

Autoimmune Pancreatitis Mimicking a Pancreatic Neuroendocrine Tumor: A Case Report with a Literature Review



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
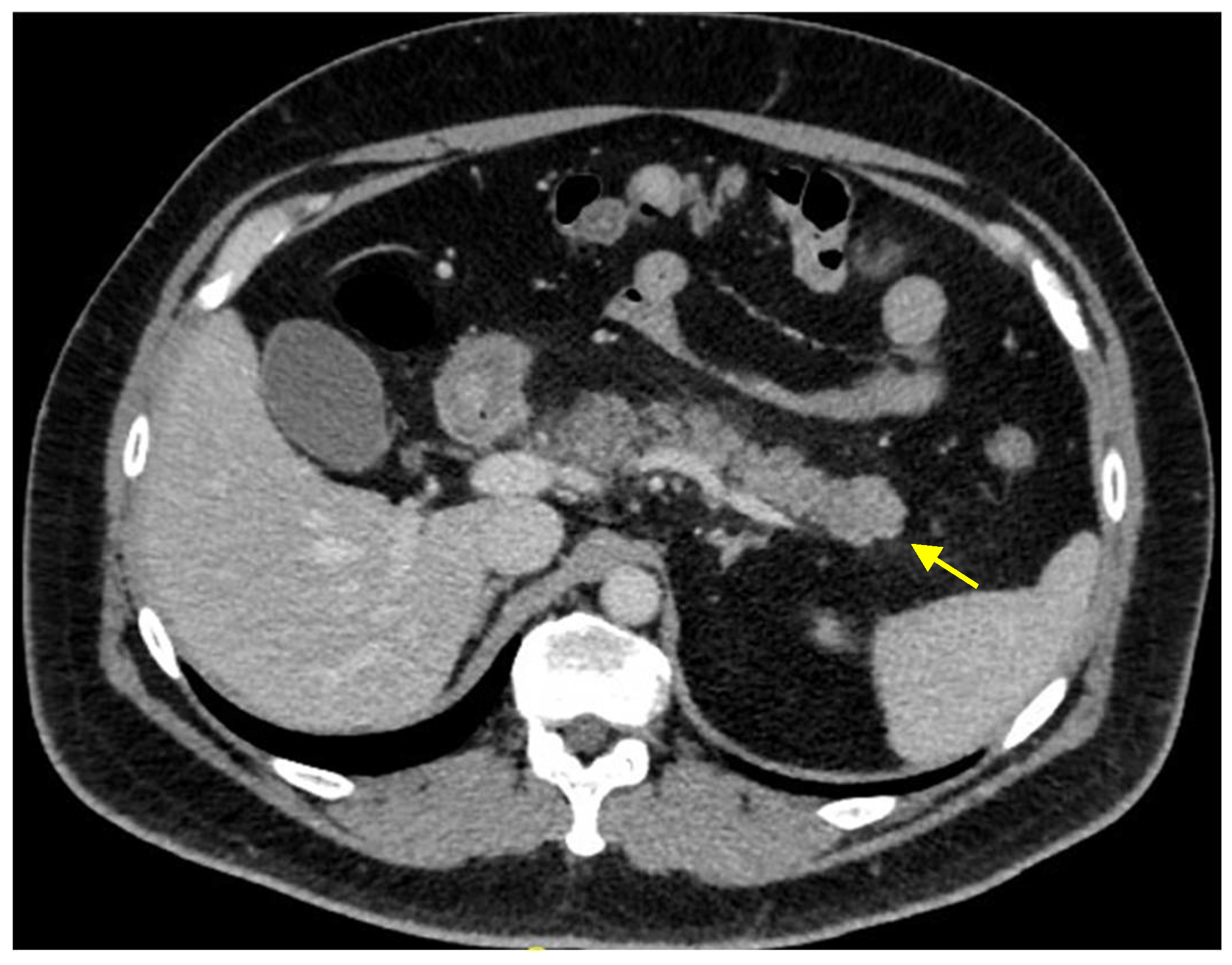
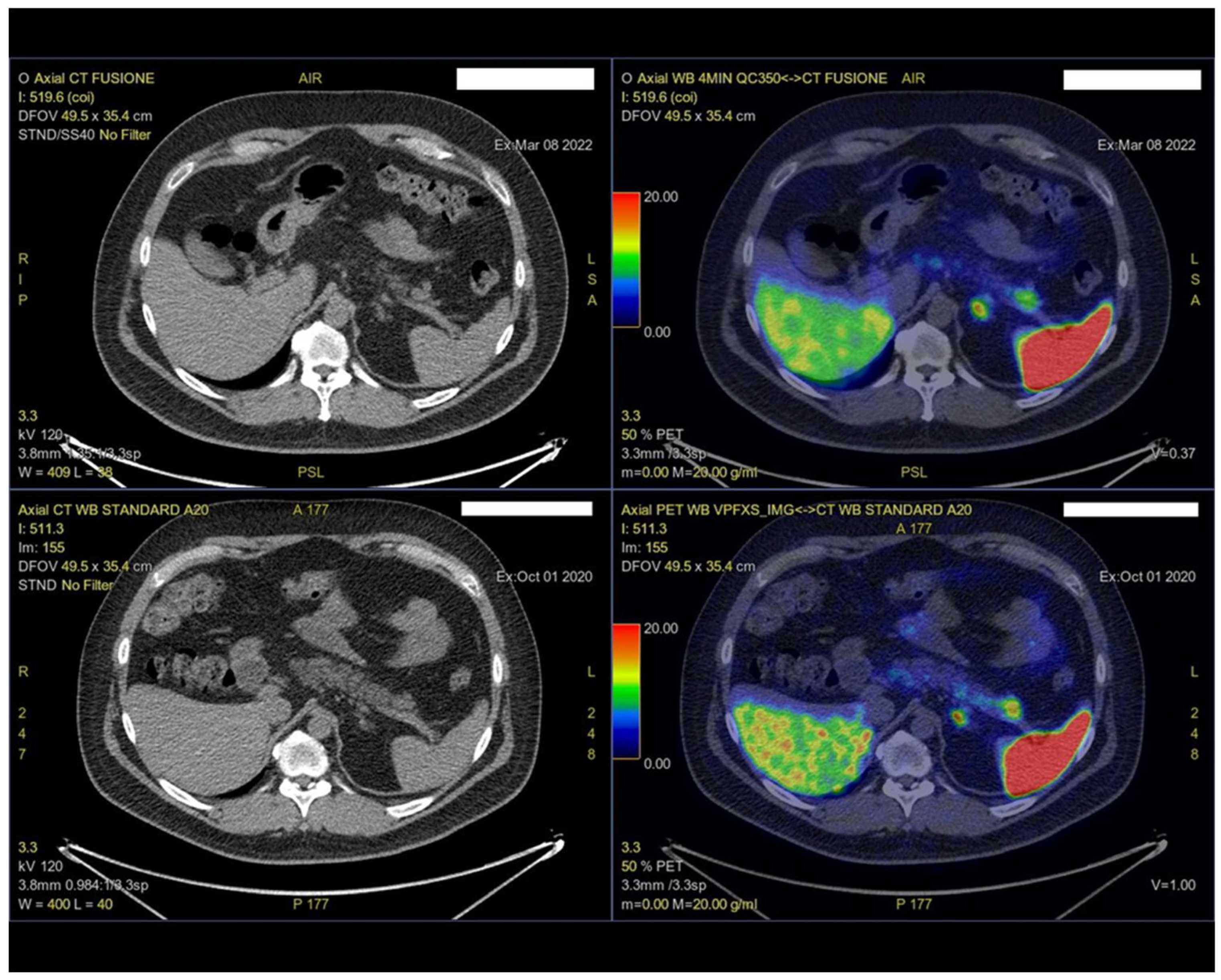
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shimosegawa, T.; Chari, S.T.; Frulloni, L.; Kamisawa, T.; Kawa, S.; Mino-Kenudson, M.; Kim, M.H.; Klöppel, G.; Lerch, M.M.; Löhr, M.; et al. International consensus diagnostic criteria for autoimmune pancreatitis: Guidelines of the International Association of Pancreatology. Pancreas 2011, 40, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Hamada, S.; Tsuji, I.; Takeyama, Y.; Shimosegawa, T.; Okazaki, K. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2016. J. Gastroenterol. 2020, 55, 462–470. [Google Scholar] [CrossRef]
- Löhr, J.M.; Beuers, U.; Vujasinovic, M.; Alvaro, D.; Frøkjær, J.B.; Buttgereit, F.; Capurso, G.; Culver, E.L.; de-Madaria, E.; Della-Torre, E.; et al. European Guideline on IgG4-related digestive disease—UEG and SGF evidence-based recommendations. United Eur. Gastroenterol. J. 2020, 8, 637–666. [Google Scholar] [CrossRef]
- Gallo, C.; Dispinzieri, G.; Zucchini, N.; Invernizzi, P.; Massironi, S. Autoimmune pancreatitis: Cornerstones and future perspectives. World J. Gastroenterol. 2024, 30, 817–832. [Google Scholar] [CrossRef] [PubMed]
- de Pretis, N.; Frulloni, L. Autoimmune pancreatitis type 2. Curr. Opin. Gastroenterol. 2020, 36, 417–420. [Google Scholar] [CrossRef]
- Chari, S.T.; Smyrk, T.C.; Levy, M.J.; Topazian, M.D.; Takahashi, N.; Zhang, L.; Clain, J.E.; Pearson, R.K.; Petersen, B.T.; Vege, S.S.; et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin. Gastroenterol. Hepatol. 2006, 4, 1010–1016, quiz 1934. [Google Scholar] [CrossRef]
- Okazaki, K.; Kawa, S.; Kamisawa, T.; Naruse, S.; Tanaka, S.; Nishimori, I.; Ohara, H.; Ito, T.; Kiriyama, S.; Inui, K.; et al. Clinical diagnostic criteria of autoimmune pancreatitis: Revised proposal. J. Gastroenterol. 2006, 41, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Kawa, S.; Kamisawa, T.; Ikeura, T.; Itoi, T.; Ito, T.; Inui, K.; Irisawa, A.; Uchida, K.; Ohara, H.; et al. Amendment of the Japanese consensus guidelines for autoimmune pancreatitis, 2020. J. Gastroenterol. 2022, 57, 225–245. [Google Scholar] [CrossRef]
