

Brain Tumors and Beyond: Multi-Compartment Microbiome and Mycobiome Analysis

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patients Characteristics
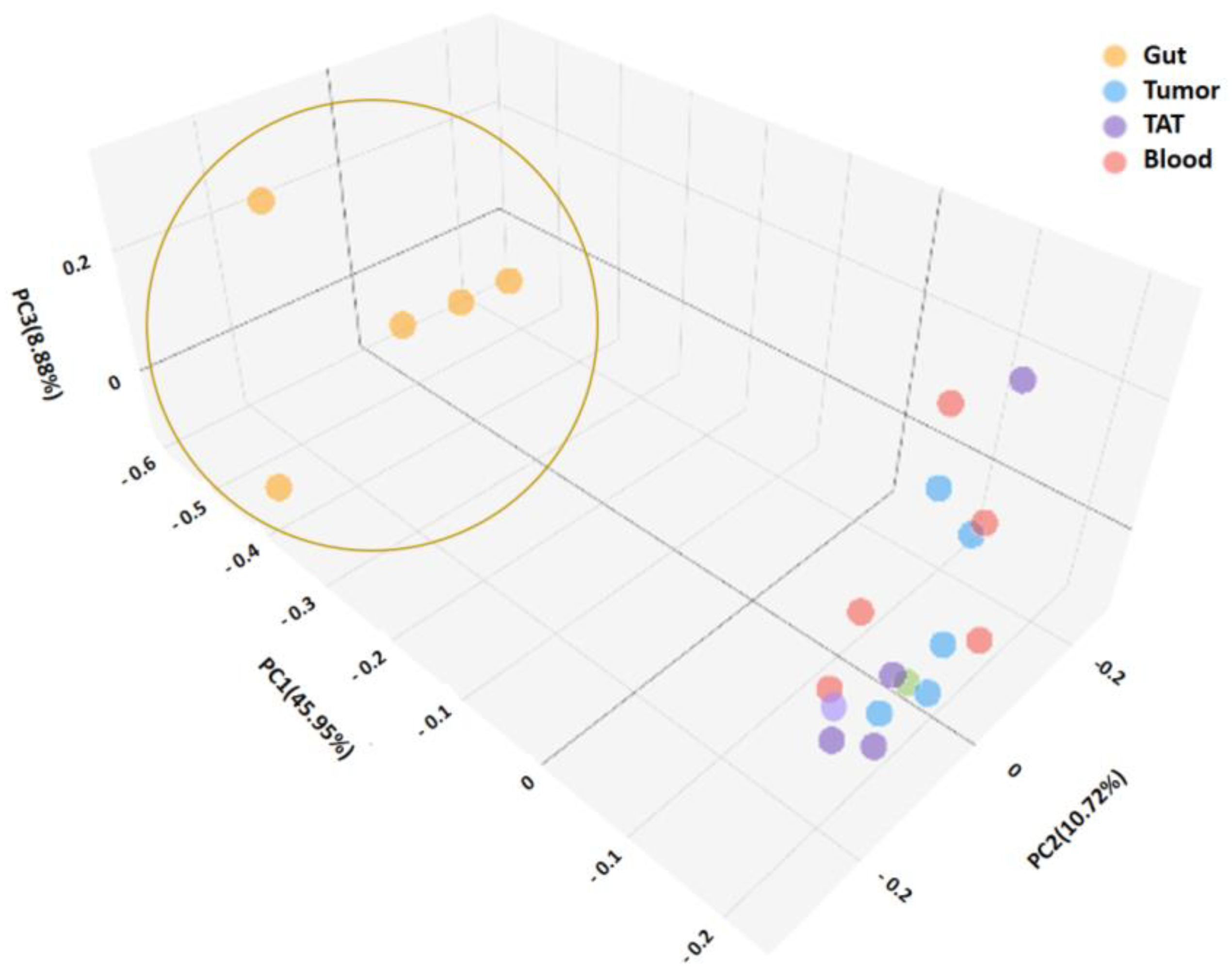
2.2. Microbiota Diversities of Patient Samples
2.2.1. Consistency of the Microbiota Composition
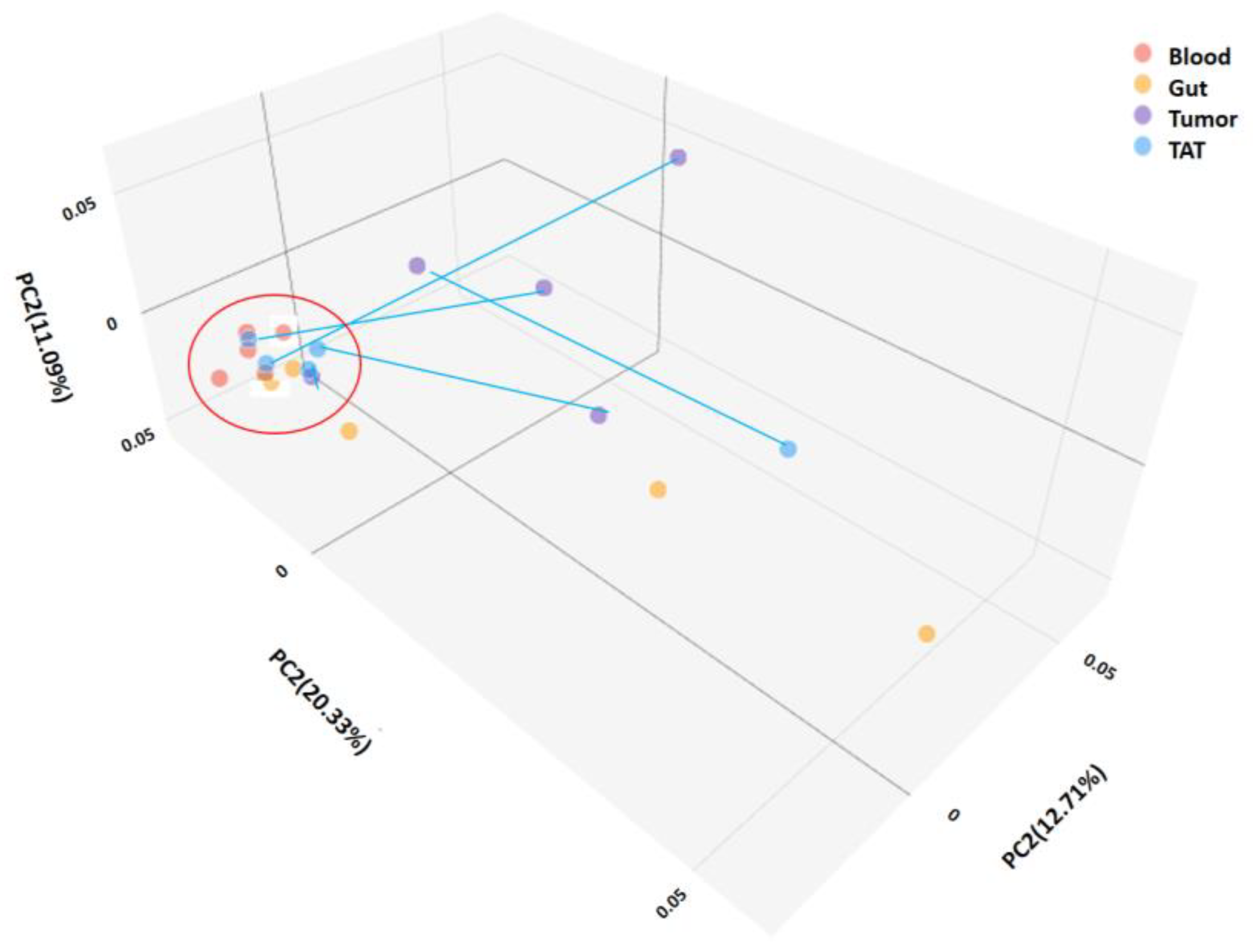
2.2.2. Mycobiota Diversity in Different Patient Samples
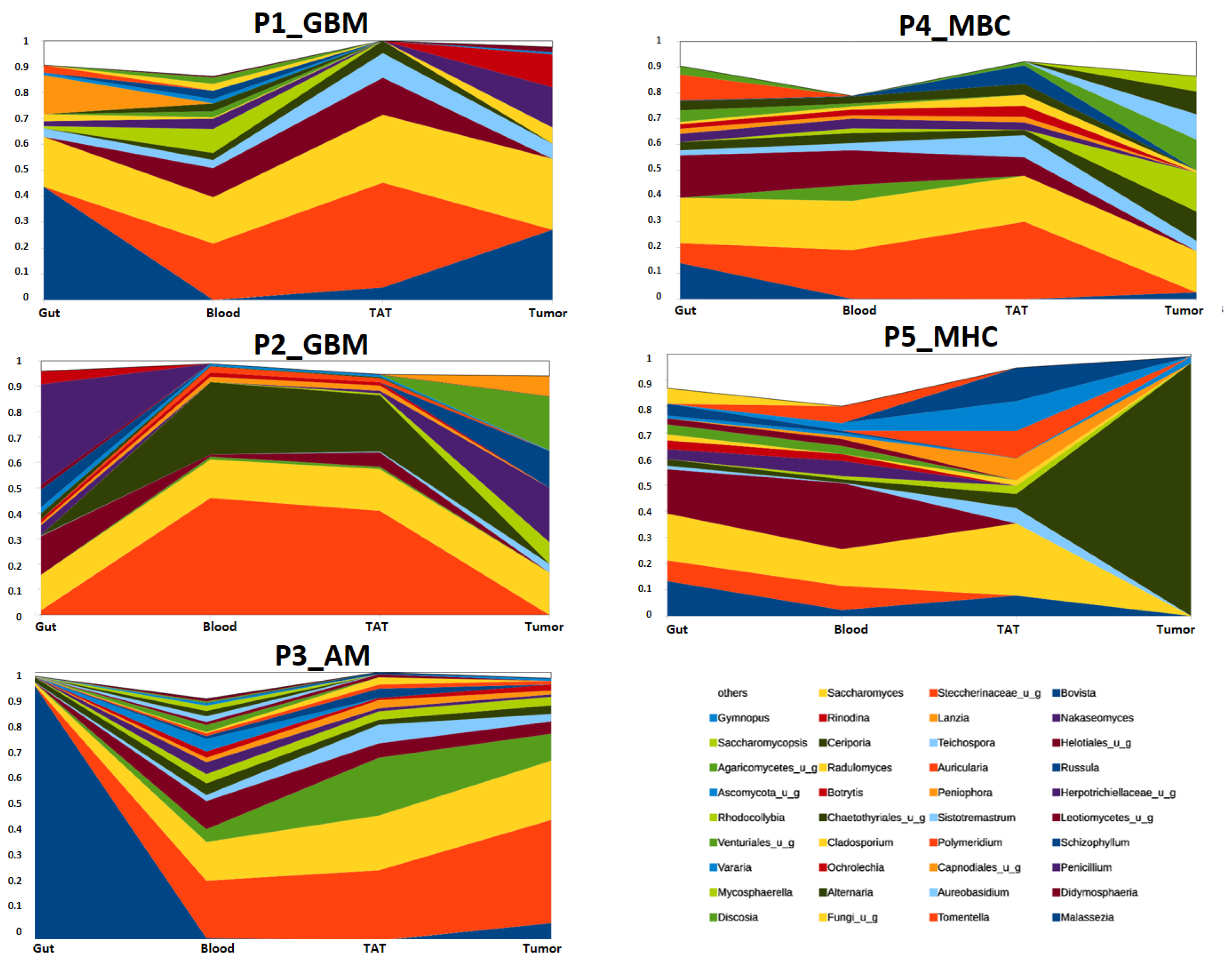
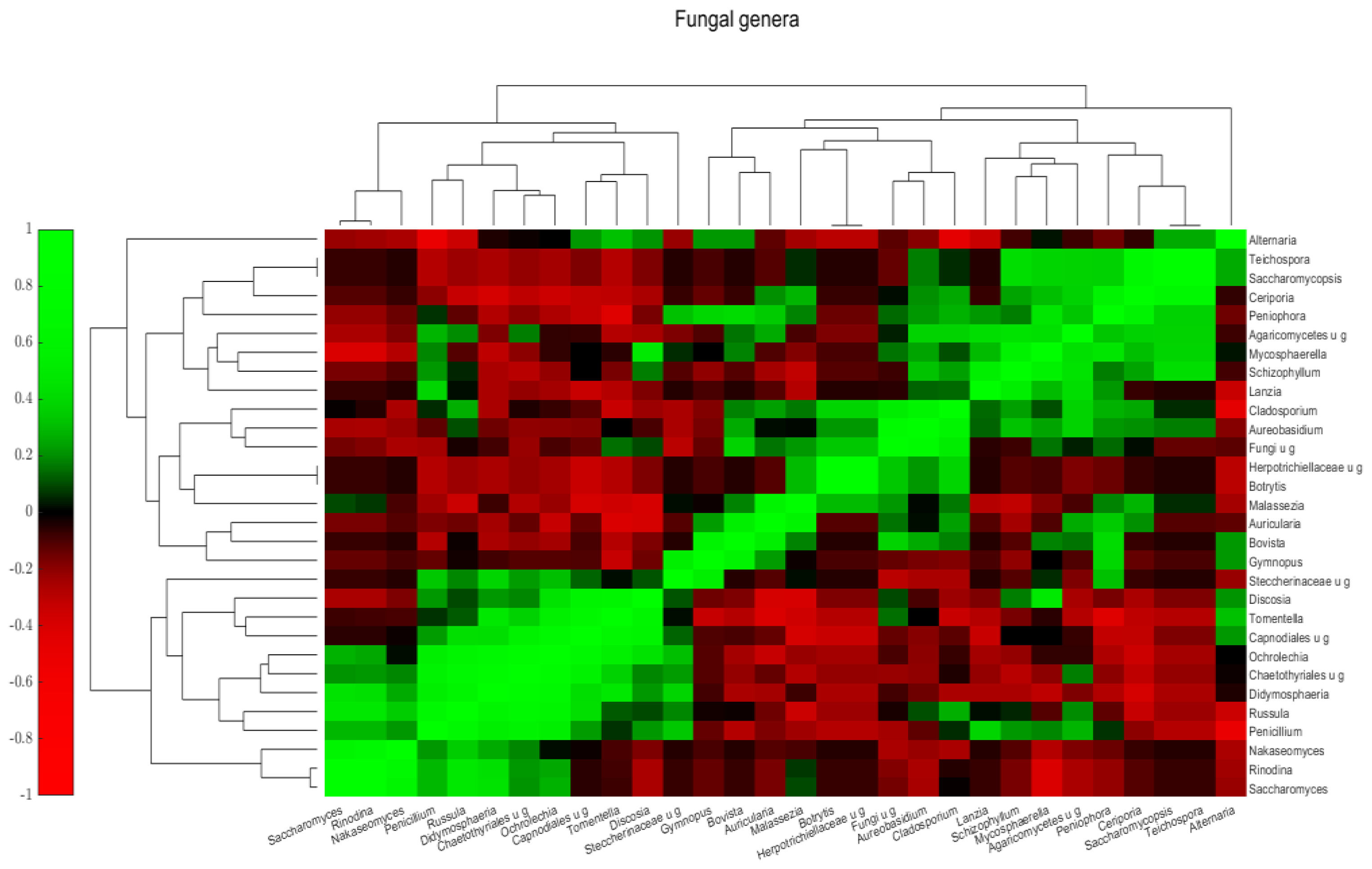
2.2.3. Mycobiota Structure of Different Samples
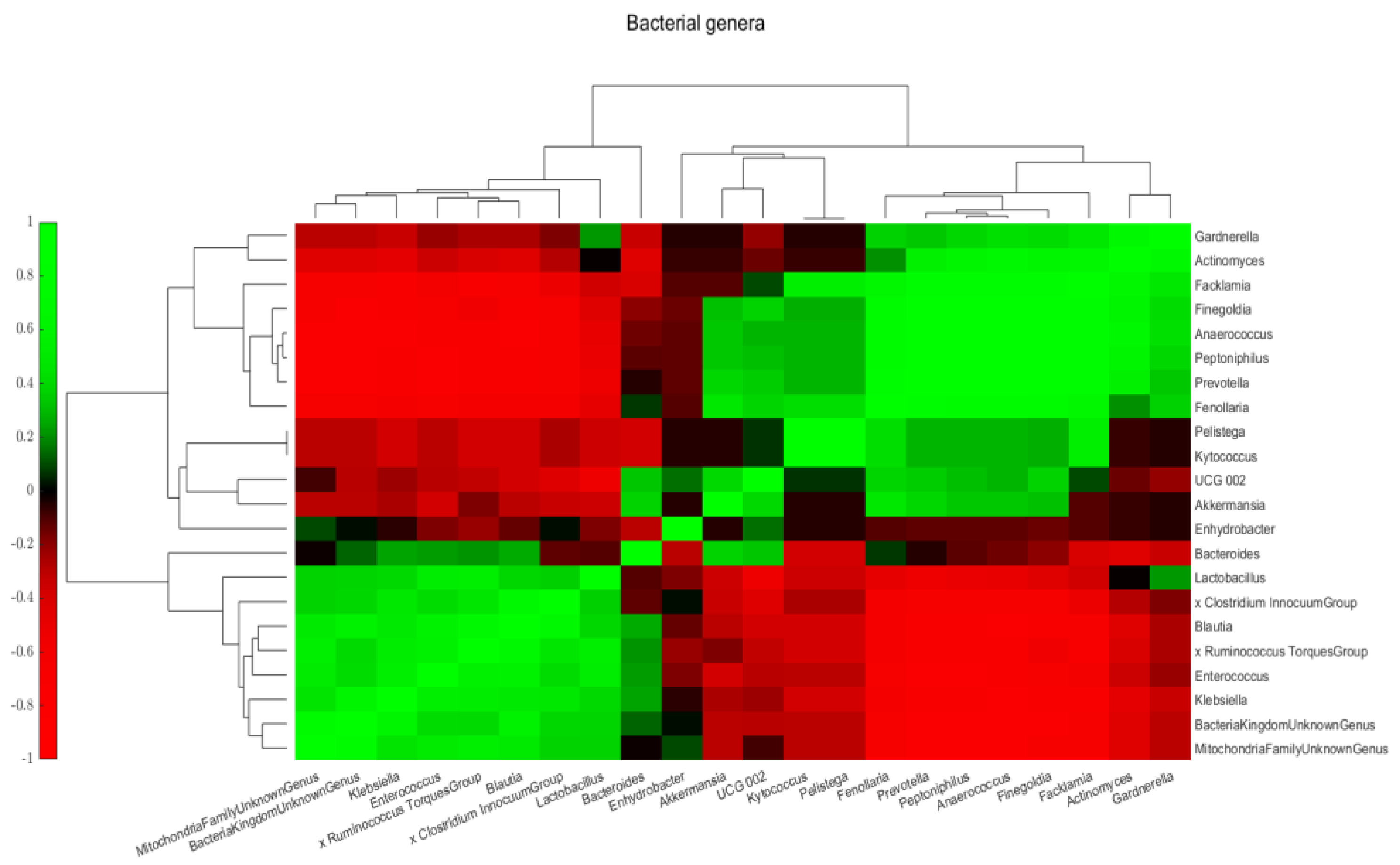
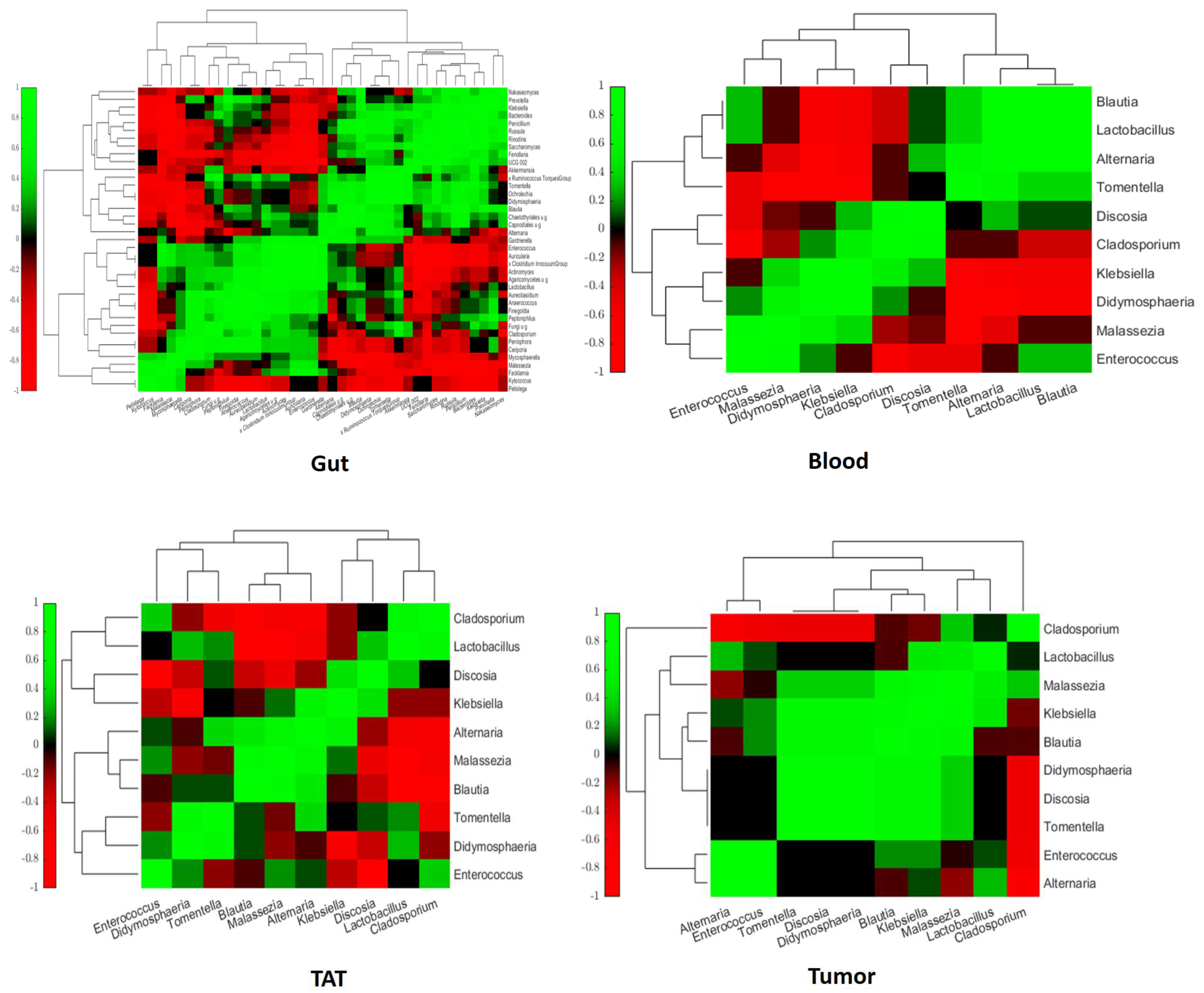
2.3. Intra- and Interkingdom Correlation Among the Most Abundant Bacterial and Fungal Genera
3. Discussion
4. Materials and Methods
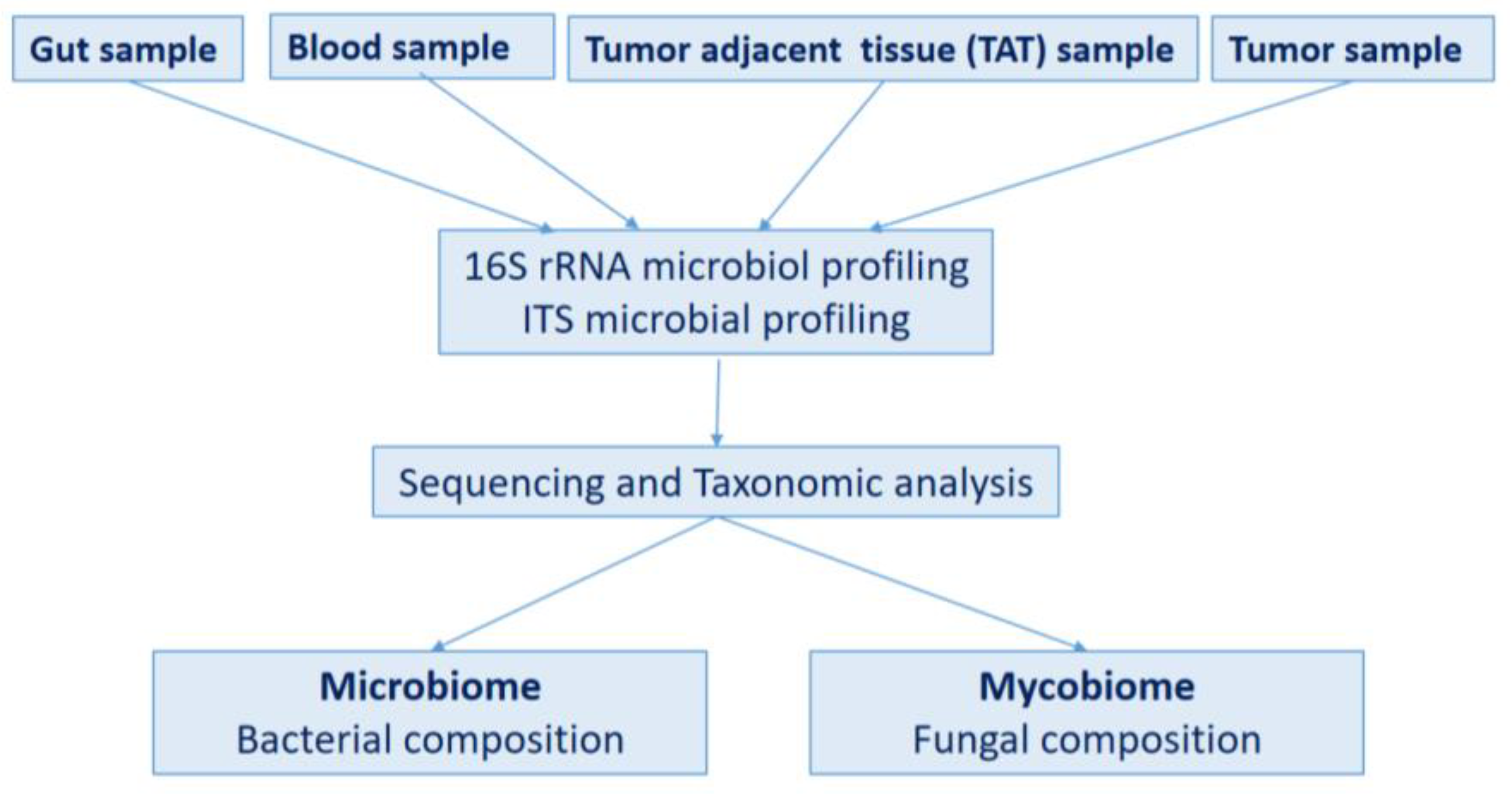
4.1. Study Design
4.2. DNA Extraction and Characteristics
4.3. 16S rRNA Microbiota Analysis
4.4. ITS Mycobiota Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. S2), iii1–iii105. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Geurts, M.; French, P.J.; Smits, M.; Capper, D.; Bromberg, J.E.C.; Chang, S.M. Primary brain tumours in adults. Lancet 2023, 402, 1564–1579. [Google Scholar] [CrossRef]
- Salari, N.; Ghasemi, H.; Fatahian, R.; Mansouri, K.; Dokaneheifard, S.; Shiri, M.H.; Hemmati, M.; Mohammadi, M. The global prevalence of primary central nervous system tumors: A systematic review and meta-analysis. Eur. J. Med. Res. 2023, 28, 39. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, S.; Akiyama, Y.; Mitsuya, K.; Ikeya, T.; Hozumi, C.; Iizuka, A.; Miyata, H.; Maeda, C.; Ashizawa, T.; Nagashima, T.; et al. Genetic and Immunological Characterization of Brain Metastases from Solid Cancers. Anticancer Res. 2024, 44, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Malviya, R.; Uniyal, P. Survival of Patients with Primary Brain Tumor: A Data Analysis of 10 Years. Curr. Pharm. Des. 2024, 30, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the gut-brain-axis: Implications for new therapeutic design in the CNS. eBioMedicine 2022, 77, 103908. [Google Scholar] [CrossRef] [PubMed]
- Dono, A.; Nickles, J.; Rodriguez-Armendariz, A.G.; McFarland, B.C.; Ajami, N.J.; Ballester, L.Y.; Wargo, J.A.; Esquenazi, Y. Glioma and the gut-brain axis: Opportunities and future perspectives. Neuro-Oncol. Adv. 2022, 4, vdac054. [Google Scholar] [CrossRef]
- Ishaq, H.M.; Yasin, R.; Mohammad, I.S.; Fan, Y.; Li, H.; Shahzad, M.; Xu, J. The gut-brain-axis: A positive relationship between gut microbial dysbiosis and glioblastoma brain tumour. Heliyon 2024, 10, e30494. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, H.; Wang, X.; Liu, X.; Huang, Y.; Wang, Z.; Ma, Q.; Dong, L.; Qi, Y.; Zhang, H.; et al. Crosstalk Between the Gut and Brain: Importance of the Fecal Microbiota in Patient With Brain Tumors. Front. Cell. Infect. Microbiol. 2022, 12, 881071. [Google Scholar] [CrossRef]
- Lin, B.; Ye, Z.; Ye, Z.; Wang, M.; Cao, Z.; Gao, R.; Zhang, Y. Gut microbiota in brain tumors: An emerging crucial player. CNS Neurosci. Ther. 2023, 29 (Suppl. S1), 84–97. [Google Scholar] [CrossRef]
