
Mechanistic Insights into CYP199A4-Catalyzed α-Hydroxyketone Formation and Hydrogen Bond-Assisted C–C Bond Cleavage Catalyzed by the CYP199A4 F182L Mutant

Abstract

1. Introduction
2. Results and Discussion
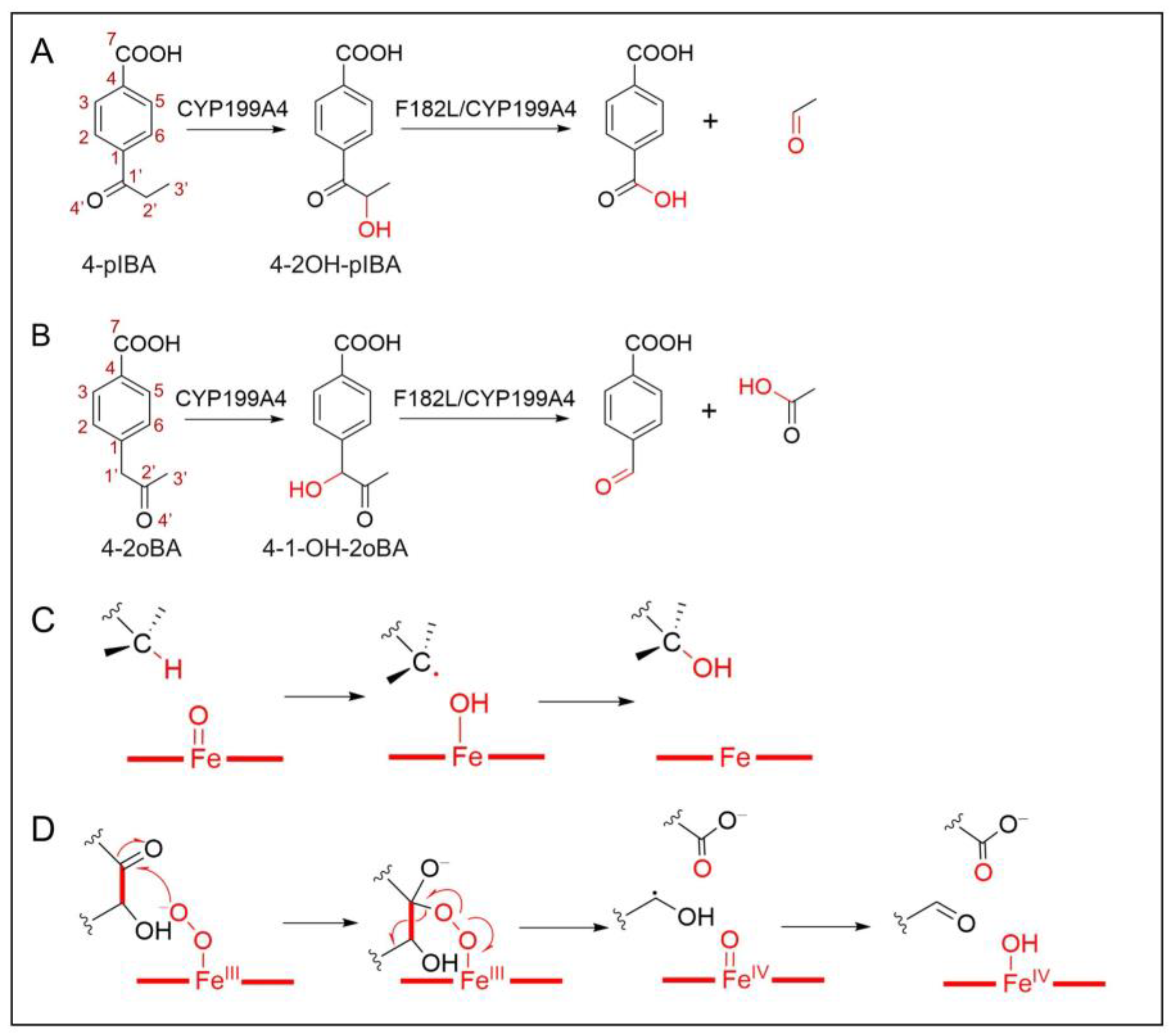
2.1. Target Systems of the C–H Bond Hydroxylation
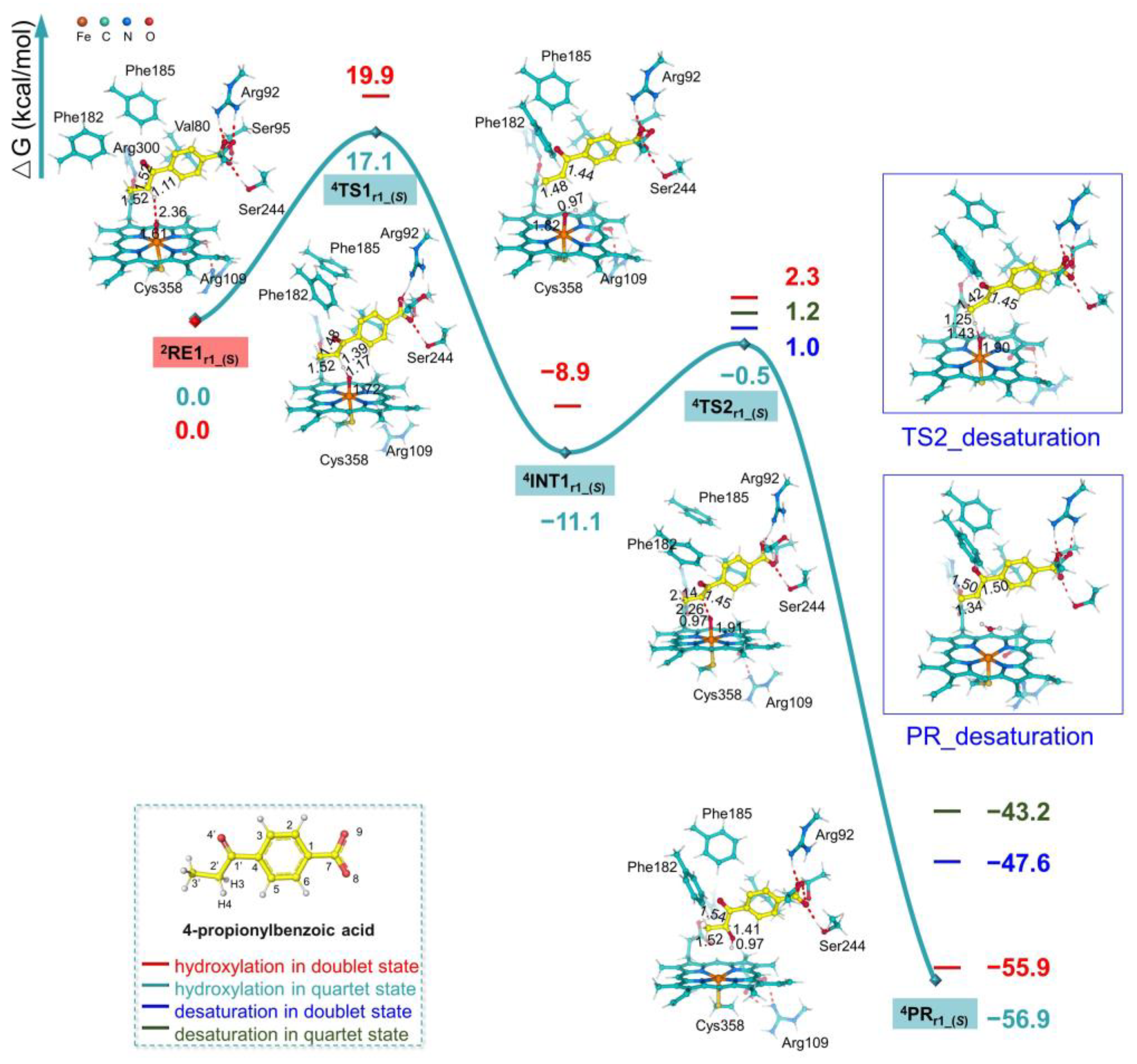
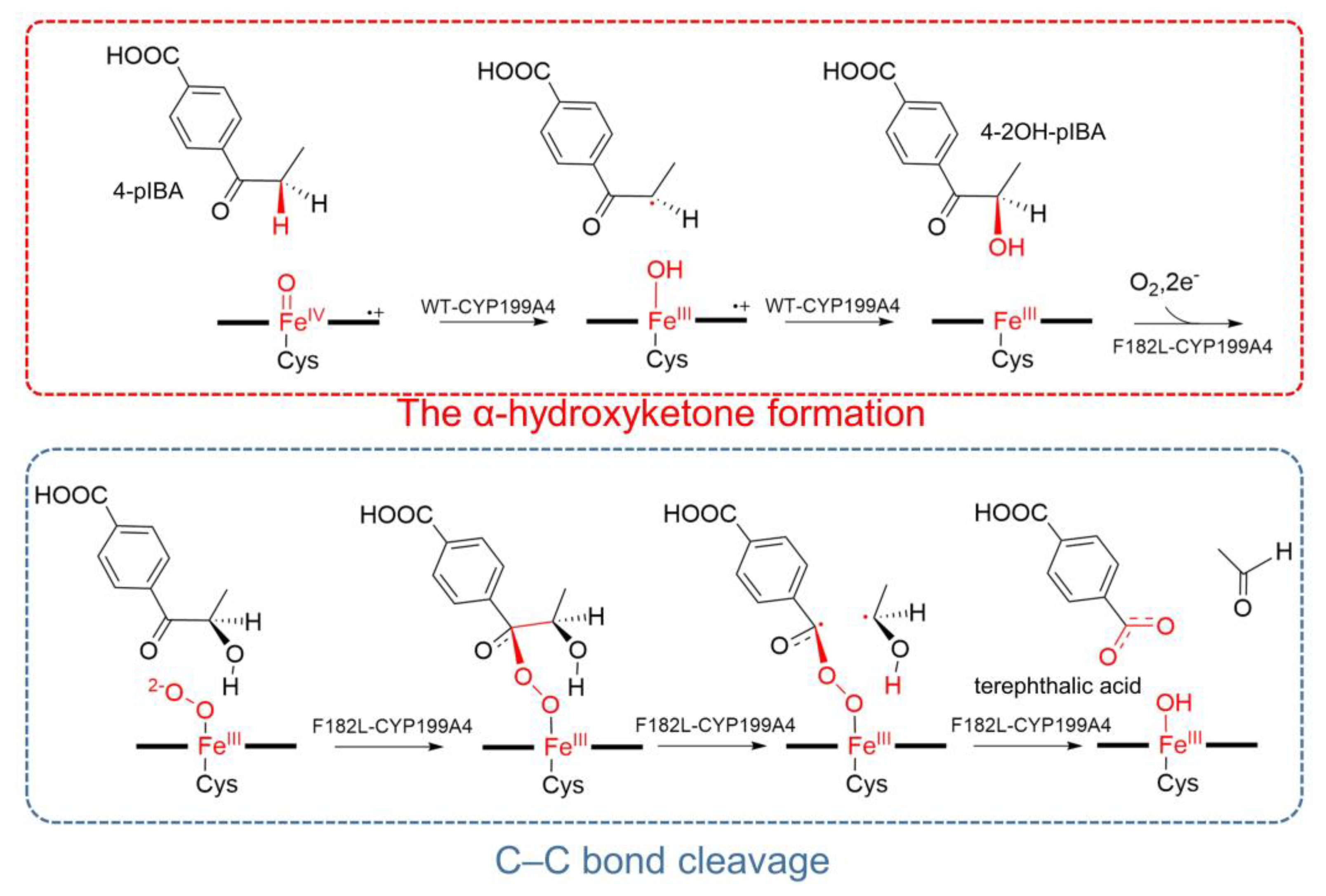
2.2. Mechanism of α-Hydroxyketone Formation from 4-pIBA Catalyzed by CYP199A4
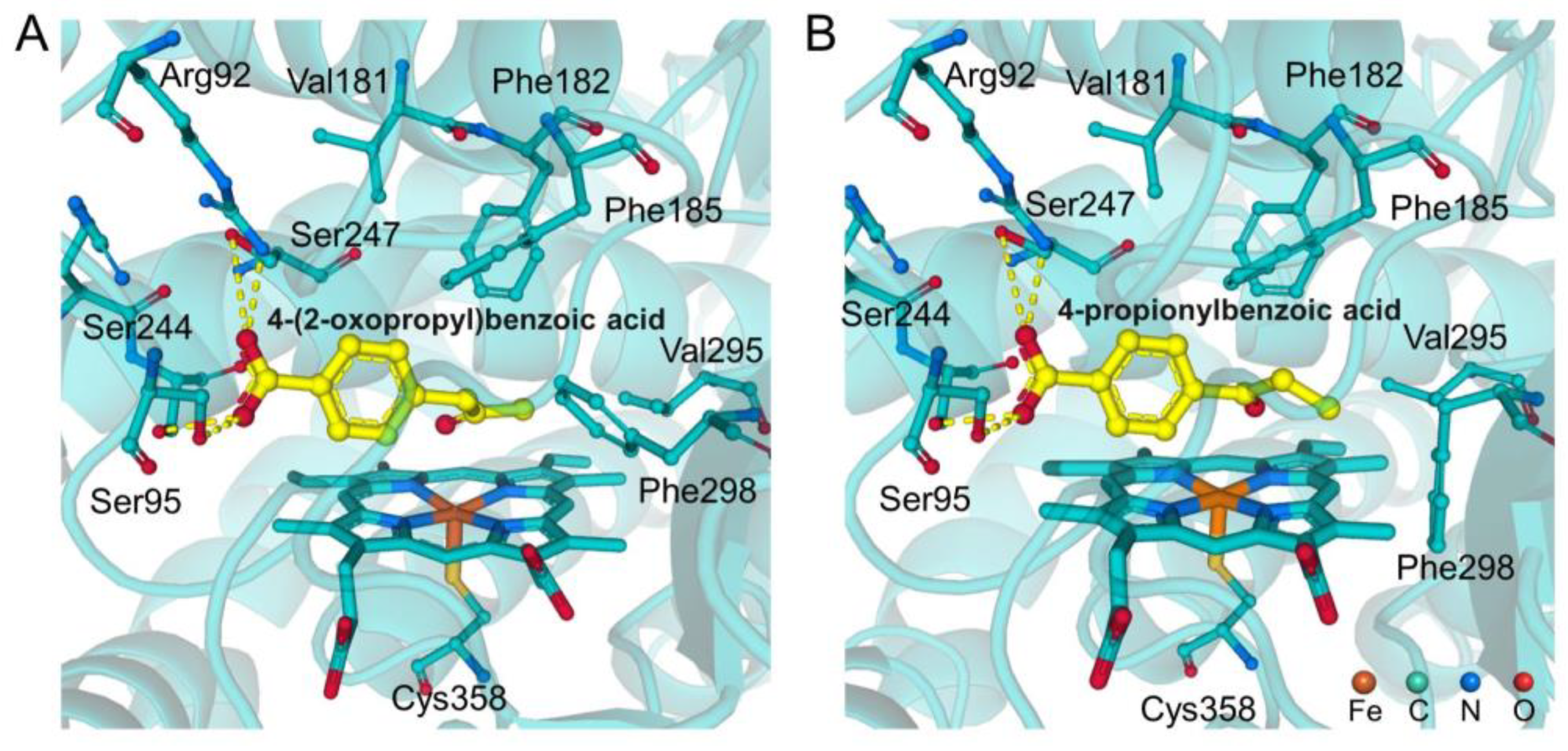
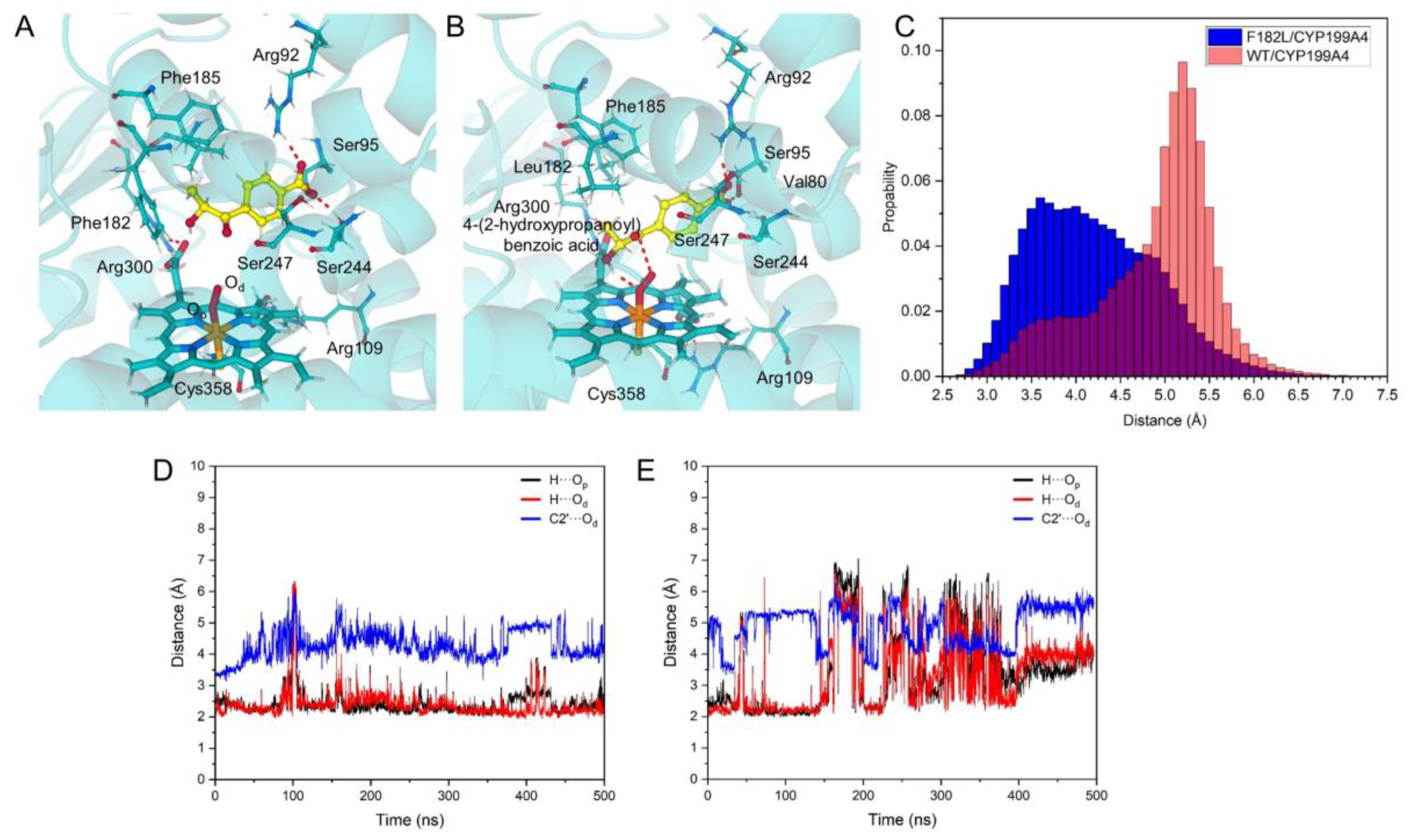
2.3. Blocking Mechanism of Phe182 in (S)-4-2OH-pIBA C–C Cleavage Reactions by CYP199A4
2.4. C–C Bond Cleavage Mechanism of 4-2OH-pIBA by CYP199A4
2.5. Substrate Design for the WT-CYP199A4 Catalyzed C–C Bond Cleavage
3. Materials and Methods
3.1. Classical MD Simulations
3.2. QM/MM MD
3.3. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liang, Y.F.; Bilal, M.; Tang, L.Y.; Wang, T.Z.; Guan, Y.Q.; Cheng, Z.; Zhu, M.; Wei, J.; Jiao, N. Carbon-Carbon Bond Cleavage for Late-Stage Functionalization. Chem. Rev. 2023, 123, 12313–12370. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Yoshimoto, F.K. Formation and Cleavage of C-C Bonds by Enzymatic Oxidation-Reduction Reactions. Chem. Rev. 2018, 118, 6573–6655. [Google Scholar] [CrossRef] [PubMed]
- Jun, C. Transition Metal-Catalyzed Carbon—Carbon Bond Activation. Chem. Soc. Rev. 2004, 33, 610–618. [Google Scholar] [CrossRef]
- Strehl, J.; Kahrs, C.; Müller, T.; Hilt, G.; Christoffers, J. Electrochemical-Induced Ring Transformation of Cyclic α-(Ortho-Iodophenyl)-β-Oxoesters. Chem.—A Eur. J. 2020, 26, 3222–3225. [Google Scholar] [CrossRef]
- Nishimoto, M.; Uetake, Y.; Yakiyama, Y.; Sakurai, H. Strain-Induced Carbon-Carbon Bond Cleavage of Bowl-Shaped Sumanenone. Chem. Commun. 2024, 60, 3982–3985. [Google Scholar] [CrossRef]
- Sivaguru, P.; Wang, Z.; Zanoni, G.; Bi, X. Cleavage of Carbon-Carbon Bonds by Radical Reactions. Chem. Soc. Rev. 2019, 48, 2615–2656. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Ortiz De Montellano, P.R. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef]
