
Recent Advances in Peptide Inhibitors Targeting Wild-Type Ras Protein Interactions in Cancer Therapy

 , and
, and
Abstract
1. Introduction
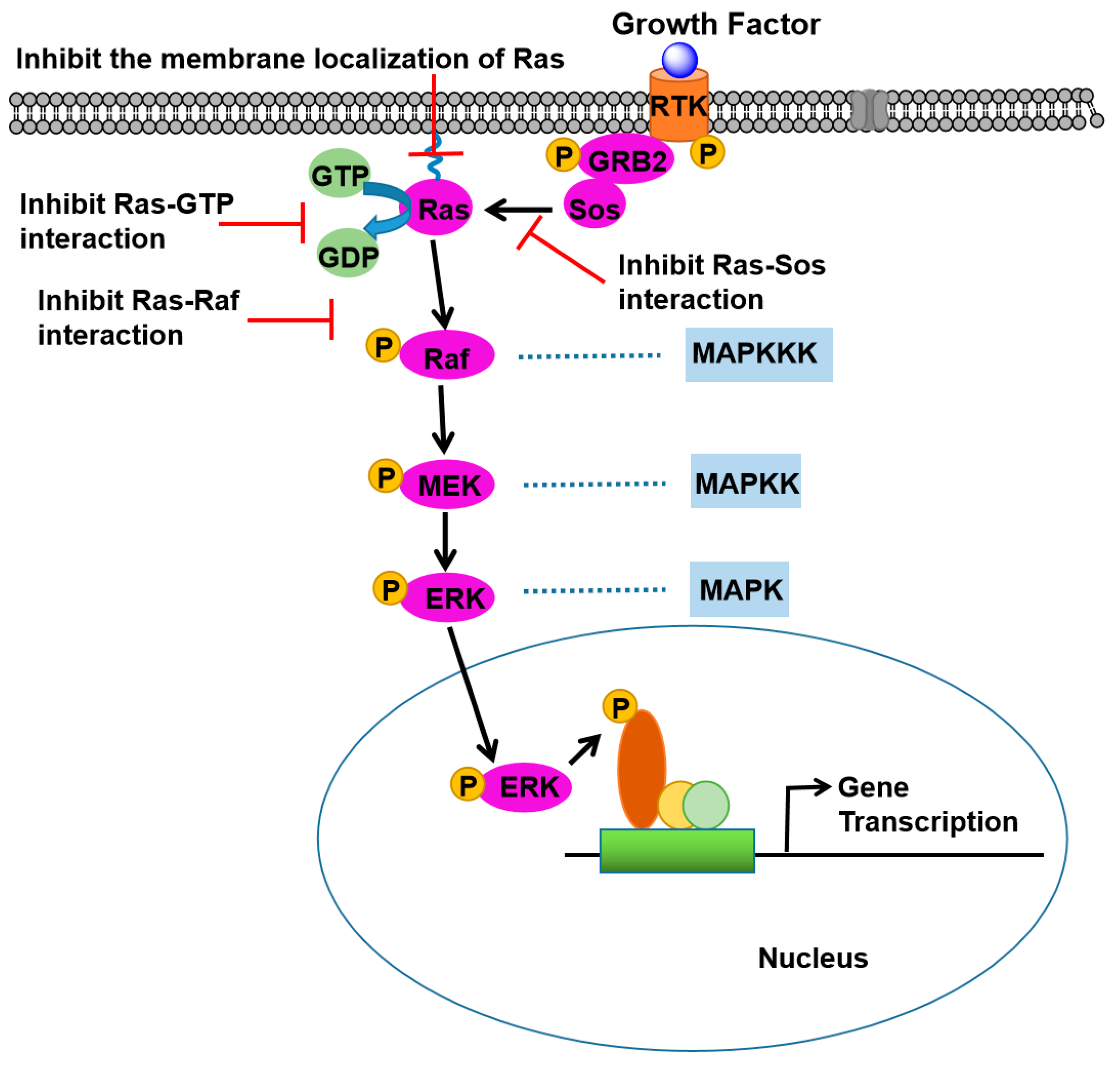
2. Ras Signaling Pathways
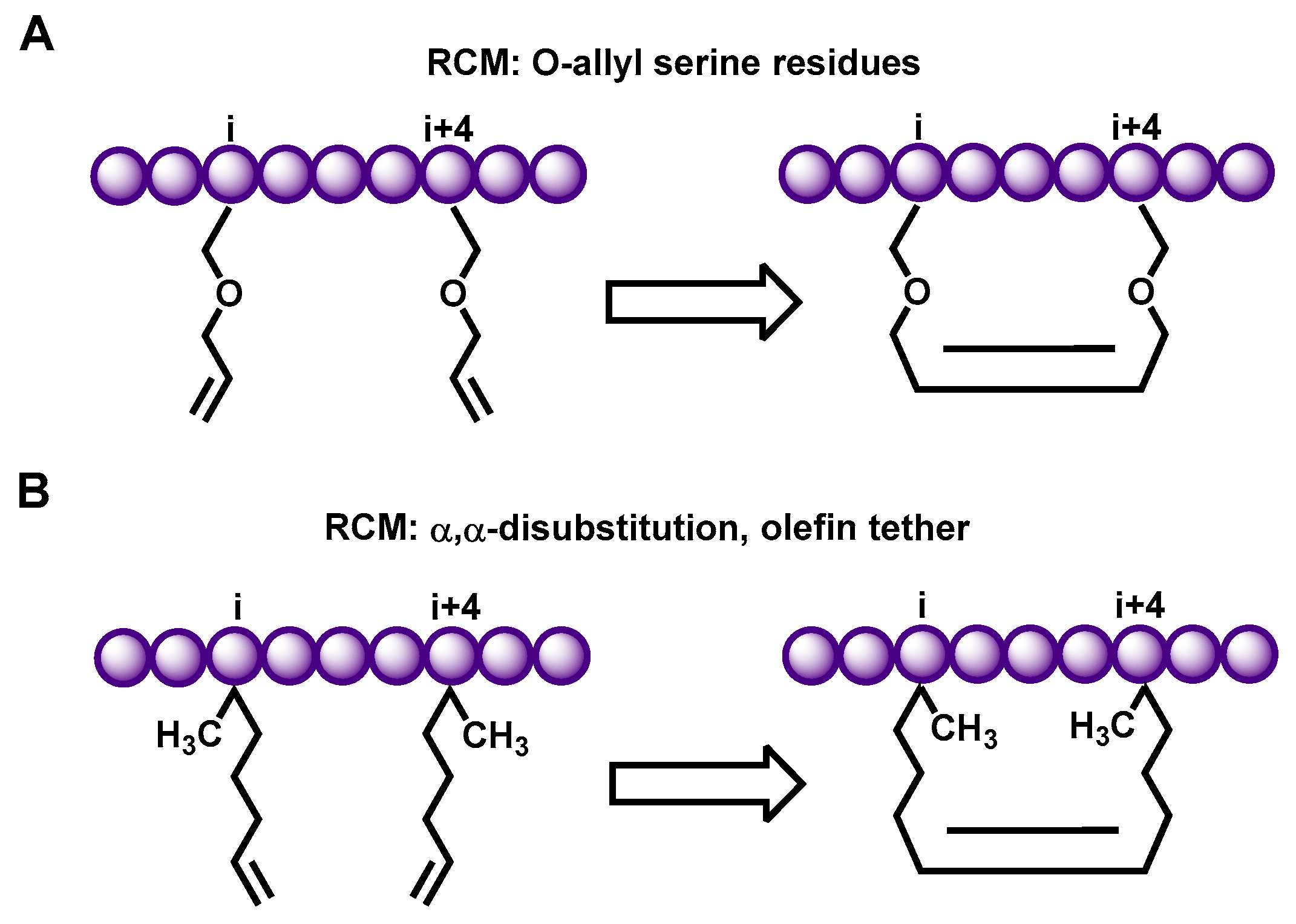
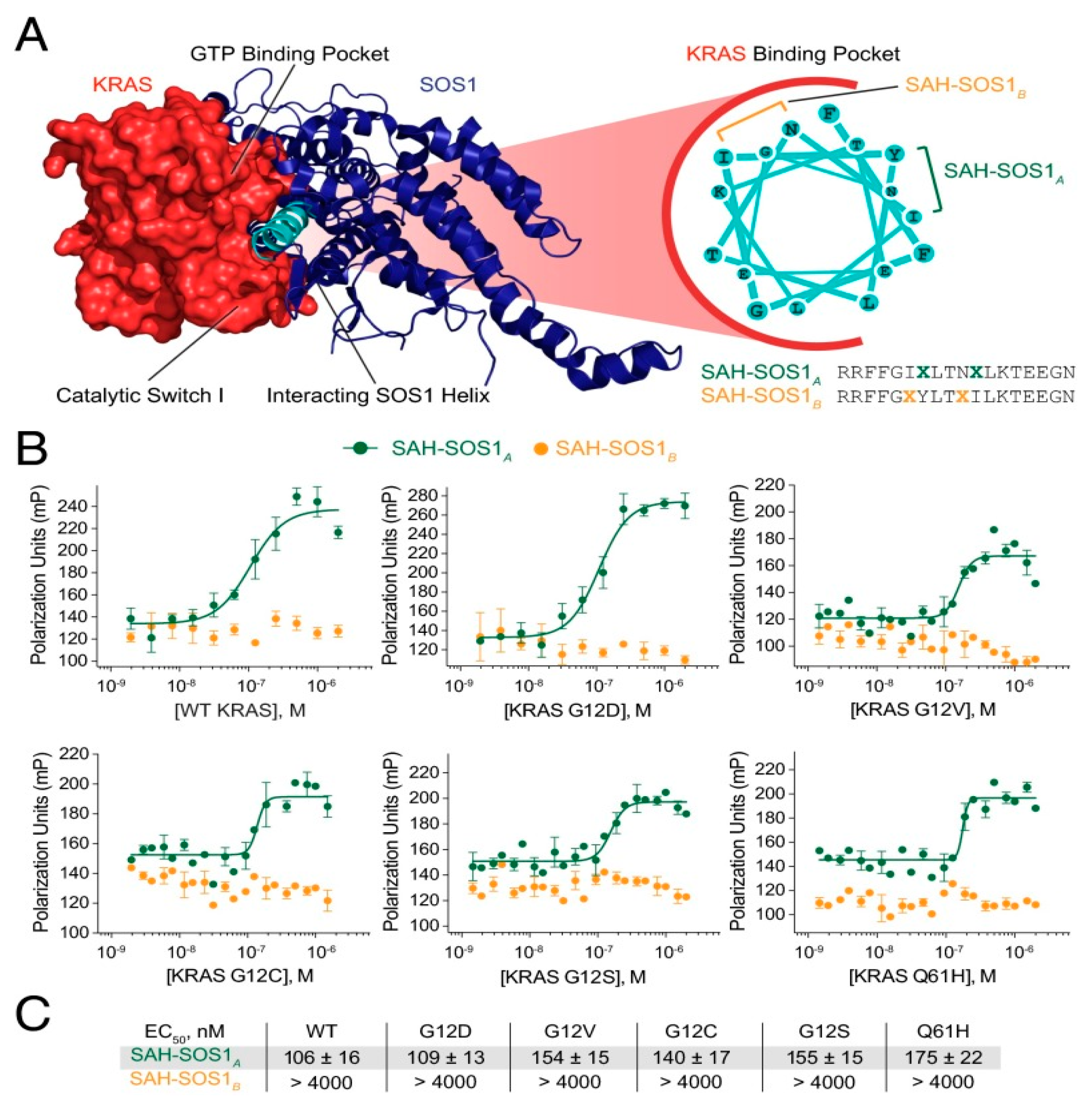
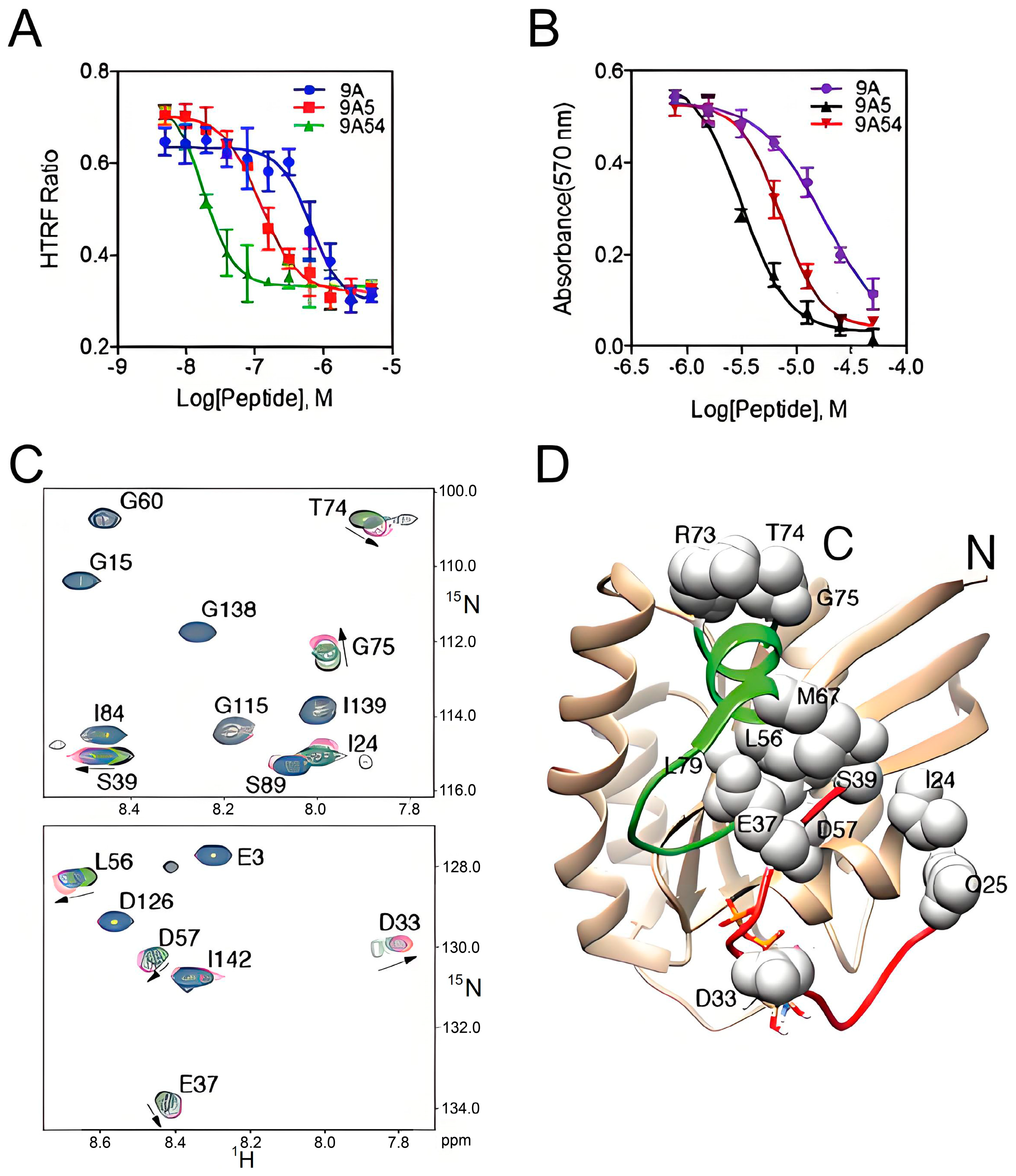
2.1. Hydrocarbon Peptide
2.2. Cyclization Peptides
2.3. Linear Peptides
2.4. Metallopeptides
2.5. N-Cap Helix Nucleation Peptides
2.6. Other Peptides
3. Conclusions
Funding
Conflicts of Interest
References
- Wu, Y.; Song, Y.Q.; Wang, R.Z.; Wang, T.L. Molecular mechanisms of tumor resistance to radiotherapy. Mol. Cancer 2023, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Urbano, M.A.; Griñán-Lisón, C.; Marchal, J.A.; Núñez, M.I. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells 2020, 9, 1651. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Jiang, X.; Wu, Y.J.; Yu, H.M. Global research landscape and trends of cancer radiotherapy plus immunotherapy: A bibliometric analysis. Heliyon 2024, 10, e27103. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.F.; Zhang, C.; Cui, K.Y.; Liang, X.; Zhu, G.H.; Hong, L. Fer-mediated activation of the Ras-MAPK signaling pathway drives the proliferation, migration, and invasion of endometrial carcinoma cells. Mol. Cell. Biochem. 2024, 479, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ro, S.W. Ras Mitogen-activated Protein Kinase Signaling and Kinase Suppressor of Ras as Therapeutic Targets for Hepatocellular Carcinoma. J. Liver Cancer 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Rauen, K.A. Defining RASopathy. Dis. Model. Mech. 2022, 15, dmm049344. [Google Scholar] [CrossRef]
