d, JC5′,F = 17.3 Hz, C-5′), 69.8 (C-4″), 69.3 (C-3/C-2/C-5), 68.1 (CHLinker), 67.9 (C-6), 67.3 (CHCbz), 63.1 (C-6″), 50.6/50.3 (NCHBn), 47.3/46.3 (CHLinker), 29.2 (CHLinker), 28.1/27.7 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 71 out of 101 carbon atoms were assigned. 19F-NMR (377 MHz, CDCl3) δ = -234.9 (td, JF,H6a = JF,H6b = 48.1 Hz, JF,H5 = 34.3 Hz). 1H-13C-coupled HSQC (CDCl3) JC1,H1 = 172 Hz, JC1′H1′ = 171 Hz, JC1″,H1″ = 164 Hz. HRMS (ESI+) calculated for C101H104O21N2F+ [M + NH4]+: 1700.7144; found: 1700. 7186.
5-Aminopentyl-(β-D-glucopyranosyl)-(1→4)-(6-deoxy-6-fluoro-α-D-glucopyrano-syl)-(1→4)-α-D-galactopyranoside (4). To a stirred solution of 42 (70 mg, 41.6 μmol, 1.0 eq.) in MeOH/THF (v/v = 1:1, 8 mL), NaOMe (1.25 mL, 0.5 M in MeOH) was added. The solution was stirred for 18 h at ambient temperature before being neutralized by the addition of Amberlite® IR120. The reaction mixture was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo and then dissolved in a mixture of MeOH/THF/H2O/AcOH (v/v = 19:5:4:1, 15 mL). Pd/C (40 mg) was added under Ar atmosphere, and the reaction was purged three times with H2. After stirring for 72 h at ambient temperature, the catalyst was removed by filtration through a short plug of Celite® Hyflo Supercel, and the solvents were removed under reduced pressure. The crude product was then dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 4 (22 mg, 37.4 μmol, 90% over 2 steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6): δ = 4.88–4.78 (m, 2H, H-1′, H-6a′), 4.64 (d, JH1,H2 = 3.7 Hz, 1H, H-1), 4.52–4.40 (m, 2H, H-6b′, H-5′), 4.13 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 3.86 (d, JH4,H3 = 3.3 Hz, 1H, H-4), 3.75 (dd, JH6a,H6b = 10.5 Hz, JH6a,H5 = 8.3 Hz, 1H, H-6a), 3.70–3.65 (m, 2H, H-6a″, H-3), 3.62 (dd, JH5,H6b = 8.3 Hz, JH5,H6a = 5.9 Hz, 1H, H-5), 3.59–3.50 (m, 3H, H-2, H-3′, CHLinker), 3.46–3.40 (m, 2H, H-6b, H-6b″), 3.33–3.25 (m, 3H, H-2′, H-4′, CHLinker), 3.19–3.13 (m, 2H, H-5″, H-3″), 3.05 (t, JH4″,H3″ = JH4″,H5″ = 9.2 Hz, 1H, H-4″), 2.99 (t, JH2″,H1″ = JH2″,H3″ = 8.5 Hz, 1H, H-2″), 2.63 (t, JCH,CH = 7.2 Hz, 2H, 2 × CHLinker), 1.56–1.43 (m, 4H, 4 × CHLinker), 1.34 (p, JCH,CH = 7.5 Hz, 2H, 2 × CHLinker). 13C NMR (200 MHz, DMSO-d6) δ = 103.6 (C-1″), 99.2 (C-1′), 99.0 (C-1), 81.8 (d, JC6′,F = 168.3 Hz, C-6′), 79.4 (d, JC4′,F = 4.3 Hz, C-4′), 76.9 (C- 3″/C-5″), 76.5 (C-3″/C-5″), 76.4 (C-4), 73.3 (C-2″), 71.9 (C-2′), 71.3 (C-3′), 71.0 (C-5), 70.1 (C-4″), 68.5 (C-3), 68.4 (d, JC5′,F = 17.4 Hz, C-5′), 68.3 (C-2), 67.0, (CHLinker), 61.0 (C-6″), 58.7 (C-6), 40.0 (CHLinker), 29.7(CHLinker), 28.8(CHLinker), 23.0 (CHLinker). 19F NMR (377 MHz, DMSO-d6) δ = −235.0 (td, JF,H6 = 47.7 Hz, JF,H5 = 33.6 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 167 Hz, JC1′H1′ = 169 Hz, JC1″,H1″ = 160 Hz. HRMS (ESI+) calculated for C23H43FNO15+ [M + H]+: 592.2611; found: 592.2608.
3.2. Synthesis of Fluorinated Tetrasaccharides 6–9
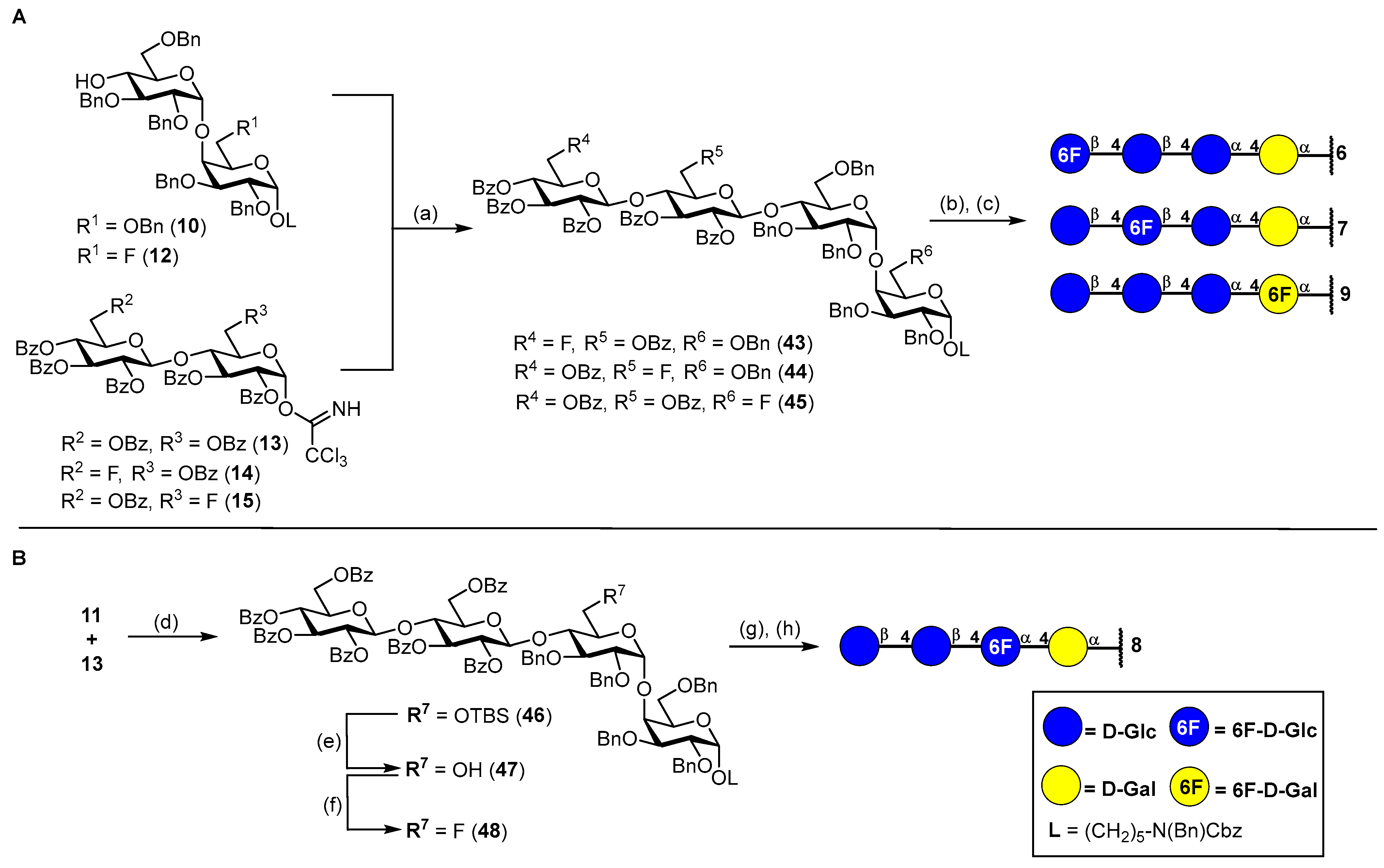
N-(Benzyl)benzyloxycarbonyl-5-aminopentyl-(2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (43). The donor 14 (200 mg, 180 μmol, 1.5 eq.) and acceptor 10 (143 mg, 120 μmol, 1.0 eq.) were combined, co-evaporated with dry toluene (15 mL) and dried under high vacuo for 1 h. Starting materials were dissolved in dry CH2Cl2 (10 mL) and stirred for 1 h over freshly activated 4 Å molecular sieves. The reaction mixture was cooled to 0 °C, and TMSOTf (2.19 μL, 12.0 μmol, 0.1 eq.) was added in one portion. The reaction was stirred 1.5 h at 0 °C before another portion of TMSOTf (3.50 μL, 19.2 μmol, 0.16 eq.) was added. After further stirring for 0.5 h, the reaction was deemed complete by TLC and was neutralized by the addition of NEt3 (100 μL). The mixture was diluted with CH2Cl2 and filtered through a pad of Celite® Hyflo Supercel. The filtrate was washed with 1 M HCl (10 mL), sat. aq. NaHCO3 (10 mL) and brine (10 mL) and was dried over MgSO4. The crude product was subjected to column chromatography (cHex/EtOAc v/v = 4:1) to obtain 43 (225 mg, 105 μmol, 87%) as a colorless oil. Rf = 0.26 (cHex/EtOAc v/v = 3:1 + 1% NEt3). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min): tR = 25.62 min, λ = 230 nm. = +12.6° (c = 0.3, CHCl3). 1H NMR (800 MHz, CD2Cl2) δ = 8.05–7.99 (m, 6H, Ar-H), 7.84–7.79 (m, 2H, Ar-H), 7.77–7.72 (m, 4H, Ar-H), 7.62–7.59 (m, 1H, Ar-H), 7.56–7.49 (m, 3H, Ar-H), 7.47–7.39 (m, 8H, Ar-H), 7.38–7.22 (m, 34H, Ar-H), 7.21–7.15 (m, 7H, Ar-H), 7.14–7.09 (m, 2H, Ar-H), 7.07–7.00 (m, 3H, Ar-H), 5.68 (t, JH3″′,H4″′ = JH3″′,H2‴ = 9.6 Hz, 1H, H-3‴), 5.49–5.46 (m, 1H, H-3″), 5.45–5.40 (m 2H, H-2‴, H-2″), 5.23–5.17 (m, 2H, H-4‴, CHBn), 5.14 (d, JCH,CH = 20.3 Hz, 2H, CHCbz), 4.90 (d, JH1′,H2′ = 3.7 Hz, 1H, H-1′), 4.81 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.68 (d, JH1″,H2″ = 8.0 Hz, 1H, H-1″), 4.66 (d, JCH,CH = 11.2 Hz, 1H, CHBn), 4.63 (d, JCH,CH = 11.3 Hz, 1H, CHBn), 4.59 (s, 1H, H-1), 4.52–4.47 (m, 3H, 4 × CHBn), 4.46 (s, 2H, NCHBn), 4.42 (dd, JH6a″,H6b″ = 11.8 Hz, JH6a″,H5″ = 1.8 Hz, 1H, H-6a″), 4.31 (d, JCH,CH = 12.5 Hz, 1H, CHBn), 4.28 (dd, JH6b″,H6a″ = 11.7 Hz, JH6b″,H5″ = 5.0 Hz, 1H, H-6b″), 4.25–4.20 (m, 3H, 3 × CHBn), 4.18 (dd, JH4″,H5″ = 9.9 Hz, JH4″,H3″ = 8.8 Hz, 1H, H-4″), 4.11 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.06 (dt, JH5′,H4′ = 10.3 Hz, JH5′,H6a′ = JH5′,H6b′ = 2.0 Hz, 1H, H-5′), 4.02–3.94 (m, 3H, H-4′, H-4, H-6a‴), 3.83–3.79 (m, 2H, H-3′, H-6a), 3.79–3.66 (m, 4H, H-2, H-3, H-5, H-6b‴), 3.63 (dddd, JH5‴,F = 20.5 Hz, JH5‴,H4‴ = 10.2 Hz, JH5‴,H6a‴ = 5.3 Hz, JH5‴,H6b‴ = 2.3 Hz, 1H, H-5‴), 3.53 (dd, JH6a′,H6b′ = 10.8 Hz, JH6a′,H5′ = 2.4 Hz, 1H, H-6a′), 3.52–3.44 (m, 1H, CHLinker), 3.43 (ddd, JH5″,H4″ = 10.0 Hz, JH5″,H6b″ = 4.9 Hz, JH5″,H6b″ = 1.8 Hz, 1H, H-5″), 3.39–3.34 (m, 2H, H-2′, H-6b), 3.32–3.24 (m, 1H, CHLinker), 3.23–3.14 (m, 2H, 2 × CHLinker), 3.01 (dd, JH6b′,H6a′ = 10.8 Hz, JH6b′,H5′ = 1.6 Hz, 1H, H-6b′), 1.55–1.43 (m, 4H, 4 × CHLinker), 1.33–1.15 (m, 2H, 2 × CHLinker). 13C-NMR (200 MHz, CD2Cl2) δ = 166.2 (C=O), 165.9 (C=O), 165.8 (C=O), 165.4 (C=O), 165.3 (2C, 2 × C=O), 157.1/156.5 (C=OCbz), 140.2, 139.4, 139.1, 139.0, 138.9, 138.8, 138.6 137.8/137.7 (8 × Cq), 134.1 (2C), 133.8 (2C), 133.7, 133.6, 130.4 (2C), 130.3, 130.2 (2C), 130.1, 129.7, 129.4, 129.2 (3C), 129.1, 129.0 (3C), 128.9 (3C), 128.8 (3C), 128.7 (2C), 128.6, 128.4, 128.3, 128.2 (2C), 128.1, 128.0 (2C), 127.9 (2C), 127.7, 127.6, 127.3 (42 × C-Ar), 101.0 (C-1‴), 100.7 (C-1″), 99.9 (C-1′), 98.3 (C-1), 81.3 (JC6‴,F = 171.3 Hz, C-6‴), 80.7 (C-3′), 80.1 (C-2′), 77.9 (C-4′), 77.8 (C-2/C-3), 77.0 (C-4), 76.7 (C-4″), 75.5 (C-2/C-3), 75.4 (CHBn), 74.8 (CHBn), 74.0 (C-3″), 73.7 (2C, 2 × CHBn), 73.6 (d, JC5‴,F = 19.8 Hz, C-5‴), 73.4 (2C, C-3‴, C-5″), 73.2 (CHBn), 72.9 (C-2″), 72.6 (CHBn), 72.3 (C-2‴), 70.9 (C-5′), 69.8 (C-5), 68.9 (d, JC4‴,F = 7.0 Hz, C-4‴), 68.4 (C-6), 68.3 (CHLinker), 67.9 (C-6′), 67.4 (CHCbz), 63.2 (C-6″), 51.0/50.6 (NCHBn), 47.7/46.9 (CHLinker), 29.7 (CHLinker), 28.5/28.0 (CHLinker), 23.9 (CHLinker). Due to signal overlap, 94 out of 128 carbon atoms were assigned. 19F NMR (377 MHz, CD2Cl2) δ = −229.95 (td, JF,H6a‴ = JF,H6b‴ = 46.7 Hz, JF,H5‴ = 20.3 Hz). 1H-13C-coupled HSQC (CD2Cl2) JC1,H1 = 169 Hz, JC1′H1′ = 170 Hz, JC1″,H1″ = 165 Hz, JC1‴,H1‴ = 161 Hz. MALDI-TOF calculated for C129H129FNO29+ [M + H + MeOH]+: 2175.87; found: 2175.90.