- Jiang, H.; Zeng, W.; Zhang, X.; Pei, Y.; Zhang, H.; Li, Y. The role of gut microbiota in patients with benign and malignant brain tumors: A pilot study. Bioengineered 2022, 13, 7847–7859. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Di Paola, M.; Stefanini, I.; Albanese, D.; Rizzetto, L.; Lionetti, P.; Calabro, A.; Jousson, O.; Donati, C.; Cavalieri, D.; et al. Age and Gender Affect the Composition of Fungal Population of the Human Gastrointestinal Tract. Front. Microbiol. 2016, 7, 1227. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef]
- Gao, R.; Kong, C.; Li, H.; Huang, L.; Qu, X.; Qin, N.; Qin, H. Dysbiosis signature of mycobiota in colon polyp and colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Sciarra, F.; Franceschini, E.; Campolo, F.; Venneri, M.A. The Diagnostic Potential of the Human Blood Microbiome: Are We Dreaming or Awake? Int. J. Mol. Sci. 2023, 24, 10422. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Tan, S.P.; Wong, D.M.K.; Koo, W.L.Y.; Wong, S.H.; Tan, N.S. The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges. Int. J. Mol. Sci. 2023, 24, 5633. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Chen, B.; Pan, H.; Wang, D.; Liu, M.; Yang, Y.; Zou, M.; Yang, J.; Xiao, K.; Zhao, R.; et al. Detection of Microbial 16S rRNA Gene in the Serum of Patients With Gastric Cancer. Front. Oncol. 2019, 9, 608. [Google Scholar] [CrossRef] [PubMed]