- Steffen, C.L.; Manoharan, G.B.; Pavic, K.; Yeste-Vázquez, A.; Knuuttila, M.; Arora, N.; Zhou, Y.; Härmä, H.; Gaigneaux, A.; Grossmann, T.N.; et al. Identification of an H-Ras nanocluster disrupting peptide. Commun. Biol. 2024, 7, 837. [Google Scholar] [CrossRef]
- Qin, W.; Wei, X.; Yang, D.; Luo, Q.; Huang, M.; Xing, S.; Wei, W.; Liang, L.; Huang, J.; Zhou, Z.; et al. Ras-Targeting Stabilized Peptide Increases Radiation Sensitivity of Cancer Cells. Bioconjug Chem. 2024, 35, 737–743. [Google Scholar] [CrossRef]
- Li, A.P.; Li, X.; Zou, J.H.; Zhuo, X.B.; Chen, S.; Chai, X.Y.; Gai, C.H.; Xu, W.H.; Zhao, Q.J.; Zou, Y. SOS1-inspired hydrocarbon-stapled peptide as a pan-Ras inhibitor. Bioorg Chem. 2023, 135, 106500. [Google Scholar] [CrossRef]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, E.S.; Parkhitko, A.; Bird, G.H.; Luccarelli, J.; Bellairs, J.A.; Escudero, S.; Opoku-Nsiah, K.; Godes, M.; Perrimon, N.; Walensky, L.D. Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl. Acad. Sci. USA 2015, 112, 1761–1766. [Google Scholar] [CrossRef]
- Han, D.; Li, A.P.; Zhu, L.; Zhuang, C.L.; Zhao, Q.J.; Zou, Y. Peptide inhibitors targeting Ras and Ras-associated protein-protein interactions. Eur. J. Med. Chem. 2024, 279, 116878. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.L.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef]
- Orgován, Z.; Keseru, G.M. Small molecule inhibitors of RAS proteins with oncogenic mutations. Cancer Metast Rev. 2020, 39, 1107–1126. [Google Scholar] [CrossRef]