5-Aminopentyl-(6-deoxy-6-fluoro-β-D-glucopyranosyl)-(1→4)-(β-D-glucopyranosyl)-(1→4)-(α-D-glucopyranosyl)-(1→4)-α-D-galactopyranoside (6). To a stirred solution of tetrasaccharide 43 (110 mg, 51.4 μmol, 1.0 eq.) in a mixture of MeOH/THF (v/v = 1:1, 6 mL), NaOMe (1.50 mL, 0.5 M in MeOH) was added. The reaction solution was stirred for 5 h at ambient temperature before being neutralized by the addition of Amberlite® IR120.The reaction mixture was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo. The crude residue was then dissolved in a mixture of MeOH/THF/H2O/AcOH (v/v = 19:5:4:1, 10 mL), and Pd/C (50 mg) was added under Ar atmosphere. The reaction was purged three times with H2 and was stirred for 40 h at ambient temperature. The catalyst was filtered off by Celite® Hyflo Supercel, and the solvents were removed under reduced pressure. The crude product was dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 6 (36 mg, 46.6 μmol, 91% over two steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6) δ = 4.82 (d, JH1′,H2′ = 3.7 Hz, 1H, H-1′), 4.65 (d, JH1,H2 = 3.6 Hz, 1H, H-1), 4.62–4.46 (m, 2H, H-6a‴, H-6b‴), 4.36 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.31 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 4.13 (dt, JH5′,H6b′ = JH5′,H4′ = 10.3 Hz, JH5′,H6a′ = 3.1 Hz, 1H, H-5′), 3.87–3.85 (m, 1H, H-4), 3.77 (d, JH6a″,H6b″ = 11.4 Hz, 1H, H-6a″), 3.72 (dd, JH6a, H6b = 10.6 Hz, JH6a, H5 = 8.0 Hz, 1H, H-6a), 3.68 (dd, JH6a′,H6b′ = 11.9 Hz, JH6a′,H5′ = 3.5 Hz, 1H, H-6a′), 3.66–3.62 (m, 2H, H-3, H-5), 3.61–3.52 (m, 5H, H-3′, H-2, H-6b′, H-6b″, CHLinker), 3.50–3.42 (m, 2H, H-5‴, H-6b), 3.39–3.29 (m, 5H, CHLinker, H-3″, H-4′, H-4″, H-5″), 3.26 (dd, JH2′,H3′ = 9.6, JH2′,H1′ = 3.7 Hz, 1H, H-2′), 3.20 (t, JH3‴,H2‴ = JH3‴,H4‴ = 9.0 Hz, 1H, H-3‴), 3.10 (t, JH4‴,H3‴ = JH4‴,H5‴ = 9.4 Hz, 1H, H-4‴), 3.06 (t, JH2″,H1″ = JH2″,H3″ = 8.1 Hz, 1H, H-2″), 3.00 (t, JH2‴,H1‴ = JH2‴,H3‴ = 8.5 Hz, 1H, H-2‴), 2.67 (s, 2H, 2 × CHLinker), 1.57–1.43 (m, 4H, 4 × CHLinker), 1.35 (p, JCH,CH = 7.6 Hz, 2H, 2 × CHLinker). 13C NMR (200 MHz, DMSO-d6) δ = 102.9 (C-1‴), 102.7 (C-1″), 99.4 (C-1′), 99.0 (C-1), 82.7 (d, JC6‴,F = 169.0 Hz, C-6‴), 79.9 (C-4′), 79.7 (C-4″), 77.2 (C-4), 76.2 (C-3‴), 74.8 (C-3″/C-5″), 74.7 (d, JC5‴,F = 17.1 Hz, C-5‴), 74.5 (C-3″/C-5″), 73.1 (2C, C-2″, C-2‴), 72.1 (C-2′), 71.4 (C-3′), 71.0 (C-3), 70.2 (C-5′), 68.8 (C-5), 68.7 (JC4‴,F = 6.6 Hz, C-4‴), 68.5 (C-2), 66.9 (CHLinker), 60.2 (C-6″), 59.7 (C-6′), 59.0 (C-6), 39.4 (CHLinker)*, 28.8 (CHLinker), 28.7 (CHLinker), 22.9 (CHLinker). *Assigned from HSQC due to signal superimposition with solvent peak. 19F NMR (377 MHz, DMSO-d6) δ = −232.1 (td, JF,H6a‴ = JF,H6b‴ = 47.8 Hz, JF,H5‴ = 23.4 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 167 Hz, JC1′H1′ = 169 Hz, JC1″,H1″ = 158 Hz, JC1‴,H1‴ = 161 Hz. HRMS (ESI+) calculated for C29H53O20NF+ [M + H]+: 754.3139; found: 754.3134.
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzoyl-6-deoxy-6-fluoro-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (44). The donor 15 (210 mg, 188 μmol, 1.5 eq.) and acceptor 10 (150 mg, 126 μmol, 1.0 eq.) were combined, co-evaporated with dry toluene (15 mL) and dried under high vacuo for 1 h. Starting materials were dissolved in dry CH2Cl2 (10 mL) and stirred for 1 h over freshly activated 4 Å molecular sieves. The reaction was cooled to 0 °C, and TMSOTf (2.30 μL, 12.6 μmol, 0.1 eq.) was added in one portion. The reaction was stirred 1.5 h at 0 °C before another portion of TMSOTf (2.30 μL, 12.6 μmol, 0.1 eq.) was added. The reaction was slowly warmed to ambient temperature and stirred until complete conversion of the donor 15 was observed by TLC. The reaction was stopped by the addition of NEt3 (100 μL) and was diluted with CH2Cl2 and filtered through Celite® Hyflo Supercel. The filtrate was washed with 1 M HCl (10 mL), sat. aq. NaHCO3 (10 mL) and brine (10 mL) and dried with MgSO4. The crude product was subjected to column chromatography (cHex/EtOAc v/v = 3:1) to obtain 44 (225 mg, 104 μmol, 83%) as a colorless oil. Rf = 0.27 (cHex/EtOAc v/v = 2:1 + 1% NEt3). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min): tR = 25.80 min, λ = 230 nm. = +7.2° (c = 0.3, CHCl3). 1H NMR (800 MHz, CDCl3) δ = 8.05–8.02 (m, 2H, Ar-H), 7.91–7.89 (m, 2H, Ar-H), 7.88–7.85 (m, 2H, Ar-H), 7.77 (ddt, JCH,CH = 10.9 Hz, JCH,CH = 6.8 Hz, JCH,CH = 1.4 Hz, 5H, Ar-H), 7.66–7.63 (m, 2H, Ar-H), 7.62–7.58 (m, 1H, Ar-H), 7.50–7.09 (m, 56H, Ar-H), 5.79 (t, JH3‴,H4‴ = JH3‴,H2‴ = 9.7 Hz, 1H, H-3‴), 5.57 (dd, JH2‴,H3‴ = 9.9 Hz, JH2‴,H1‴ = 7.9 Hz, 1H, H-2‴), 5.44 (t, JH4‴,H3‴ = JH4‴,H5‴ = 9.6 Hz, 1H, H-4‴), 5.32 (t, JH3″,H4″ = JH3″,H2″ = 9.3 Hz, 1H, H-3″), 5.29 (dd, JH2″,H3″ = 9.9 Hz, JH2″,H1″ = 7.8 Hz, 1H, H-2″), 5.15 (d, JCH,CH =18.6 Hz, 2H, CHCbz), 5.05 (d, JCH,CH = 10.1 Hz, 1H, CHBn), 4.92 (d, JH1′,H2′ = 3.7 Hz, 1H, H-1′), 4.89 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.77 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.68 (d, JCH,CH = 10.1 Hz, 1H, CHBn), 4.61 (d, JCH,CH = 11.9 Hz, 1H, CHBn), 4.59 (d, JCH,CH = 12.1 Hz, 1H, CHBn), 4.53 (t, JCH,CH = 11.7 Hz, 2H 2 × CHBn), 4.50–4.42 (m, 4H, NCHBn, H-1, H-6a″), 4.42–4.34 (m, 1H, H-6b″), 4.31 (d, JH1″,H2″ = 7.8 Hz, 1H, H-1″), 4.28 (d, JCH,CH = 12.5 Hz, 1H, CHBn), 4.18–4.10 (m, 4H, H-4″, 3 × CHBn), 4.03 (dt, JH5′,H4′ = 10.0 Hz, JH5′,H6′ = 2.0 Hz, 1H, H-5′), 3.98 (s, 1H, H-4), 3.93–3.78 (m, 7H, H-3′, H-4′, H-5‴, H-6a, H-6a‴, H-6b‴, CHBn), 3.74–3.66 (m, 3H, H-2, H-3, H-5), 3.48–3.38 (m, 3H, H-2′, H-6a′, CHLinker), 3.38–3.33 (m, 1H, H-6b), 3.34–3.23 (m, 1H, CHLinker), 3.19 (t, JCH,CH = 7.7 Hz, 1H, CHLinker), 3.11 (t, JCH,CH = 7.6 Hz, 1H, CHLinker), 2.96–2.85 (m, 2H, H-5″, H-6b′), 1.56–1.44 (m, 4H, 4 × CHLinker), 1.32–1.08 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, CDCl3) δ = 165.8 (2C, 2 × C=O), 165.4 (C=O), 165.1 (C=O), 164.8 (C=O), 164.7 (C=O), 156.8/156.3 (C=OCbz), 139.2, 139.0, 138.6, 138.4, 138.3, 138.1, 138.0, 137.7, 137.0/136.9 (9 × Cq), 133.8, 133.5, 133.4, 133.2, 133.1, 129.9, 129.8, 129.7 (2C), 129.6, 129.5, 129.2, 129.1 (2C), 129.0, 128.8 (2C), 128.7 (2C), 128.5, 128.4 (4C), 128.3, 128.2, 128.0, 127.9, 127.7, 127.6, 127.5, 127.4 (2C), 127.3 (34 × C-Ar), 101.2 (C-1‴), 100.2 (C-1″), 99.9 (C-1′), 97.9 (C-1), 80.6 (d, JC6″,F = 173.7 Hz, C-6″), 80.1 (C-3′), 79.6 (C-2′), 77.5 (C-3/C-5), 77.2 (C-4)*, 77.1 (C-4′)*, 75.7 (CHBn), 75.4 (d, JC4″,F = 6.1 Hz, C-4″), 74.9 (C-2), 74.2 (CHBn), 73.7 (CHBn), 73.6 (d, JC5″,F = 19.3 Hz, C-5″), 73.5 (CHBn), 73.2 (CHBn), 73.1 (C-3‴/C-3″), 73.0 (C-3‴/C-3″), 72.5 (CHBn), 72.3 (C-2″), 72.2 (C-5‴), 72.0 (C-2‴), 70.5 (C-5′), 69.5 (C-4‴), 69.4 (C-3/C-5), 68.1/68.0 (CHLinker), 67.9 (C-6), 67.3/67.2 (CHCbz), 67.0 (C-6′), 62.7 (C-6‴), 50.6/50.3 (NCHBn), 47.3/46.3 (CHLinker), 29.2/29.1 (CHLinker), 28.0/27.6 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 87 out of 128 carbon atoms were assigned. *Assigned from HSQC due to signal superimposition with solvent peaks. 19F NMR (377 MHz, CDCl3) δ = −230.5 (td, JF,H6″ = 47.8 Hz, JF,H5″ = 30.8 Hz). 1H-13C-coupled HSQC (CD2Cl2) JC1,H1 = 170 Hz, JC1′H1′ = 169 Hz, JC1″,H1″ = 164 Hz, JC1‴,H1‴ = 162 Hz. MALDI-TOF calculated for C128H124FNO28+ [M + NH4]+: 2160.87; found: 2160.95.
5-Aminopentyl-(β-D-glucopyranosyl)-(1→4)-(6-deoxy-6-fluoro-β-D-glucopyranosyl)-(1→4)-(α-D-glucopyranosyl)-(1→4)-α-D-galactopyranoside (7). To a stirred solution of 44 (100 mg, 46.7 μmol, 1.0 eq.) in MeOH/THF (v/v = 1:1, 10 mL), NaOMe (1.40 mL, 0.5 M in MeOH) was added. The reaction solution was stirred for 17 h at ambient temperature before being neutralized by the addition of Amberlite® IR120. The reaction was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo. The crude residue was then dissolved in a mixture of MeOH/THF/H2O/AcOH (v/v = 19:5:4:1, 40 mL), and Pd/C (50 mg) was added under Ar atmosphere. The reaction was purged three times with H2 and was stirred for 40 h at ambient temperature. The catalyst was filtered off by Celite® Hyflo Supercel, and the solvents were removed under reduced pressure. The crude product was dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 7 (33 mg, 44.0 μmol, 94% over two steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6) δ = 4.82 (d, JH1′,H2′ = 3.9 Hz, 1H, H-1′), 4.78–4.62 (m, 3H, H-6a″, H-6b″, H-1), 4.40 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 4.19 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.16 (dt, JH5′H4′ = 10.1 Hz, JH5′H6a′ = JH5′H6b′ = 3.2 Hz, 1H, H-5′), 3.86 (s, 1H, H-4), 3.75–3.62 (m, 6H, H-6a, H-6a′, H-6a‴,H-3, H-5, H-5″), 3.61–3.52 (m, 4H, CHLinker, H-2, H-3′, H-6b‴), 3.44 (dd, JH6b,H6a = 10.6 Hz, JH6b,H5 = 5.8 Hz, 1H, H-6b), 3.42–3.37 (m, 3H, H-6b′, H-4′, H-3″), 3.35–3.29 (m, 2H, H-4″, CHLinker), 3.26 (dd, JH2′,H3′ = 9.7 Hz, JH2′,H1′ = 3.6 Hz, 1H, H-2′), 3.21–3.12 (m, 2H, H-5‴, H-3‴), 3.07 (t, JH2″,H1″ = JH2″,H3″ = 8.4 Hz, 1H, H-2″), 3.04 (t, JH4‴,H3‴ = JH4‴,H5‴ = 9.2 Hz, 1H, H-4‴), 2.99 (t, JH2‴,H1‴ = JH2‴,H3‴ = 8.5 Hz, 1H, H-2‴), 2.67 (s, 2H, 2 × CHLinker), 1.55–1.46 (m, 4H, 4 × CHLinker), 1.38–1.32 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, DMSO-d6) δ = 103.4 (C-1‴), 102.6 (C-1″), 99.4 (C-1′), 99.0 (C-1), 82.1 (d, JC6″,F = 167.5 Hz, C-6″), 79.8 (C-4′), 79.1(d, JC4″,F = 5.5 Hz, C-4″), 77.2 (C-4), 77.0 (C-3‴/C-5‴), 76.4 (C-3‴/C-5‴), 74.6 (C-3″), 73.2 (C-2‴), 72.9–72.8 (2C, C-2″, C-5″ [d, JC5″,F = 16.7 Hz]), 72.2 (C-2′), 71.3 (C-3′), 71.0 (C-3/C-5), 70.1 (C-4‴), 70.0 (C-5′), 68.8 (C-3/C-5), 68.5 (C-2), 66.9 (CHLinker), 61.1 (C-6′), 59.5 (C-6‴), 59.0 (C-6), 39.4 (CHLinker)*, 28.7 (CHLinker), 28.6 (CHLinker), 22.9 (CHLinker). *Assigned by HSQC due to signal superimposition with solvent peak. 19F NMR (377 MHz, DMSO-d6) δ = −231.9 (td, JF,H6a″ = JF,H6b″ = 47.5 Hz, JF,H5″ = 23.2 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 166 Hz, JC1′H1′ = 170 Hz, JC1″,H1″ = 160 Hz, JC1‴,H1‴ = 161 Hz. HRMS (ESI+) calculated for C29H53O20NF+ [M + H]+: 754.3139; found: 754.3127.