- Bennis, R.; Roy, T.; Atto, Y.N.; Colin, M.I. La maladie systémique à IgG4, une cause rare de pancréatite aigüe sévère. Louvain Med. 2020, 139, 185–191. [Google Scholar]
- Nikolic, S.; Lanzillotta, M.; Panic, N.; Brismar, T.B.; Moro, C.F.; Capurso, G.; Della Torre, E.; Löhr, J.M.; Vujasinovic, M. Unraveling the relationship between autoimmune pancreatitis type 2 and inflammatory bowel disease: Results from two centers and systematic review of the literature. United Eur. Gastroenterol. J. 2022, 10, 496–506. [Google Scholar] [CrossRef]
- Tan, B.; Zhang, B.; Chen, H. Gastroenteropancreatic neuroendocrine neoplasms: Epidemiology, genetics, and treatment. Front. Endocrinol. 2024, 15, 1424839. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Massironi, S. The Increasing Incidence of Neuroendocrine Neoplasms Worldwide: Current Knowledge and Open Issues. J. Clin. Med. 2022, 11, 3794. [Google Scholar] [CrossRef]
- Kos-Kudła, B.; Castaño, J.P.; Denecke, T.; Grande, E.; Kjaer, A.; Koumarianou, A.; de Mestier, L.; Partelli, S.; Perren, A.; Stättner, S.; et al. European Neuroendocrine Tumour Society (ENETS) 2023 guidance paper for nonfunctioning pancreatic neuroendocrine tumours. J. Neuroendocrinol. 2023, 35, e13343. [Google Scholar] [CrossRef]
- Hallet, J.; Law, C.H.; Cukier, M.; Saskin, R.; Liu, N.; Singh, S. Exploring the rising incidence of neuroendocrine tumors: A population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer 2015, 121, 589–597. [Google Scholar] [CrossRef] [PubMed]
- McKenna, L.R.; Edil, B.H. Update on pancreatic neuroendocrine tumors. Gland Surg. 2014, 3, 258–275. [Google Scholar] [PubMed]
- Hofland, J.; Falconi, M.; Christ, E.; Castaño, J.P.; Faggiano, A.; Lamarca, A.; Perren, A.; Petrucci, S.; Prasad, V.; Ruszniewski, P.; et al. European Neuroendocrine Tumor Society 2023 guidance paper for functioning pancreatic neuroendocrine tumour syndromes. J. Neuroendocrinol. 2023, 35, e13318. [Google Scholar] [CrossRef]
- Ro, C.; Chai, W.; Yu, V.E.; Yu, R. Pancreatic neuroendocrine tumors: Biology, diagnosis, and treatment. Chin. J. Cancer 2013, 32, 312–324. [Google Scholar] [CrossRef]
