
Enhanced Analysis of Low-Abundance Proteins in Soybean Seeds Using Advanced Mass Spectrometry

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Evaluating Proteomic Methods: DDA, DIA, and BoxCar Workflow
2.2. Spectral Library Construction for Soybean Proteomic Analysis
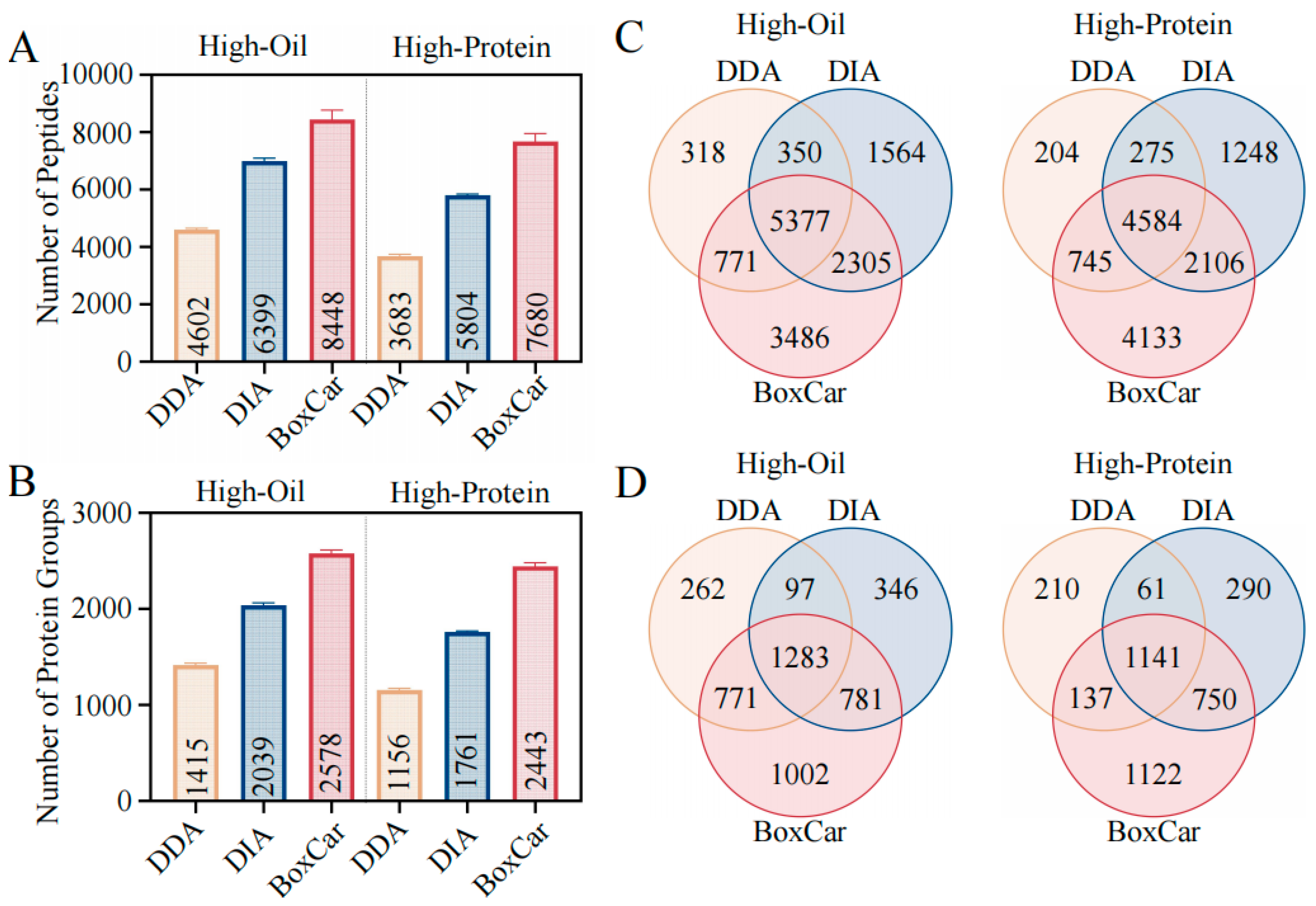
2.3. Comparative Analysis of DDA, DIA and BoxCar Methods
2.4. Evaluating the Reproducibility of DDA, DIA, and BoxCar Methods
2.5. Evaluating the Abundance Range of Proteins Identified by DDA, DIA, and BoxCar Methods
2.6. Application of BoxCar in Proteomic Analysis of Soybean Seeds
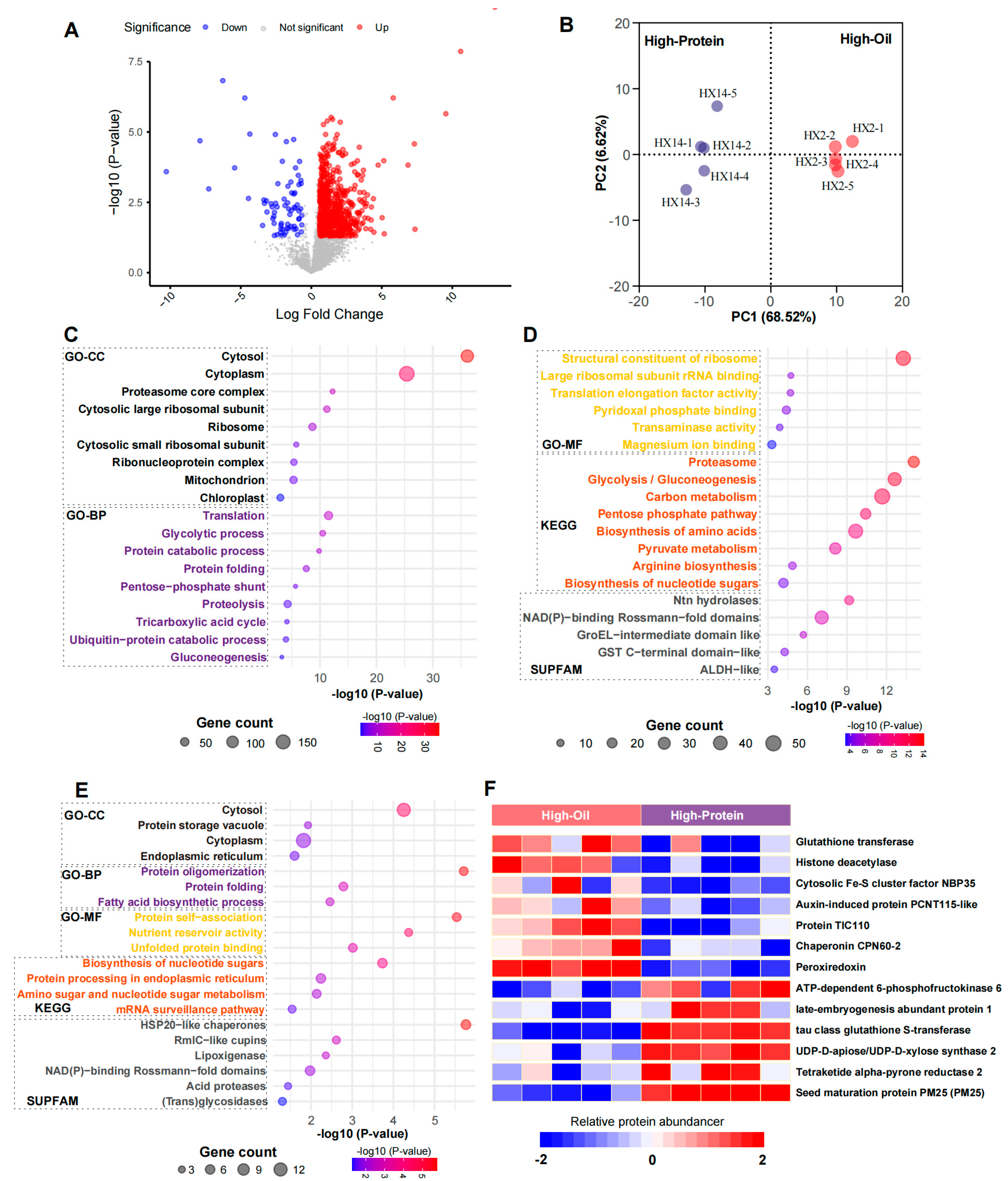
2.7. Major Difference Between High-Oil and High-Protein Groups
3. Discussion
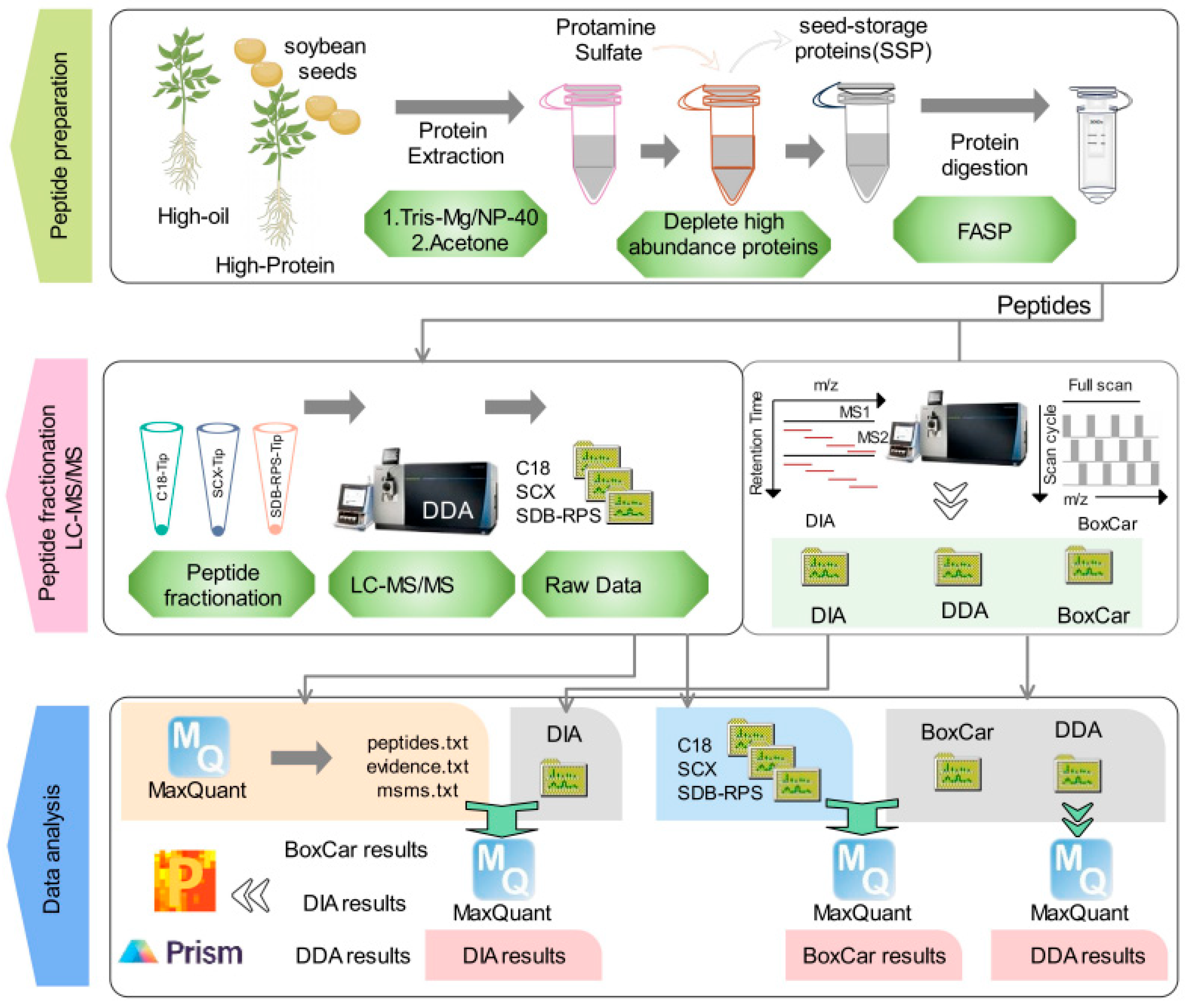
4. Materials and Methods
4.1. Soybean Seed Collection and Protein Extraction
4.2. Peptide Preparation Using the FASP Method
4.3. Peptide Fractionation Using C18-Tip, SCX-Tip and SDB-RPS-Tip Methods
4.4. LC-MS/MS Analysis
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Duan, Z.; Li, Q.; Wang, H.; He, X.; Zhang, M. Genetic regulatory networks of soybean seed size, oil and protein contents. Front. Plant Sci. 2023, 14, 1160418. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.-C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.-Q.; et al. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Messina, M. Perspective: Soybeans Can Help Address the Caloric and Protein Needs of a Growing Global Population. Front. Nutr. 2022, 9, 909464. [Google Scholar] [CrossRef]