- Krest, C.M.; Onderko, E.L.; Yosca, T.H.; Calixto, J.C.; Karp, R.F.; Livada, J.; Rittle, J.; Green, M.T. Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem. 2013, 288, 17074–17081. [Google Scholar] [CrossRef]
- Groves, J.T. Cytochrome P450 Enzymes: Understanding the Biochemical Hieroglyphs. F1000Research 2015, 4, 1–8. [Google Scholar] [CrossRef]
- Bernhardt, R. Cytochromes P450 as Versatile Biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Goldberg, M. The P-450 System. Clin. Rev. 1996, 5, 1–23. [Google Scholar]
- Dawson, J.H.; Sono, M. Cytochrome P-450 and Chloroperoxidase: Thiolate-Ligated Heme Enzymes. Spectroscopic Determination of Their Active Site Structures and Mechanistic Implications of Thiolate Ligation. Chem. Rev. 1987, 87, 1255–1276. [Google Scholar] [CrossRef]
- Podgorski, M.N.; Harbort, J.S.; Coleman, T.; Stok, J.E.; Yorke, J.A.; Wong, L.L.; Bruning, J.B.; Bernhardt, P.V.; De Voss, J.J.; Harmer, J.R.; et al. Biophysical Techniques for Distinguishing Ligand Binding Modes in Cytochrome P450 Monooxygenases. Biochemistry 2020, 59, 1038–1050. [Google Scholar] [CrossRef]
- Coleman, T.; Wong, S.H.; Podgorski, M.N.; Bruning, J.B.; De Voss, J.J.; Bell, S.G. Cytochrome P450 CYP199A4 from Rhodopseudomonas Palustris Catalyzes Heteroatom Dealkylations, Sulfoxidation, and Amide and Cyclic Hemiacetal Formation. ACS Catal. 2018, 8, 5915–5927. [Google Scholar] [CrossRef]
- Podgorski, M.N.; Coleman, T.; Chao, R.R.; De Voss, J.J.; Bruning, J.B.; Bell, S.G. Investigation of the Requirements for Efficient and Selective Cytochrome P450 Monooxygenase Catalysis across Different Reactions. J. Inorg. Biochem. 2020, 203, 110913. [Google Scholar] [CrossRef]
- Bell, S.G.; Zhou, R.; Yang, W.; Tan, A.B.H.; Gentleman, A.S.; Wong, L.L.; Zhou, W. Investigation of the Substrate Range of CYP199A4: Modification of the Partition between Hydroxylation and Desaturation Activities by Substrate and Protein Engineering. Chem.—A Eur. J. 2012, 18, 16677–16688. [Google Scholar] [CrossRef]
- Coleman, T.; Stok, J.E.; Podgorski, M.N.; Bruning, J.B.; De Voss, J.J.; Bell, S.G. Structural Insights into the Role of the Acid-Alcohol Pair of Residues Required for Dioxygen Activation in Cytochrome P450 Enzymes. J. Biol. Inorg. Chem. 2020, 25, 583–596. [Google Scholar] [CrossRef]