- Franz, M.; Mörchen, B.; Degenhart, C.; Gülden, D.; Shkura, O.; Wolters, D.; Koch, U.; Klebl, B.; Stoll, R.; Helfrich, I.; et al. Sequence-Selective Covalent CaaX-Box Receptors Prevent Farnesylation of Oncogenic Ras Proteins and Impact MAPK/PI3 K Signaling. Chemmedchem 2021, 16, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Navarro, A.; Pourfarjam, Y.; Hu, F.; Rodriguez, D.J.; Vides, A.; Sang, B.; Fan, S.J.; Goldgur, Y.; de Stanchina, E.; Lito, P. Pharmacological restoration of GTP hydrolysis by mutant RAS. Nature 2025, 637, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Lee, B.J.; Jiang, J.J.; Tomlinson, A.; Seamon, K.J.; Mira, A.; Patrucco, E.; Goodhart, G.; Dilly, J.; Gindin, Y.; et al. Concurrent inhibition of oncogenic and wild-type RAS-GTP for cancer therapy. Nature 2024, 628, E4. [Google Scholar] [CrossRef]
- Gentile, D.R.; Rathinaswamy, M.K.; Jenkins, M.L.; Moss, S.M.; Siempelkamp, B.D.; Renslo, A.R.; Burke, J.E.; Shokat, K.M. Ras Binder Induces a Modified Switch-II Pocket in GTP and GDP States. Cell Chem. Biol. 2017, 24, 1455–1466. [Google Scholar] [CrossRef]
- O’Bryan, J.P. Pharmacological Targeting of RAS: Recent Success with Direct Inhibitors. Pharmacol Res. 2019, 139, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Pincus, M.R.; Lin, B.; Patel, P.; Gabutan, E.; Zohar, N.; Bowne, W.B. Peptides That Block RAS-p21 Protein-Induced Cell Transformation. Biomedicines 2023, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, N.S.; Scopton, A.P.; Dar, A.C. Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling. Nature 2016, 537, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Rensland, H.; Schlichting, I.; Vetter, I.; Borasio, G.D.; Goody, R.S.; Wittinghofer, A. Kinetic and structural analysis of the Mg(2+)-binding site of the guanine nucleotide-binding protein p21H-ras. J. Biol. Chem. 1993, 268, 923–929. [Google Scholar] [CrossRef]