- Islam, N.; Krishnan, H.B.; Natarajan, S. Proteomic Profiling of Fast Neutron-Induced Soybean Mutant Unveiled Pathways Associated with Increased Seed Protein Content. J. Proteome Res. 2020, 19, 3936–3944. [Google Scholar] [CrossRef]
- Min, C.W.; Hyeon, H.; Gupta, R.; Park, J.; Cheon, Y.E.; Lee, G.H.; Jang, J.W.; Ryu, H.W.; Lee, B.W.; Park, S.U.; et al. Integrated Proteomics and Metabolomics Analysis Highlights Correlative Metabolite-Protein Networks in Soybean Seeds Subjected to Warm-Water Soaking. J. Agric. Food Chem. 2020, 68, 8057–8067. [Google Scholar] [CrossRef]
- Lyu, X.; Sun, C.; Zhang, J.; Wang, C.; Zhao, S.; Ma, C.; Li, S.; Li, H.; Gong, Z.; Yan, C. Integrated proteomics and metabolomics analysis of nitrogen system regulation on soybean plant nodulation and nitrogen fixation. Int. J. Mol. Sci. 2022, 23, 2545. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, W.; Sun, J.; Zhang, Z.-R.; Yang, W.-C. Quantitative proteomics reveals key pathways in the symbiotic interface and the likely extracellular property of soybean symbiosome. J. Genet. Genom. 2023, 50, 7–19. [Google Scholar] [CrossRef]
- Kim, W.; Nott, J.; Kim, S.; Krishnan, H.B. Soybean seed proteomics: Methods for the isolation, detection, and identification of low abundance proteins. In Biochemical Pathways and Environmental Responses in Plants: Part A; Elsevier: Amsterdam, The Netherlands, 2022; pp. 325–345. [Google Scholar]