- Bell, S.G.; Tan, A.B.H.; Johnson, E.O.D.; Wong, L.L. Selective Oxidative Demethylation of Veratric Acid to Vanillic Acid by CYP199A4 from Rhodopseudomonas Palustris HaA2. Mol. Biosyst. 2009, 6, 206–214. [Google Scholar] [CrossRef]
- Bell, S.G.; Yang, W.; Tan, A.B.H.; Zhou, R.; Johnson, E.O.D.; Zhang, A.; Zhou, W.; Rao, Z.; Wong, L.L. The Crystal Structures of 4-Methoxybenzoate Bound CYP199A2 and CYP199A4: Structural Changes on Substrate Binding and the Identification of an Anion Binding Site. Dalt. Trans. 2012, 41, 8703–8714. [Google Scholar] [CrossRef]
- Lee, J.H.Z.; Coleman, T.; Mclean, M.A.; Podgorski, M.N.; Hayball, E.F.; Stone, I.S.J.; Bruning, J.B.; Whelan, F.; De Voss, J.J.; Sligar, S.G.; et al. Selective α-Hydroxyketone Formation and Subsequent C-C Bond Cleavage by Cytochrome P450 Monooxygenase Enzymes. ACS Catal. 2024, 14, 8958–8971. [Google Scholar] [CrossRef] [PubMed]
- Chao, R.R.; De Voss, J.J.; Bell, S.G. The Efficient and Selective Catalytic Oxidation of Para-Substituted Cinnamic Acid Derivatives by the Cytochrome P450 Monooxygenase, CYP199A4. RSC Adv. 2016, 6, 55286–55297. [Google Scholar] [CrossRef]
- Coleman, T.; Chao, R.R.; De Voss, J.J.; Bell, S.G. Biochimica et Biophysica Acta The Importance of the Benzoic Acid Carboxylate Moiety for Substrate Recognition by CYP199A4 from Rhodopseudomonas Palustris HaA2. BBA—Proteins Proteom. 2016, 1864, 667–675. [Google Scholar] [CrossRef]
- Mokkawes, T.; de Visser, S.P. Caffeine Biodegradation by Cytochrome P450 1A2. What Determines the Product Distributions? Chem.—A Eur. J. 2023, 29, e202203875. [Google Scholar] [CrossRef]
- Miller, J.C.; Lee, J.H.Z.; Mclean, M.A.; Chao, R.R.; Stone, I.S.J.; Pukala, T.L.; Bruning, J.B.; De Voss, J.J.; Schuler, M.A.; Sligar, S.G.; et al. Engineering C-C Bond Cleavage Activity into a P450 Monooxygenase Enzyme. J. Am. Chem. Soc. 2023, 145, 9207–9222. [Google Scholar] [CrossRef]
- Bonomo, S.; Jørgensen, F.S.; Olsen, L. Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J. Chem. Inf. Model. 2017, 57, 1123–1133. [Google Scholar] [CrossRef]