- Massironi, S. The diagnostic challenges of functioning neuroendocrine tumors: Balancing accuracy, availability, and personalized care. Expert Rev. Endocrinol. Metab. 2024, 19, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Massironi, S.; Franchina, M.; Ippolito, D.; Elisei, F.; Falco, O.; Maino, C.; Pagni, F.; Elvevi, A.; Guerra, L.; Invernizzi, P. Improvements and future perspective in diagnostic tools for neuroendocrine neoplasms. Expert Rev. Endocrinol. Metab. 2024, 19, 349–366. [Google Scholar] [CrossRef]
- Zilli, A.; Arcidiacono, P.G.; Conte, D.; Massironi, S. Clinical impact of endoscopic ultrasonography on the management of neuroendocrine tumors: Lights and shadows. Dig. Liver Dis. 2018, 50, 6–14. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, R.; Cingarlini, S.; Tinazzi Martini, P.; Ortolani, S.; Butturini, G.; Landoni, L.; Regi, P.; Girelli, R.; Capelli, P.; Gobbo, S.; et al. Pancreatic neuroendocrine neoplasms: Magnetic resonance imaging features according to grade and stage. World J. Gastroenterol. 2017, 23, 275–285. [Google Scholar] [CrossRef]
- Lee, L.; Ito, T.; Jensen, R.T. Imaging of pancreatic neuroendocrine tumors: Recent advances, current status, and controversies. Expert Rev. Anticancer. Ther. 2018, 18, 837–860. [Google Scholar] [CrossRef]
- Khanna, L.; Prasad, S.R.; Sunnapwar, A.; Kondapaneni, S.; Dasyam, A.; Tammisetti, V.S.; Salman, U.; Nazarullah, A.; Katabathina, V.S. Pancreatic Neuroendocrine Neoplasms: 2020 Update on Pathologic and Imaging Findings and Classification. Radiographics 2020, 40, 1240–1262. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Poon, R.; Wong, R.; Metser, U. 68Ga PET Imaging in Patients With Neuroendocrine Tumors: A Systematic Review and Meta-analysis. Clin. Nucl. Med. 2018, 43, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Kan, Y.; Yang, J.G. Clinical value of (68)Ga-DOTA-SSTR PET/CT in the diagnosis and detection of neuroendocrine tumors of unknown primary origin: A systematic review and meta-analysis. Acta Radiol. 2021, 62, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, N.; Osoegawa, T.; Lee, L.; Tachibana, Y.; Aso, A.; Kubo, H.; Kawabe, K.; Igarashi, H.; Nakamura, K.; Oda, Y.; et al. Efficacy of endoscopic ultrasonography and endoscopic ultrasonography-guided fine-needle aspiration for the diagnosis and grading of pancreatic neuroendocrine tumors. Scand. J. Gastroenterol. 2016, 51, 245–252. [Google Scholar] [CrossRef]