- Min, C.W.; Gupta, R.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Concepts and strategies of soybean seed proteomics using the shotgun proteomics approach. Expert Rev. Proteom. 2019, 16, 795–804. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Meng, B.; Luo, M.; Li, G.; Liu, F.; Chang, C.; Dai, X.; Fang, X. A Novel Integrated Pipeline for Site-Specific Quantification of N-glycosylation. Phenomics 2024, 4, 213–226. [Google Scholar] [CrossRef]
- Zhou, G.; Ma, S.; Yang, M.; Yang, Y. Global Quantitative Proteomics Analysis Reveals the Downstream Signaling Networks of Msx1 and Msx2 in Myoblast Differentiation. Phenomics 2022, 2, 201–210. [Google Scholar] [CrossRef]
- Zhao, Y.; Xue, Q.; Wang, M.; Meng, B.; Jiang, Y.; Zhai, R.; Zhang, Y.; Dai, X.; Fang, X. Evolution of Mass Spectrometry Instruments and Techniques for Blood Proteomics. J. Proteome Res. 2023, 22, 1009–1023. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, M.; Meng, B.; Gao, Y.; Xue, Z.; He, M.; Jiang, Y.; Dai, X.; Yan, D.; Fang, X. Identification of dysregulated complement activation pathways driven by N-glycosylation alterations in T2D patients. Front. Chem. 2021, 9, 677621. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-independent acquisition mass spectrometry-based proteomics and software tools: A glimpse in 2020. Proteomics 2020, 20, 1900276. [Google Scholar] [CrossRef]