N-(Benzyl)benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3-di-O-benzyl-6-deoxy-6-fluoro-α-D-galactopyranoside (45). The donor 13 (165 mg, 136 μmol, 1.5 eq.) and acceptor 12 (100 mg, 96.6 μmol, 1.0 eq.) were combined, co-evaporated with dry toluene (5 mL) and dried under high vacuo for 1 h. The starting materials were then dissolved in dry CH2Cl2 (10 mL), and freshly dried 4 Å molecular sieves were added. The mixture was stirred for 40 min at ambient temperature, cooled to 0 °C, and TMSOTf (4.00 μL, 23.0 μmol, 0.1 eq.) was added. The reaction was slowly warmed to room temperature and stirred for 1.5 h. Another portion of TMSOTf (10.0 μL, 57.5 μmol, 0.25 eq.) was added, and the reaction was stirred further 15 min before being stopped by the addition of NEt3 (100 μL). The mixture was diluted with CH2Cl2 and filtered through a short plug of Celite® Hyflo Supercel. The filtrate was washed with 1 M HCl (10 mL), sat. aq. NaHCO3 (10 mL) and brine (10 mL) and was dried over MgSO4. The crude product was subjected to column chromatography (cHex/EtOAc v/v = 3:1) to obtain 45 (165 mg, 76.4 μmol, 84%) as a colorless oil. Rf = 0.39 (cHex/EtOAc v/v = 3:1 + 1% NEt3). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min): tR = 23.76 min, λ = 230 nm. [α]D22 = +30.0° (c = 0.33; CHCl3). 1H NMR (800 MHz, CDCl3) δ = 8.07–8.03 (m, 2H, Ar-H), 7.94–7.88 (m, 6H, Ar-H), 7.75–7.71 (m, 4H, Ar-H), 7.69–7.64 (m, 2H, Ar-H), 7.51 (t, JCH,CH = 7.1 Hz, 2H, Ar-H), 7.49–7.12 (m, 49H, Ar-H), 7.11 (dd, JCH,CH = 7.2, JCH,CH = 2.3 Hz, 2H, Ar-H), 7.02–6.94 (m, 3H, Ar-H), 5.65 (t, JH3‴,H4‴ = JH3‴,H2‴ = 9.6 Hz, 1H, H-3‴), 5.53 (dd, JH2‴,H3‴ = 9.7 Hz, JH2‴,H1‴ = 7.9 Hz, 1H, H-2‴), 5.35–5.27 (m, 3H, H-4‴, H-2″,H-3″), 5.15 (d, JCH,CH = 19.9 Hz, 2H, CHCbz), 5.11 (d, JCH,CH = 11.0 Hz, 1H, CHBn), 4.78 (d, JH1′,H2′ = 3.6 Hz, 1H, H-1′), 4.73–4.62 (m, 3H, H-1‴, H-6a, CHBn), 4.62–4.54 (m, 3H, 3 × CHBn), 4.51–4.48 (m, 2H, 2 × CHBn), 4.48–4.43 (m, 3H, NCHBn, H-1), 4.39–4.35 (m, 1H, H-1″), 4.35–4.31 (m, 2H, H-6a″, CHBn), 4.32–4.18 (m, 2H, H-6b″, H-6b), 4.10 (d, JCH,CH = 12.0 Hz, 1H, CHBn), 4.06–4.01 (m, 1H, H-4″), 3.95–3.89 (m, 4H, H-5′, H-4, H-6a‴, CHBn), 3.88 (t, JH4′,H3′ = JH4′,H5′ = 9.5 Hz, 1H, H-4′), 3.85–3.78 (m, 1H, H-5), 3.76 (t, JH3′,H4′ = JH3′,H2′ = 9.3 Hz, 1H, H-3′), 3.73–3.68 (m, 1H, H-3), 3.65 (dd, JH2,H3 = 10.0 Hz, JH2,H1 = 3.8 Hz, 1H, H-2), 3.63–3.58 (m, 2H, H-5‴, H-6b‴), 3.49–3.39 (m, 2H, H-6a′, CHLinker), 3.37 (dd, JH2′,H3′ = 9.8 Hz, JH2′,H1′ = 3.6 Hz, 1H, H-2′), 3.34–3.24 (m, 1H, CHLinker), 3.23–3.18 (m, 2H, H-5″, CHLinker), 3.15–3.11 (m, 1H, CHLinker), 2.88 (dd, JH6b′,H6a′ = 10.7 Hz, JH6b′,H5′ = 1.9 Hz, 1H, H-6b′), 1.65–1.40 (m, 4H, 4 × CHLinker), 1.25–1.09 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, CDCl3) δ = 165.8 (C=O), 165.7 (2C, 2 × C=O), 165.4 (C=O), 165.1(C=O), 164.9 (2C, 2 × C=O), 156.8/156.3 (C=OCbz), 139.5, 138.9, 138.3, 138.2, 138.1, 138.0, 137.7, 137.0/136.9 (8 × Cq), 133.6, 133.5, 133.3 (2C), 133.1, 129.9 (2C), 129.8 (2C), 129.7, 129.6, 129.2 (2C), 128.9, 128.8, 128.7 (2C), 128.6, 128.5 (3C), 128.4 (3C), 128.0, 127.8, 127.6, 127.5, 127.3 (29 × C-Ar), 100.9 (C-1‴), 100.3 (C-1′), 100.1 (C-1″), 98.0 (C-1), 80.8 (d, JC6,F = 160.8 Hz, C-6), 80.2 (C-3′), 78.9 (C-2′), 77.3 (C-4′)*, 77.1 (C-3)*, 77.0 (C-4)*, 76.3 (C-4″), 75.0 (CHBn), 74.9 (C-2), 74.3 (CHBn), 73.8 (CHBn), 73.6 (CHBn), 73.3 (C-3″), 72.9 (C-3‴), 72.7 (C-5″), 72.6 (CHBn), 72.3 (2C, C-5‴, C-2″), 71.9 (C-2‴), 70.6 (C-5′), 69.7 (C-4‴), 68.7 (d, JC5,F = 25.3 Hz, C-5), 68.2 (CHLinker), 67.3 (CHCbz), 66.8 (C-6′), 62.8 (C-6″), 62.7 (C-6‴), 50.6/50.3 (NCHBn), 47.2/46.3 (CHLinker), 29.2/29.1 (CHLinker), 28.0/27.6 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 81 out of 128 carbon atoms were assigned. *Assigned by HSQC due to signal superimposition with solvent peak. 19F NMR (377 MHz, CDCl3) δ = −230.6 – −231.1 (m). 1H-13C-coupled HSQC (CDCl3) JC1,H1 = 170 Hz, JC1′H1′ = 168 Hz, JC1″,H1″ = 163 Hz, JC1‴,H1‴ = 163 Hz. MALDI-TOF calculated for C129H129FNO29+ [M + H + MeOH]+: 2189.85; found: 2189.95.