- Kopra, K.; van Adrichem, A.J.; Salo-Ahen, O.M.H.; Peltonen, J.; Wennerberg, K.; Härmä, H. High-Throughput Dual Screening Method for Ras Activities and Inhibitors. Anal. Chem. 2017, 89, 4508–4516. [Google Scholar] [CrossRef] [PubMed]
- Maurer, T.; Garrenton, L.S.; Oha, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.L.; Hu, L.Z.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Samani, A.; Downward, J. Drugging the Undruggable: Advances on RAS Targeting in Cancer. Genes 2021, 12, 899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, J.; Gao, Y.T.; Li, Y.F.; Li, Y.Z. Strategies for targeting undruggable targets. Expert. Opin. Drug Dis. 2022, 17, 55–69. [Google Scholar] [CrossRef]
- Tomazini, A.; Shifman, J.M. Targeting Ras with protein engineering. Oncotarget 2023, 14, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Goldfinger, L.E. Concepts and advances in cancer therapeutic vulnerabilities in RAS membrane targeting. Semin. Cancer Biol. 2019, 54, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Upadhyaya, P.; Villalona-Calero, M.A.; Briesewitz, R.; Pei, D. Inhibition of Ras-Effector Interaction by Cyclic Peptides. Medchemcomm 2013, 4, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, P.; Qian, Z.; Selner, N.G.; Clippinger, S.R.; Wu, Z.; Briesewitz, R.; Pei, D. Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Angew. Chem. Int. Ed. Engl. 2015, 54, 7602–7606. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.B.; Upadhyaya, P.; Qian, Z.Q.; Pei, D.H. Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci. 2016, 18, 75–85. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Gao, R.; Hu, Q.; Peacock, H.; Peacock, D.M.; Dai, S.Z.; Shokat, K.M.; Suga, H. GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Central Sci. 2020, 6, 1753–1761. [Google Scholar] [CrossRef]