- Rice, S.J.; Belani, C.P. Optimizing data-independent acquisition (DIA) spectral library workflows for plasma proteomics studies. Proteomics 2022, 22, 2200125. [Google Scholar] [CrossRef]
- Sato, H.; Inoue, Y.; Kawashima, Y.; Nakajima, D.; Ishikawa, M.; Konno, R.; Nakamura, R.; Kato, D.; Mitsunaga, K.; Yamamoto, T. In-depth serum proteomics by DIA-MS with in silico spectral libraries reveals dynamics during the active phase of systemic juvenile idiopathic arthritis. ACS Omega 2022, 7, 7012–7023. [Google Scholar] [CrossRef]
- Yu, Y.; Tan, P.; Zhuang, Z.; Wang, Z.; Zhu, L.; Qiu, R.; Xu, H. DIA proteomics analysis through serum profiles reveals the significant proteins as candidate biomarkers in women with PCOS. BMC Med. Genom. 2021, 14, 125. [Google Scholar] [CrossRef]
- Lou, R.; Shui, W.J.M.; Proteomics, C. Acquisition and analysis of DIA-based proteomic data: A comprehensive survey in 2023. Mol. Cell. Proteom. 2024, 23, 100712. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Ye, S.; Zhai, L.; Hu, H.; Tan, M.; Du, S. BoxCar increases the depth and reproducibility of diabetic urinary proteome analysis. Proteom. Clin. Appl. 2021, 15, 2000092. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q.; Du, Z.; Liu, J.; Zhang, Y.; Zhang, W.; Qin, W. Synthesis of Surface-Functionalized Molybdenum Disulfide Nanomaterials for Efficient Adsorption and Deep Profiling of the Human Plasma Proteome by Data-Independent Acquisition. Anal. Chem. 2022, 94, 14956–14964. [Google Scholar] [CrossRef]