5-Aminopentyl-(β-D-glucopyranosyl)-(1→4)-(β-D-glucopyranosyl)-(1→4)-(α-D-glucopyranosyl)-(1→4)-6-deoxy-6-fluoro-α-D-galactopyranoside (9). To a stirred solution of tetrasaccharide 45 (100 mg, 46.4 μmol, 1.0 eq.) in a mixture of MeOH/THF (v/v = 1:1, 10 mL), NaOMe (1.40 mL, 0.5 M in MeOH) was added. The reaction solution was stirred for 8 h at ambient temperature before being neutralized by the addition of Amberlite® IR120. The reaction mixture was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo. The crude residue was dissolved in a mixture of MeOH/THF/H2O/AcOH (v/v = 19:5:4:1, 20 mL), and Pd/C (50 mg) was added under Ar atmosphere. The reaction was purged three times with H2 and stirred for 48 h at ambient temperature. The catalyst was removed by filtration through Celite® Hyflo Supercel and the solvents were removed under reduced pressure. The crude product was dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 9 (30 mg, 39.5 μmol, 85% over two steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6) δ = 4.78–4.57 (m, 4H, H-1, H-1′, H-6a, H-6b), 4.30 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 4.24 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.10–4.07 (m, 1H, H-5′), 3.96–3.90 (m, 1H, H-5), 3.83 (bs, 1H, H-4), 3.77 (d, JH-6a″;H-6b″ = 11.2 Hz, 1H, H-6a″), 3.71–3.65 (m, 3H, H-3, H-6a′,H-6a‴), 3.61–3.54 (m, 4H, H-2, H-6b′, H-6b″, CHLinker), 3.51 (t, JH3′,H2′ = JH3′,H4′ = 9.1 Hz, 1H, H-3′), 3.41 (dd, JH6b‴,H6a‴ = 11.7, JH6b‴,H5‴ = 6.6 Hz, 1H, H-6b‴), 3.38–3.31 (m, 5H, H-4′, H-3″, H-4″, H-5″, CHLinker), 3.26 (dd, JH2′,H3′ = 9.6 Hz, JH2′,H1′ = 3.7 Hz, 1H, H-2′), 3.21–3.13 (m, 2H, H-3‴,H-5‴), 3.07–3.03 (m, 2H, H-4‴, H-2″), 2.98 (t, JH2‴,H1‴ = JH2‴,H3‴ = 8.5 Hz, 1H, H-2‴), 2.65 (s, 2H, 2 × CHLinker), 1.56–1.46 (m, 4H, 4 × CHLinker), 1.42–1.29 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, DMSO-d6) δ = 103.3 (C-1‴), 102.7 (C-1″), 100.1 (C-1′), 99.0 (C-1), 82.8 (d, JC6,F = 163.2Hz, C-6), 80.3 (C-4″), 79.8 (C-4′), 78.4 (d, JC4,F = 5.7 Hz, C-4), 76.9 (C-5‴), 76.5 (C-3‴), 74.8 (2C, C-3″, C-5″), 73.3 (C-2‴), 73.0 (C-4‴/C-2″), 72.0 (C-2′), 71.3 (C-3′), 70.4 (C-5′), 70.0 (C-4‴/C-2″), 69.7 (d, JC5,F = 21.1 Hz, C-5), 68.3 (2C, C-2, C-3), 67.2 (CHLinker), 61.0 (C-6‴), 60.3 (C-6″), 59.6 (C-6′), 39.6 (CHLinker)*, 29.2 (CHLinker), 28.6 (CHLinker), 22.9 (CHLinker). *Assigned by HSQC due to signal superimposition with solvent peak. 19F NMR (377 MHz, DMSO-d6) δ = −227.4 (td, JF,H6a = JF,H6b = 47.1 Hz, JF,H5 = 14.4 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 169 Hz, JC1′H1′ = 168 Hz, JC1″,H1″ = 160 Hz, JC1‴,H1‴ = 161 Hz. HRMS (ESI+) calculated for C29H53FNO20+ [M + H]+: 754.3139; found: 754.3128.
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (46). The donor 13 (420 mg, 345 μmol, 1.5 eq.) and TBS-protected acceptor 11 (280 mg, 230 μmol, 1.0 eq.) were combined, co-evaporated with dry toluene (5 mL) and dry CH2Cl2 (5 mL) and dried under high vacuo for 1 h. The starting materials were then dissolved in dry CH2Cl2 (10 mL), and freshly dried 4 Å molecular sieves were added to the reaction solution. The mixture was stirred for 1 h at ambient temperature and cooled to −20 °C before TMSOTf (4.00 μL, 23.0 μmol, 0.1 eq.) was added. The reaction was allowed to slowly warm to room temperature before another portion of TMSOTf (4.00 μL, 23.0 μmol, 0.1 eq.) was added after 0.5 h. The reaction was stirred for further 1.5 h before being stopped by addition of NEt3 (100 μL). The mixture was diluted with CH2Cl2 and filtered through a short plug of Celite® Hyflo Supercel. The organic phase was washed with 1 M HCl (10 mL), sat. aq. NaHCO3 (10 mL) and brine (10 mL) and was dried over MgSO4. The crude product was subjected to column chromatography (cHex/EtOAc v/v = 4:1) to obtain the TBS-protected tetrasaccharide intermediate 46 as an inseparable mixture, together with a decomposition product of the cellobiose donor 13. The product was dissolved in CH2Cl2/MeOH (20 mL, v/v = 1:1), and p-TsOH (44.0 mg, 231 μmol, 1.0 eq.) was added. The reaction was warmed to 50 °C and stirred 6 h before being neutralized by the addition of NEt3 (200 μL). The solvents were removed under reduced pressure, and the crude product was subjected to column chromatography (cHex/EtOAc v/v = 3:1) to obtain 47 (150 μmol, 65% over 2 steps) as an amorphous solid. Rf = 0.30 (cHex/EtOAc v/v = 2:1). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min): tR = 22.72 min, λ = 230 nm. = +30.1° (c = 0.5, CHCl3). 