- Woerner, J.; Huang, Y.; Hutter, S.; Gurnari, C.; Sanchez, J.M.H.; Wang, J.; Huang, Y.; Schnabel, D.; Aaby, M.; Xu, W.; et al. Circulating microbial content in myeloid malignancy patients is associated with disease subtypes and patient outcomes. Nat. Commun. 2022, 13, 1038. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, X.; Zhou, X.; Zhao, J.; Yang, H.; Wang, S.; Morse, M.A.; Wu, J.; Yuan, Y.; Li, S.; et al. Blood microbiota diversity determines response of advanced colorectal cancer to chemotherapy combined with adoptive T cell immunotherapy. Oncoimmunology 2021, 10, 1976953. [Google Scholar] [CrossRef]
- Ouaknine Krief, J.; Helly de Tauriers, P.; Dumenil, C.; Neveux, N.; Dumoulin, J.; Giraud, V.; Labrune, S.; Tisserand, J.; Julie, C.; Emile, J.F.; et al. Role of antibiotic use, plasma citrulline and blood microbiome in advanced non-small cell lung cancer patients treated with nivolumab. J. Immunother. Cancer 2019, 7, 176. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Kwon, H.; Lim, W.; Moon, B.I. Staphylococcus aureus-Derived Extracellular Vesicles Enhance the Efficacy of Endocrine Therapy in Breast Cancer Cells. J. Clin. Med. 2022, 11, 2030. [Google Scholar] [CrossRef]
- Cho, E.J.; Leem, S.; Kim, S.A.; Yang, J.; Lee, Y.B.; Kim, S.S.; Cheong, J.Y.; Cho, S.W.; Kim, J.W.; Kim, S.M.; et al. Circulating Microbiota-Based Metagenomic Signature for Detection of Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 7536. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Belvoncikova, P.; Splichalova, P.; Videnska, P.; Gardlik, R. The Human Mycobiome: Colonization, Composition and the Role in Health and Disease. J. Fungi 2022, 8, 1046. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Livyatan, I.; Nejman, D.; Shental, N.; Straussman, R. Characterization of the human tumor microbiome reveals tumor-type specific intra-cellular bacteria. Oncoimmunology 2020, 9, 1800957. [Google Scholar] [CrossRef]