- Learte-Aymami, S.; Martin-Malpartida, P.; Roldan-Martin, L.; Sciortino, G.; Couceiro, J.R.; Marechal, J.D.; Macias, M.J.; Mascarenas, J.L.; Vazquez, M.E. Controlling oncogenic KRAS signaling pathways with a Palladium-responsive peptide. Commun. Chem. 2022, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Yoo, D.Y.; Conway, L.; Richards-Corke, K.C.; Parker, C.G.; Arora, P.S. A Sos proteomimetic as a pan-Ras inhibitor. Proc. Natl. Acad. Sci. USA 2021, 118, e2101027118. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Grubbs, R.H. Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem. Int. Ed. Engl. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Julia Po, A.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 186P–187P. [Google Scholar] [CrossRef]
- Joy, S.T.; Arora, P.S. An optimal hydrogen-bond surrogate for α-helices. Chem. Commun. 2016, 52, 5738–5741. [Google Scholar] [CrossRef]
- Schott, M.E.; Wells, D.T.; Schlom, J.; Abrams, S.I. Comparison of linear and branched peptide forms (MAPs) in the induction of T helper responses to point-mutated ras immunogens. Cell. Immunol. 1996, 174, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Q.S.; Geng, H.; Tian, Y.; Cheng, M.; Jiang, Y.H.; Xie, M.S.; Niu, X.G.; Jiang, F.; Zhang, Y.O.; et al. Crosslinked Aspartic Acids as Helix-Nucleating Templates. Angew. Chem. Int. Edit 2016, 55, 12088–12093. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhao, H.; Liu, Q.; Zhu, Y.; Yin, F.; Liang, Y.; Jiang, Y.; Wang, D.; Hu, K.; Qin, X.; et al. Structural Basis of Inhibition of ERalpha-Coactivator Interaction by High-Affinity N-Terminus Isoaspartic Acid Tethered Helical Peptides. J. Med. Chem. 2017, 60, 8731–8740. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, W.; Zhao, R.; Chen, L.; Liu, N.; Tian, Y.; Zhao, H.; Xie, M.; Lu, F.; Fang, Q.; et al. Stabilized Peptide HDAC Inhibitors Derived from HDAC1 Substrate H3K56 for the Treatment of Cancer Stem-Like Cells In Vivo. Cancer Res. 2019, 79, 1769–1783. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Chen, H.; Tu, L.; Ma, Y.; Liu, N.; Zhang, H.; Li, D.; Riedl, B.; Bierer, D.; Yin, F.; et al. Potent Inhibition of HIF1α and p300 Interaction by a Constrained Peptide Derived from CITED2. J. Med. Chem. 2021, 64, 13693–13703. [Google Scholar] [CrossRef] [PubMed]
- Pallara, C.; Cabot, D.; Rivas, J.; Brun, S.; Seco, J.; Abuasaker, B.; Tarragó, T.; Jaumot, M.; Prades, R.; Agell, N. Peptidomimetics designed to bind to RAS effector domain are promising cancer therapeutic compounds. Sci. Rep. 2022, 12, 15810. [Google Scholar] [CrossRef]
- Wolny, M.; Batchelor, M.; Bartlett, G.J.; Baker, E.G.; Kurzawa, M.; Knight, P.J.; Dougan, L.; Woolfson, D.N.; Paci, E.; Peckham, M. Characterization of long and stable de novo single alpha-helix domains provides novel insight into their stability. Sci. Rep. 2017, 7, 44341. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Peptide Sequence | Binding Affinity KRAS (μM) | Binding Affinity NRAS (μM) | Binding Affinity HRAS (μM) | Reference |
|---|---|---|---|---|---|
| HBS 3 | XFE*GIYRLELLKAEEAN-NH2 | N.D | N.D | 28 ± 4.5 | [11] |
| SAH-SOS1A | RRFFGI{Aaa}LTN{Aaa}LKTEEGN (Covalent bridge:Aaa7-Aaa11) | 5–15 | N.D | N.D | [12] |
| SSOSH-5 | Ac-WIGRLLTS5IRHRS5RNGN-NH2 | N.D | N.D | 3.92 | [10] |
| compound 12 | 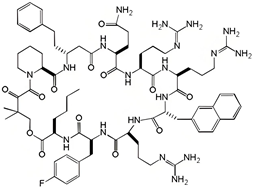 | 0.70 | N.D | N.D | [33] |
| 9A5 | Cyclo({d-Ala}-RRRARF-{Nal}-QWT) | 0.12 | N.D | N.D | [34] |