- Kalita, S.; Shaik, S.; Dubey, K.D. Mechanistic Conundrum of C-C Bond Cleavage by CYP51. ACS Catal. 2022, 12, 5673–5683. [Google Scholar] [CrossRef]
- de Montellano, P.R.O. Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed.; Springer Science & Business Media: Dordrecht, The Netherlands, 2015; ISBN 9783319121086. [Google Scholar]
- Henry, K.M.; Townsend, C.A. Ordering the Reductive and Cytochrome P450 Oxidative Steps in Demethylsterigmatocystin Formation Yields General Insights into the Biosynthesis of Aflatoxin and Related Fungal Metabolites. J. Am. Chem. Soc. 2005, 127, 3724–3733. [Google Scholar] [CrossRef]
- Mizutani, M.; Sato, F. Unusual P450 Reactions in Plant Secondary Metabolism. Arch. Biochem. Biophys. 2011, 507, 194–203. [Google Scholar] [CrossRef]
- Liu, S.H.; Sun, J.L.; Hu, Y.L.; Zhang, L.; Zhang, X.; Yan, Z.Y.; Guo, X.; Guo, Z.K.; Jiao, R.H.; Zhang, B.; et al. Biosynthesis of Sordarin Revealing a Diels–Alderase for the Formation of the Norbornene Skeleton. Angew. Chem. 2022, 134, 1–7. [Google Scholar] [CrossRef]
- Moraga, J.; Dalmais, B.; Izquierdo-Bueno, I.; Aleu, J.; Hanson, J.R.; Hernández-Galán, R.; Viaud, M.; Collado, I.G. Genetic and Molecular Basis of Botrydial Biosynthesis: Connecting Cytochrome P450-Encoding Genes to Biosynthetic Intermediates. ACS Chem. Biol. 2016, 11, 2838–2846. [Google Scholar] [CrossRef] [PubMed]
- Sen, K.; Hackett, J.C.; Street, E.L. Peroxo—Iron Mediated Deformylation in Sterol 14α-Demethylase Catalysis. J. Am. Chem. Soc. 2010, 132, 10293–10305. [Google Scholar] [CrossRef]
- Dias, A.H.S.; Cao, Y.; Skaf, M.S.; de Visser, S.P. Machine Learning-Aided Engineering of a Cytochrome P450 for Optimal Bioconversion of Lignin Fragments. Phys. Chem. Chem. Phys. 2024, 26, 17577–17587. [Google Scholar] [CrossRef]
- Cantú Reinhard, F.G.; Lin, Y.T.; Stańczak, A.; de Visser, S.P. Bioengineering of Cytochrome P450 OleTJE: How Does Substrate Positioning Affect the Product Distributions? Molecules 2020, 25, 2675. [Google Scholar] [CrossRef] [PubMed]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic Structure of Compound I in Human Isoforms of Cytochrome P450 from QM/MM Modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Ouyang, Q.; Wang, X.; Li, X.; Tan, H.; Chen, G. Interactive Regulation between Aliphatic Hydroxylation and Aromatic Hydroxylation of Thaxtomin D in TxtC: A Theoretical Investigation. Inorg. Chem. 2021, 60, 6433–6445. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, T.; Nishimi, T.; Yoshizawa, K. A New Understanding on How Heme Metabolism Occurs in Heme Oxygenase: Water-Assisted Oxo Mechanism. Dalt. Trans. 2012, 41, 11642–11650. [Google Scholar] [CrossRef]
- Field, M.J.; Oyala, P.H.; Green, M.T. 17O Electron Nuclear Double Resonance Analysis of Compound I: Inverse Correlation between Oxygen Spin Population and Electron Donation. J. Am. Chem. Soc. 2022, 144, 19272–19283. [Google Scholar] [CrossRef]
- Kamachi, T.; Yoshizawa, K. A Theoretical Study on the Mechanism of Camphor Hydroxylation by Compound I of Cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652–4661. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Wang, Y.; Yang, C.; Yang, L.; Han, K. Theoretical Study of the Mechanism of Acetaldehyde Hydroxylation by Compound i of CYP2E1. J. Phys. Chem. B 2006, 110, 6154–6159. [Google Scholar] [CrossRef] [PubMed]
- Pickl, M.; Kurakin, S.; Cantú Reinhard, F.G.; Schmid, P.; Pöcheim, A.; Winkler, C.K.; Kroutil, W.; De Visser, S.P.; Faber, K. Mechanistic Studies of Fatty Acid Activation by CYP152 Peroxygenases Reveal Unexpected Desaturase Activity. ACS Catal. 2019, 9, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Isobe, H.; Yamaguchi, K.; Okumura, M.; Shimada, J. Role of Perferryl-Oxo Oxidant in Alkane Hydroxylation Catalyzed by Cytochrome P450: A Hybrid Density Functional Study. J. Phys. Chem. B 2012, 116, 4713–4730. [Google Scholar] [CrossRef]
- Zhou, T.P.; Feng, J.; Wang, Y.; Li, S.; Wang, B. Substrate Conformational Switch Enables the Stereoselective Dimerization in P450 NascB: Insights from Molecular Dynamics Simulations and Quantum Mechanical/Molecular Mechanical Calculations. JACS Au 2024, 4, 1591–1604. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Han, K. Recent Density Functional Theory Model Calculations of Drug Metabolism by Cytochrome P450. Coord. Chem. Rev. 2012, 256, 1137–1150. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; De Visser, S.P. A Valence Bond Modeling of Trends in Hydrogen Abstraction Barriers and Transition States of Hydroxylation Reactions Catalyzed by Cytochrome P450 Enzymes. J. Am. Chem. Soc. 2008, 130, 14016. [Google Scholar] [CrossRef]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; Van Eldik, R.; Liu, W.; De Visser, S.P. Drug Metabolism by Cytochrome P450 Enzymes: What Distinguishes the Pathways Leading to Substrate Hydroxylation over Desaturation? Chem.—A Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef]
- Kumar, D.; De Visser, S.P.; Shaik, S. Oxygen Economy of Cytochrome P450: What Is the Origin of the Mixed Functionality as a Dehydrogenase-Oxidase Enzyme Compared with Its Normal Function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef]
- Coleman, T.; Doherty, D.Z.; Zhang, T.; Podgorski, M.N.; Qiao, R.; Lee, J.H.Z.; Bruning, J.B.; De Voss, J.J.; Zhou, W.; Bell, S.G. Exploring the Factors Which Result in Cytochrome P450 Catalyzed Desaturation Versus Hydroxylation. Chem.—Asian J. 2022, 17, e202200986. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Update to the General Amber Force Field for Small Solutes with an Emphasis on Free Energies of Hydration. J. Phys. Chem. B 2014, 118, 3793–3804. [Google Scholar] [CrossRef]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. Quantum Mechanically Derived AMBER-Compatible Heme Parameters for Various States of the Cytochrome P450 Catalytic Cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Gill, P.M.W.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The Performance of the Becke-Lee-Yang-Parr (B-LYP) Density Functional Theory with Various Basis Sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An Efficient Real Space Multigrid QM/MM Electrostatic Coupling. J. Chem. Theory Comput. 2005, 1, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; Vandevondele, J. Cp2k: Atomistic Simulations of Condensed Matter Systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Himo, F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef]