- Narunsky-Haziza, L.; Sepich-Poore, G.D.; Livyatan, I.; Asraf, O.; Martino, C.; Nejman, D.; Gavert, N.; Stajich, J.E.; Amit, G.; Gonzalez, A.; et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 2022, 185, 3789–3806.e17. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.L.; Tomkovich, S.; Yang, Y.; Jobin, C. Microbiota as a mediator of cancer progression and therapy. Transl. Res. 2017, 179, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Cummins, J.; Tangney, M. Bacteria and tumours: Causative agents or opportunistic inhabitants? Infect. Agent. Cancer 2013, 8, 11. [Google Scholar] [CrossRef]
- Baban, C.K.; Cronin, M.; O’Hanlon, D.; O’Sullivan, G.C.; Tangney, M. Bacteria as vectors for gene therapy of cancer. Bioeng. Bugs 2010, 1, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Dohlman, A.B.; Klug, J.; Mesko, M.; Gao, I.H.; Lipkin, S.M.; Shen, X.; Iliev, I.D. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 2022, 185, 3807–3822.e12. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Zhou, F.; Zhang, L. The fungal mycobiome: A new hallmark of cancer revealed by pan-cancer analyses. Signal Transduct. Target. Ther. 2023, 8, 50. [Google Scholar] [CrossRef]