1H NMR (800 MHz, CDCl3) δ = 8.00 –7.97 (m, 4H, Ar-H), 7.96–7.92 (m, 4H, Ar-H), 7.84 (d, JCH,CH = 7.7 Hz, 2H, Ar-H), 7.74 (t, JCH,CH = 7.6 Hz, 4H, Ar-H), 7.55–7.50 (m, 2H, Ar-H), 7.47 (t, JCH,CH = 7.3 Hz, 1H, Ar-H), 7.45–7.11 (m, 50H, Ar-H), 6.99 (t, JCH,CH = 7.6 Hz, 2H, Ar-H), 6.90 (t, JCH,CH = 7.3 Hz, 1H, Ar-H), 5.72 (t, JH3″,H4″ = JH3″,H2″ = 9.4 Hz, 1H, H-3″), 5.63 (t, JH3‴,H4‴ = JH3‴,H2‴ = 9.6 Hz, 1H, H-3‴), 5.50 (dd, JH2″,H3″ = 9.8 Hz, JH2″,H1″ = 8.1 Hz, 1H, H-2″), 5.45 (dd, JH2‴,H3‴ = 9.6 Hz, JH2‴,H1‴ = 7.9 Hz, 1H, H-2‴), 5.29 (t, JH4‴,H3‴ = JH4‴,H5‴ = 9.6 Hz, 1H, H-4‴), 5.16 (d, JCH,CH = 18.7 Hz, 2H, CHCbz), 5.07 (d, JCH,CH = 11.4 Hz, 1H, CHBn), 5.01 (d, JH1″,H2″ = 8.1 Hz, 1H, H-1″), 4.82–4.77 (m, 3H, H-1‴, H-1′, CHBn), 4.63–4.55 (m, 3H, H-1, 2 × CHBn), 4.53–4.48 (m, 2H, 2 × CHBn), 4.48–4.41 (m, 3H, NCHBn, CHBn), 4.34–4.28 (m, 2H, CHBn, H-6″/H-6‴), 4.26–4.16 (m, 4H, H-4″, H-6″/H-6‴, 2 × CHBn), 4.02–3.96 (m, 2H, H-5′, H-6″/H-6‴), 3.91 (t, JH3′,H4′ = JH3′,H2′ = 9.2 Hz, 1H, H-3′), 3.88 (s, 1H, H-4), 3.82–3.62 (m, 7H, H-5‴, H-6a, H-6″/H-6‴, H-4′, H-2, H-3, H-5), 3.61–3.56 (m, 1H, H-5″), 3.50–3.44 (m, 2H, H-6a′, CHLinker), 3.41 (dd, JH6b,H6a = 9.4 Hz, JH6b,H5 = 6.1 Hz, 1H, H-6b), 3.36 (dd, JH2′,H3′ = 9.8 Hz, JH2′,H1′ = 3.5 Hz, 1H, H-2′), 3.34–3.25 (m, 2H, H-6b′, CHLinker), 3.23–3.19 (m, 1H, CHLinker), 3.16–3.10 (m, 1H, CHLinker), 1.57–1.40 (m, 4H, 4 × CHLinker), 1.26–1.11 (m, 2H, 2 × CHLinker).13C NMR (200 MHz, CDCl3) δ = 165.8 (C=O), 165.7 (2C, 2 × C=O), 165.5 (C=O), 165.2 (C=O), 165.1 (C=O), 164.7 (C=O), 156.8/156.3 (C=OCbz), 139.4, 138.6, 138.5, 138.3, 138.1, 138.0, 137.0/136.9 (7 × Cq), 133.5 (2C), 133.3, 133.2, 130.1, 129.9, 129.8 (3C), 129.7, 129.6, 129.1, 128.8, 128.7, 128.6 (3C), 128.5, 128.4 (2C), 128.3 (2C), 128.1, 128.0, 127.9 (3C), 127.7, 127.6 (2C), 127.5, 127.4, 127.3, 126.9, 126.5 (35 × C-Ar), 101.4 (C-1″), 100.8 (C-1‴), 99.5 (C-1′), 97.7 (C-1), 80.4 (C-3′), 79.9 (C-2′), 78.3 (C-4′), 77.8 (C-4), 77.4 (C-5)*, 76.0 (C-4″), 75.3 (C-2), 74.8 (CHBn), 74.0 (CHBn), 73.5 (CHBn), 73.2 (2C, C-3″, C-5″), 73.0 (2C, CHBn, C-3‴), 72.6 (2C, CHBn, C-2″),72.4 (C-5‴), 71.9 (C-2‴), 71.0 (C-5′), 69.6 (C-3), 69.5 (C-4‴), 68.5 (C-6), 68.1/68.0 (CHLinker), 67.3/67.2 (CHCbz), 62.7 (C-6″/C-6‴), 62.5 (C-6″/C-6‴), 60.5 (C-6′), 50.6/50.3 (NCHBn), 47.2/46.3 (CHLinker), 29.2/29.1 (CHLinker), 28.0/27.6 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 86 out of 128 carbon atoms were assigned. *Assigned by HSQC due to signal superimposition with solvent peak. 1H-13C-coupled HSQC (CDCl3) JC1,H1 = 169 Hz, JC1′H1′ = 168 Hz, JC1″,H1″ = 160 Hz, JC1‴,H1‴ = 162 Hz. MALDI-TOF calculated for C129H128FNO31+ [M + H + MeOH]+: 2187.85; found: 2188.76.
N-(Benzyl)-benzyloxycarbonyl-5-aminopentyl-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3,6-tri-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-6-deoxy-6-fluoro-α-D-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-D-galactopyranoside (48). In a microwave vessel equipped with a stirring bar, 47 (140 mg, 69.6 μmol, 1.0 eq.) was dissolved in dry CH2Cl2 (2 mL). Subsequently, 2,4,6-collidine (28.0 μL, 0.21 mmol, 3.0 eq.) and DAST (14.0 μL, 0.10 mmol, 1.5 eq.) were added. The reaction solution was heated for 1 h in a microwave oven (80 °C, 100 W). Since the reaction was not deemed complete by TLC, another portion of 2,4,6-collidine (28.0 μL, 0.21 mmol, 3.0 eq.) and DAST (14.0 μL, 0.10 mmol, 1.5 eq.) were added, and microwave heating was continued for another 1 h. Then, the reaction was cooled to 0 °C and stopped by the addition of MeOH (2 mL). The organic solvents were removed under reduced pressure. The crude residue was dissolved in CH2Cl2, and the solution was washed with sat. aq. NaHCO3 solution (10 mL) and brine (10 mL) and was dried with MgSO4. The crude product was subjected to column chromatography (cHex/EtOAc v/v = 4:1) to obtain 48 (112 mg, 51.9 μmol, 75%) as a colorless foam. Rf = 0.43 (cHex/EtOAc v/v = 2:1). RP HPLC (Luna, 0.1% TFA; 0 min 50% B → 10 min 100% B, flow: 1 mL/min): tR = 24.07 min, λ = 230 nm. = +30.6° (c = 0.3, CHCl3). 1H NMR (800 MHz, CDCl3) δ = 7.97–7.94 (m, 4H, Ar-H), 7.93–7.90 (m, 4H, Ar-H), 7.81–7.79 (m, 2H, Ar-H), 7.75–7.71 (m, 4H, Ar-H), 7.53–7.49 (m, 2H, Ar-H), 7.49–7.09 (m, 51H, Ar-H), 6.98 (t, JCH,CH = 7.6 Hz, 2H, Ar-H), 6.87 (t, JCH,CH = 7.3 Hz, 1H, Ar-H), 5.69 (t, JH3″,H4″ = JH3″,H2″ = 9.5 Hz, 1H, H-3″), 5.64–5.58 (m, 1H, H-3‴), 5.51–5.46 (m, 1H, H-2″), 5.45–5.42 (m, 1H, H-2‴), 5.28 (t, JH4‴,H3‴ = JH4‴,H5‴ = 9.1 Hz, 1H, H-4‴), 5.15 (d, JCH,CH = 19.1 Hz, 2H, CHCbz), 5.05 (d, JCH,CH = 11.5 Hz, 1H, CHBn), 4.95 (d, JH1″,H2″ = 8.0 Hz, 1H, H-1″), 4.88 (d, JH1′,H2′ = 3.6 Hz, 1H, H-1′), 4.83 (d, JCH,CH = 11.6 Hz, 1H, CHBn), 4.76 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.64 (d, JCH,CH = 12.3 Hz, 1H, CHBn), 4.61–4.58 (m, 2H, H-1, CHBn), 4.53–4.49 (m, 2H, 2 × CHBn), 4.45 (d, JCH,CH = 25.2 Hz, 2H, NCHBn), 4.41 (d, JCH,CH = 12.0 Hz, 1H, CHBn), 4.39–4.36 (m, 1H, CHBn), 4.28–4.16 (m, 5H, 2 × CHBn, H-6a′, H-4″, H-6a″), 4.13 (dd, JH6b″,H6a″ = 12.0 Hz, JH6b″,H5″ = 4.0 Hz, 1H, H-6b″), 4.07 (dd, JH5′,F = 34.1 Hz, JH5′,H4 = 10.3 Hz, 1H, H-5′), 3.97–3.94 (m, 2H, H-6a‴, H-4), 3.90 (t, JH3′,H4′ = JH3′,H2′ = 9.3 Hz, 1H, H-3′), 3.85–3.68 (m, 6H, H-6b′, H-4′, H-2, H-3, H-5, H-6a), 3.66–3.62 (m, 2H, H-5‴, H-6b‴), 3.53 (ddd, JH5″,H4″ = 9.9, JH5″,H6b″ = 3.9, JH5″,H6a″ = 1.8 Hz, 1H, H-5″), 3.50–3.41 (m, 1H, CHLinker), 3.41–3.35 (m, 2H, H-2′, H-6b), 3.35–3.26 (m, 1H, CHLinker), 3.23–3.16 (m, 1H, CHLinker), 3.15–3.10 (m, 1H, CHLinker), 1.60–1.40 (m, 4H, 4 × CHLinker), 1.31–1.11 (m, 2H, 2 × CHLinker). 