- Himo, F.; de Visser, S.P. Status Report on the Quantum Chemical Cluster Approach for Modeling Enzyme Reactions. Commun. Chem. 2022, 5, 20–23. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Gan, Z.; Feng, J.; Yin, J.; Huang, J.; Wang, B.; Zhang, J.Z.H. Diverse Mechanisms for the Aromatic Hydroxylation: Insights into the Mechanisms of the Coumarin Hydroxylation by CYP2A6. ACS Catal. 2024, 14, 16277–16286. [Google Scholar] [CrossRef]
- Li, N.; Yan, S.; Wu, P.; Li, J.; Wang, B. Local Electric Fields Drives the Proton-Coupled Electron Transfer within Cytochrome P450 Reductase. ACS Catal. 2024, 14, 7893–7900. [Google Scholar] [CrossRef]
- Xiao, F.; Zhou, T.P.; Dong, S.; Li, T.; Yun, C.H.; Feng, Y.; Cui, Q.; Hong, K.; Wang, B.; Li, W. Molecular Basis for the P450-Catalyzed Sp3 C-N Glycosidic Bond Formation in Staurosporine Biosynthesis. ACS Catal. 2024, 14, 14274–14284. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision B.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spin Densities | |||||
| 2RE1r1_(S) | 4TS1r1_(S) | 4INT1r1_(S) | 4TS2r1_(S) | 4PR1r1_(S) | |
| Porphrin | −0.25 | 0.17 | −0.14 | −0.14 | −0.05 |
| Fe | 1.29 | 1.16 | 1.78 | 2.12 | 2.56 |
| oxo | 0.81 | 0.54 | 0.16 | 0.02 | 0.00 |
| 4-pIBA | 0.00 | 0.47 | 1.00 | 0.83 | 0.00 |
| C2’ | 0.00 | 0.36 | 0.72 | 0.56 | 0.00 |
| O4’ | 0.00 | 0.10 | 0.27 | 0.23 | 0.00 |
| H4 | 0.00 | −0.04 | 0.00 | 0.00 | 0.00 |
| Cys358 | −0.85 | 0.71 | 0.19 | 0.17 | 0.49 |
| Mulliken Charges | |||||
| 2RE1r1_(S) | 2TS1r1_(S) | 2INT1r1_(S) | 2TS2r1_(S) | 2PR1r1_(S) | |
| Porphrin | −0.95 | −0.43 | −0.39 | −0.32 | −0.78 |
| Fe | 0.61 | −0.02 | −0.21 | −0.19 | 0.15 |
| oxo | −0.33 | −0.51 | −0.56 | −0.58 | −0.45 |
| 4-pIBA | −0.67 | −0.89 | −0.74 | −0.67 | −0.58 |
| C2’ | −0.05 | −0.27 | −0.12 | −0.02 | 0.12 |
| O4’ | −0.21 | −0.38 | −0.41 | −0.40 | −0.38 |
| H4 | 0.07 | 0.29 | 0.30 | 0.32 | 0.28 |
| Cys358 | 0.02 | 0.01 | −0.06 | −0.08 | −0.24 |
| Spin Densities | |||||||
| 2RE2r2 | 2TS1r2 | 2INT1r2 | 2TS2r2 | 2INT2r2 | 2TS3r2 | 2PR2r2 | |
| Porphrin | −0.10 | −0.11 | −0.10 | −0.09 | −0.09 | −0.10 | −0.10 |
| Fe | 1.08 | 0.77 | 0.82 | 1.03 | 1.07 | 0.37 | 0.96 |
| Op | −0.44 | 0.26 | 0.23 | −0.08 | 0.03 | −0.15 | 0.10 |
| Od | −0.63 | 0.12 | 0.09 | −0.11 | 0.00 | −0.10 | 0.00 |