- Banerjee, S.; Alwine, J.C.; Wei, Z.; Tian, T.; Shih, N.; Sperling, C.; Guzzo, T.; Feldman, M.D.; Robertson, E.S. Microbiome signatures in prostate cancer. Carcinogenesis 2019, 40, 749–764. [Google Scholar] [CrossRef]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Alwine, J.C.; Coukos, G.; Robertson, E.S. The ovarian cancer oncobiome. Oncotarget 2017, 8, 36225–36245. [Google Scholar] [CrossRef]
- Zhu, F.; Willette-Brown, J.; Song, N.Y.; Lomada, D.; Song, Y.; Xue, L.; Gray, Z.; Zhao, Z.; Davis, S.R.; Sun, Z.; et al. Autoreactive T Cells and Chronic Fungal Infection Drive Esophageal Carcinogenesis. Cell Host Microbe 2017, 21, 478–493.e7. [Google Scholar] [CrossRef]
- Luan, C.; Xie, L.; Yang, X.; Miao, H.; Lv, N.; Zhang, R.; Xiao, X.; Hu, Y.; Liu, Y.; Wu, N.; et al. Dysbiosis of fungal microbiota in the intestinal mucosa of patients with colorectal adenomas. Sci. Rep. 2015, 5, 7980. [Google Scholar] [CrossRef]
- Lin, Y.; Lau, H.C.; Liu, Y.; Kang, X.; Wang, Y.; Ting, N.L.; Kwong, T.N.; Han, J.; Liu, W.; Liu, C.; et al. Altered Mycobiota Signatures and Enriched Pathogenic Aspergillus rambellii Are Associated With Colorectal Cancer Based on Multicohort Fecal Metagenomic Analyses. Gastroenterology 2022, 163, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Peck, K.N.; DeMichele, A.M.; Alwine, J.C.; Robertson, E.S. Distinct Microbial Signatures Associated With Different Breast Cancer Types. Front. Microbiol. 2018, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Makinen, A.; Nawaz, A.; Makitie, A.; Meurman, J.H. Role of Non-Albicans Candida and Candida albicans in Oral Squamous Cell Cancer Patients. J. Oral Maxillofac. Surg. 2018, 76, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Levanduski, E.; Denz, P.; Villavicencio, H.S.; Bhatta, M.; Alhorebi, L.; Zhang, Y.; Gomez, E.C.; Morreale, B.; Senchanthisai, S.; et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell 2022, 40, 153–167.e11. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Hogan, D.A.; Mylonakis, E. Medically important bacterial-fungal interactions. Nat. Rev. Microbiol. 2010, 8, 340–349. [Google Scholar] [CrossRef]
- Baena-Monroy, T.; Moreno-Maldonado, V.; Franco-Martinez, F.; Aldape-Barrios, B.; Quindos, G.; Sanchez-Vargas, L.O. Candida albicans, Staphylococcus aureus and Streptococcus mutans colonization in patients wearing dental prosthesis. Med. Oral Patol. Oral Cir. Bucal 2005, 10 (Suppl. S1), E27–E39. [Google Scholar]
- Rivera, M.; Norman, S.; Sehgal, R.; Juthani, R. Updates on Surgical Management and Advances for Brain Tumors. Curr. Oncol. Rep. 2021, 23, 35. [Google Scholar] [CrossRef]
- Apra, C.; Bemora, J.S.; Palfi, S. Achieving Gross Total Resection in Neurosurgery: A Review of Intraoperative Techniques and Their Influence on Surgical Goals. World Neurosurg. 2024, 185, 246–253. [Google Scholar] [CrossRef]
- Yan, Q.; Wi, Y.M.; Thoendel, M.J.; Raval, Y.S.; Greenwood-Quaintance, K.E.; Abdel, M.P.; Jeraldo, P.R.; Chia, N.; Patel, R. Evaluation of the CosmosID Bioinformatics Platform for Prosthetic Joint-Associated Sonicate Fluid Shotgun Metagenomic Data Analysis. J. Clin. Microbiol. 2019, 57, e01182-18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender | Age | Diagnosis | Abbreviation |
|---|---|---|---|---|