- Onda, S.; Okamoto, T.; Kanehira, M.; Fujioka, S.; Harada, T.; Hano, H.; Fukunaga, M.; Yanaga, K. Histopathologically proven autoimmune pancreatitis mimicking neuroendocrine tumor or pancreatic cancer. Case Rep. Gastroenterol. 2012, 6, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Nada, R.; Dey, P.; Basher, R.K.; Gupta, R.; Rana, S.S. Autoimmune Pancreatitis Mimicking Neuroendocrine Tumor of Pancreas. Dig. Dis. Sci. 2023, 68, 3479–3481. [Google Scholar] [CrossRef]
- Łapińska, G.; Bryszewska, M.; Fijołek-Warszewska, A.; Kozłowicz-Gudzińska, I.; Ochman, P.; Sackiewicz-Słaby, A. The diagnostic role of 68Ga-DOTATATE PET/CT in the detection of neuroendocrine tumours. Nucl. Med. Rev. Cent East Eur. 2011, 14, 16–20. [Google Scholar] [CrossRef]
- Chang, C.Y.; Chuang, S.C.; Ma, Y.C. A rare case of type 1 autoimmune pancreatitis combined with a pancreatic neuroendocrine tumor. Kaohsiung J. Med. Sci. 2023, 39, 191–192. [Google Scholar] [CrossRef]
- Salahshour, F.; Taslimi, R.; Moosavi, N.S.; Yazdi, N.A.; Esfandbod, M. Pancreatic Neuroendocrine Tumor presenting as a diffuse pancreatic enlargement, case report and review of literature. J. Radiol. Case Rep. 2021, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franchina, M.; Dell’Oro, L.; Massironi, S. Autoimmune Pancreatitis Mimicking a Pancreatic Neuroendocrine Tumor: A Case Report with a Literature Review. Int. J. Mol. Sci. 2025, 26, 1536. https://doi.org/10.3390/ijms26041536
Franchina M, Dell’Oro L, Massironi S. Autoimmune Pancreatitis Mimicking a Pancreatic Neuroendocrine Tumor: A Case Report with a Literature Review. International Journal of Molecular Sciences. 2025; 26(4):1536. https://doi.org/10.3390/ijms26041536
Chicago/Turabian StyleFranchina, Marianna, Liliana Dell’Oro, and Sara Massironi. 2025. "Autoimmune Pancreatitis Mimicking a Pancreatic Neuroendocrine Tumor: A Case Report with a Literature Review" International Journal of Molecular Sciences 26, no. 4: 1536. https://doi.org/10.3390/ijms26041536
APA StyleFranchina, M., Dell’Oro, L., & Massironi, S. (2025). Autoimmune Pancreatitis Mimicking a Pancreatic Neuroendocrine Tumor: A Case Report with a Literature Review. International Journal of Molecular Sciences, 26(4), 1536. https://doi.org/10.3390/ijms26041536