13C NMR (200 MHz, CDCl3) δ = 165.8 (2C, 2 × C=O), 165.7 (C=O), 165.5 (C=O), 165.3 (C=O), 165.1 (C=O), 164.7 (C=O), 156.8/156.3 (C=OCbz), 139.3, 138.6, 138.3 (2C), 138.2 (2C), 138.1, 138.03, 137.0/136.9 (9 × Cq), 133.5 (2C), 133.3, 133.2, 130.1, 129.9, 129.8 (2C), 129.7, 129.6, 129.1, 128.8, 128.7 (2C), 128.6 (2C), 128.5, 128.4 (3C), 128.3, 128.1, 128.0 (2C), 127.7 (2C), 127.6, 127.5, 127.4, 127.3, 126.9, 126.2 (32 × C-Ar), 101.6 (C-1″), 100.9 (C-1‴), 99.5 (C-1′), 97.7 (C-1), 81.0 (d, JC6′,F = 170.9 Hz, C-6′), 80.4 (C-3′), 79.7 (C-2′), 77.7 (d, JC4′,F = 3.8 Hz, C-4′), 77.2 (C-3*), 77.1 (C-4*), 76.0 (C-4″), 75.1 (C-2), 74.6 (CHBn), 74.1 (CHBn), 73.3 (2C, C-5″, CHBn), 73.1 (2C, 2 × CHBn),73.0 (C-3″), 72.8 (C-3‴), 72.6 (C-2″), 72.4 (C-5‴), 71.9 (C-2‴), 69.9 (d, JC5′,F = 16.3 Hz, C-5′), 69.6 (C-4‴), 69.3 (C-5), 68.1/68.0 (CHLinker), 67.9 (C-6), 67.3 (CHCbz), 62.7 (C-6‴), 62.4 (C-6″), 50.6/50.3 (NCHBn), 47.3/46.3 (CHLinker), 29.2/29.1 (CHLinker), 28.1/27.6 (CHLinker), 23.5 (CHLinker). Due to signal overlap, 85 out of 128 carbon atoms were assigned. *Assigned by HSQC-Spectrum due to signal superimposition with solvent peak. 19F NMR (377 MHz, CDCl3) δ = −234.9 – −235.4 (m). 1H, 13C-coupled HSQC (CDCl3) JC1,H1 = 169 Hz, JC1′H1′ = 170 Hz, JC1″,H1″ = 160 Hz, JC1‴,H1‴ = 158 Hz. MALDI-TOF calculated for C128H122FNO29Na+ [M + Na]+: 2179.80; found: 2179.60.
5-Aminopentyl-(β-D-glucopyranosyl)-(1→4)-(β-D-glucopyranosyl)-(1→4)-(6-deoxy-6-fluoro-α-D-gluco-pryranosyl)-(1→4)-α-D-galactopyranoside (8). To a stirred solution of 48 (46 mg, 21.3 μmol, 1.0 eq.) in MeOH/THF (v/v = 1:1, 8 mL), NaOMe (640 μL, 0.5 M in MeOH) was added. The reaction solution was stirred for 18 h at ambient temperature before being neutralized by the addition of Amberlite® IR120. The reaction mixture was filtered, and the solvents were removed under reduced pressure. The crude residue was dried for 17 h under high vacuo before it was dissolved in a mixture of MeOH/THF/H2O/AcOH (v/v = 19:5:4:1, 20 mL), and Pd/C (40 mg) was added under Ar atmosphere. The reaction was purged three times with H2 and stirred for 48 h at ambient temperature. The catalyst was filtered off by Celite® Hyflo Supercel, and the solvents were removed under reduced pressure. The crude product was dissolved in H2O/MeOH (v/v = 4:1) and subjected to RP column chromatography (H2O/MeOH, v/v = 4:1) to obtain 8 (15.0 mg, 19.9 μmol, 93% over two steps) after lyophilization. 1H NMR (800 MHz, DMSO-d6) δ = 4.88–4.78 (m, 2H, H-1′, H-6a′), 4.64 (d, JH1,H2 = 3.6 Hz, 1H, H-1), 4.52–4.40 (m, 2H, H-6b′, H-5′), 4.25 (d, JH1‴,H2‴ = 7.9 Hz, 1H, H-1‴), 4.22 (d, JH1″,H2″ = 7.9 Hz, 1H, H-1″), 3.86 (d, JH4,H3 = 3.2 Hz, 1H, H-4), 3.78 (d, JH6a″,H6b″ = 11.3 Hz, 1H, H-6a″), 3.75 (t, JH6a,H6b = JH6a,H5 = 9.4 Hz, 1H, H-6a), 3.70–3.64 (m, 2H, H-3, H-6a‴), 3.62 (t, JH5,H6a = JH5,H6b = 7.1 Hz, 1H, H-5), 3.60–3.54 (m, 4H, H-6b″, CHLinker, H-2, H-3′), 3.46–3.39 (m, 2H, H-6b, H-6b‴), 3.38–3.28 (m, 5H, H-3″, CHLinker, H-4′, H-4″, H-5″), 3.27 (dd, JH2′,H3′ = 9.6 Hz, JH1′,H2′ = 3.6 Hz, 1H, H-2′), 3.19–3.14 (m, 2H, H-3‴, H-5‴), 3.08–3.03 (m, 2H, H-4‴, H-2″), 2.98 (t, JH2″,H1″ = JH2″,H3″ = 8.5 Hz, 1H, H-2″), 2.62 (s, 2H, 2 × CHLinker), 1.56–1.42 (m, 4H, 4 × CHLinker), 1.35 (q, JCH,CH = 7.5 Hz, 2H, 2 × CHLinker). 13C NMR (200 MHz, DMSO-d6) δ =103.2 (C-1‴), 103.1 (C-1″), 99.1 (C-1′), 99.0 (C-1), 81.8 (d, JC6′,F = 167.7 Hz, C-6′), 79.5 (C-4″), 79.4 (d, JC4′,F = 5.2 Hz, C-4′), 76.9 (C-5‴), 76.4 (C-4), 76.3 (C-3‴), 74.9 (C-3″/C-5″), 74.8 (C-3″/C-5″), 73.3 (C-2‴), 73.0 (C-2″), 71.9 (C-2′), 71.3 (C-3′), 71.0 (C-5), 70.0 (C-4‴), 68.5 − 68.3 (3C, C-2, C-3, C-5′), 67.0 (CHLinker), 61.0 (C-6‴), 60.3 (C-6″), 58.7 (C-6), 40.0 (CHLinker), 29.8/28.8 (CHLinker), 24.1/24.0 (CHLinker), 23.0 (CHLinker). 19F-NMR (377 MHz, DMSO-d6) δ = -235.0 (td, JF,H6a′ = JF,H6b′ = 47.6 Hz, JF,H5′ = 33.3 Hz). 1H-13C-coupled HSQC (DMSO-d6) JC1,H1 = 167 Hz, JC1′H1′ = 169 Hz, JC1″,H1″ = 160 Hz, JC1‴,H1‴ = 161 Hz. HRMS (ESI+) calculated for C29H53FNO20+ [M + H]+: 754.3139; found: 754.3142.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}