| bicyclic peptides(peptides 49) | A-L-F-R-N-Pra*-I-D | 0.21 ± 0.10 | N.D | N.D | [35] |
| bicyclic peptides(peptides 54) | A-L-F-R-N-Pra-I-D | 17 ± 11 | N.D | N.D | [35] |
| KD2 | 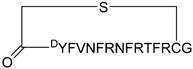 | 12.4 | N.D | N.D | [36] |
| αh-His2 | TMR-Ahx-RRFFGIHLTNHLKTEEGN | 0.345 | N.D | N.D | [37] |
| H2 | H-WRR-cyclo(isoDFFDap)-IYLTNILKTEEGN-NH2 | N.D | N.D | 0.53 | [9] |
| H5 | H-WRR-cyclo(isoDFFDap)-IYLTNILKTQEGN-NH2 | N.D | N.D | 0.13 | [9] |
| CHDSos-5 | 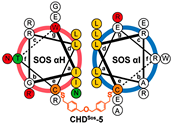 | 1.9 ± 0.3 | N.D | N.D | [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, W.; Liu, Z.; Huang, M.; Liang, L.; Gan, Y.; Huang, Z.; Huang, J.; Wei, X. Recent Advances in Peptide Inhibitors Targeting Wild-Type Ras Protein Interactions in Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 1425. https://doi.org/10.3390/ijms26041425
Qin W, Liu Z, Huang M, Liang L, Gan Y, Huang Z, Huang J, Wei X. Recent Advances in Peptide Inhibitors Targeting Wild-Type Ras Protein Interactions in Cancer Therapy. International Journal of Molecular Sciences. 2025; 26(4):1425. https://doi.org/10.3390/ijms26041425
Chicago/Turabian StyleQin, Weirong, Zijian Liu, Mingyu Huang, Lin Liang, Yuxin Gan, Zubei Huang, Jin Huang, and Xiangzan Wei. 2025. "Recent Advances in Peptide Inhibitors Targeting Wild-Type Ras Protein Interactions in Cancer Therapy" International Journal of Molecular Sciences 26, no. 4: 1425. https://doi.org/10.3390/ijms26041425
APA StyleQin, W., Liu, Z., Huang, M., Liang, L., Gan, Y., Huang, Z., Huang, J., & Wei, X. (2025). Recent Advances in Peptide Inhibitors Targeting Wild-Type Ras Protein Interactions in Cancer Therapy. International Journal of Molecular Sciences, 26(4), 1425. https://doi.org/10.3390/ijms26041425