| 4-2OH-pIBA | 0.01 | 0.00 | 0.00 | −0.72 | −0.98 | 0.97 | 0.00 |
| C1’ | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 |
| C2’ | 0.00 | 0.00 | 0.00 | −0.26 | −0.85 | 0.80 | 0.00 |
| Cys358 | 0.08 | −0.04 | −0.04 | 0.05 | −0.02 | 0.00 | 0.04 |
| Mulliken Charges | |||||||
| 2RE2r2 | 2TS1r2 | 2INT1r2 | 2TS2r2 | 2INT2r2 | 2TS3r2 | 2PR2r2 | |
| Porphrin | −0.77 | −0.93 | −0.88 | −0.72 | −1.19 | −1.06 | −0.90 |
| Fe | −0.15 | −0.14 | −0.14 | −0.14 | 0.53 | −0.15 | −0.08 |
| Op | −0.26 | −0.34 | −0.36 | −0.42 | −0.34 | −0.47 | −0.63 |
| Od | −0.26 | −0.28 | −0.24 | −0.17 | −0.17 | −0.30 | −0.52 |
| 4-2OH-pIBA | −0.80 | −1.14 | −0.91 | −0.83 | −0.64 | −0.81 | −0.48 |
| C1’ | 0.22 | 0.36 | 0.31 | 0.37 | 0.28 | 0.36 | 0.31 |
| C2’ | 0.22 | 0.21 | 0.21 | 0.15 | −0.03 | −0.04 | 0.15 |
| Cys358 | −0.44 | −0.61 | −0.60 | −0.45 | −0.23 | −0.59 | −0.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, C.; Xu, J.; Wang, S.; Fang, Y.-G.; Tan, H. Mechanistic Insights into CYP199A4-Catalyzed α-Hydroxyketone Formation and Hydrogen Bond-Assisted C–C Bond Cleavage Catalyzed by the CYP199A4 F182L Mutant. Int. J. Mol. Sci. 2025, 26, 1526. https://doi.org/10.3390/ijms26041526
Yuan C, Xu J, Wang S, Fang Y-G, Tan H. Mechanistic Insights into CYP199A4-Catalyzed α-Hydroxyketone Formation and Hydrogen Bond-Assisted C–C Bond Cleavage Catalyzed by the CYP199A4 F182L Mutant. International Journal of Molecular Sciences. 2025; 26(4):1526. https://doi.org/10.3390/ijms26041526
Chicago/Turabian StyleYuan, Chang, Jiaqi Xu, Shun Wang, Ye-Guang Fang, and Hongwei Tan. 2025. "Mechanistic Insights into CYP199A4-Catalyzed α-Hydroxyketone Formation and Hydrogen Bond-Assisted C–C Bond Cleavage Catalyzed by the CYP199A4 F182L Mutant" International Journal of Molecular Sciences 26, no. 4: 1526. https://doi.org/10.3390/ijms26041526
APA StyleYuan, C., Xu, J., Wang, S., Fang, Y.-G., & Tan, H. (2025). Mechanistic Insights into CYP199A4-Catalyzed α-Hydroxyketone Formation and Hydrogen Bond-Assisted C–C Bond Cleavage Catalyzed by the CYP199A4 F182L Mutant. International Journal of Molecular Sciences, 26(4), 1526. https://doi.org/10.3390/ijms26041526