| P1 | male | 48 years | Glioblastoma grade 4 (NOS) | P1_GBM |
| P2 | female | 74 years | Glioblastoma grade 4 (NOS), IDH mutant | P2_GBM |
| P3 | male | 47 years | Anaplastic meningeoma grade 3 | P3_AM |
| P4 | female | 63 years | Metastatic breast cancer, ER, HR, Her2 positive | P4_MBC |
| P5 | male | 72 years | Metastatic hepatic cancer | P5_MHC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sipos, L.; Banczerowski, P.; Juhász, J.; Fedorcsák, I.; Berényi, G.; Makra, N.; Dunai, Z.A.; Szabó, D.; Erőss, L. Brain Tumors and Beyond: Multi-Compartment Microbiome and Mycobiome Analysis. Int. J. Mol. Sci. 2025, 26, 991. https://doi.org/10.3390/ijms26030991
Sipos L, Banczerowski P, Juhász J, Fedorcsák I, Berényi G, Makra N, Dunai ZA, Szabó D, Erőss L. Brain Tumors and Beyond: Multi-Compartment Microbiome and Mycobiome Analysis. International Journal of Molecular Sciences. 2025; 26(3):991. https://doi.org/10.3390/ijms26030991
Chicago/Turabian StyleSipos, László, Péter Banczerowski, János Juhász, Imre Fedorcsák, György Berényi, Nóra Makra, Zsuzsanna A. Dunai, Dóra Szabó, and Loránd Erőss. 2025. "Brain Tumors and Beyond: Multi-Compartment Microbiome and Mycobiome Analysis" International Journal of Molecular Sciences 26, no. 3: 991. https://doi.org/10.3390/ijms26030991
APA StyleSipos, L., Banczerowski, P., Juhász, J., Fedorcsák, I., Berényi, G., Makra, N., Dunai, Z. A., Szabó, D., & Erőss, L. (2025). Brain Tumors and Beyond: Multi-Compartment Microbiome and Mycobiome Analysis. International Journal of Molecular Sciences, 26(3), 991. https://doi.org/10.3390/ijms26030991