- Mehta, D.; Scandola, S.; Uhrig, R.G. BoxCar and library-free data-independent acquisition substantially improve the depth, range, and completeness of label-free quantitative proteomics. Anal. Chem. 2022, 94, 793–802. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Y.; Meng, B.; Lu, A.; Wei, Y.; Dong, L.; Fang, X.; An, D.; Dai, X. SEAOP: A statistical ensemble approach for outlier detection in quantitative proteomics data. Brief. Bioinform. 2024, 25, bbae129. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef]
- Lu, G.; Zhang, X.; Apsokardu, M. Using the Empore™ C18 StageTips for High pH Peptide Fractionation in Global Proteomics. 2021. Available online: https://www.chromatographyonline.com/view/using-the-empore-c18-stagetips-for-high-ph-peptide-fractionation-in-global-proteomics (accessed on 17 February 2024).
- Wu, Z.; Li, J.; Huang, L.; Zhang, X. Basic pH reversed-phase liquid chromatography (bRPLC) in combination with tip-based strong cation exchange (SCX-Tip), ReST, an efficient approach for large-scale cross-linked peptide analysis. Anal. Chim. Acta 2021, 1179, 338838. [Google Scholar] [CrossRef]
- Min, C.W.; Park, J.; Bae, J.W.; Agrawal, G.K.; Rakwal, R.; Kim, Y.; Yang, P.; Kim, S.T.; Gupta, R. In-depth investigation of low-abundance proteins in matured and filling stages seeds of Glycine max employing a combination of protamine sulfate precipitation and TMT-based quantitative proteomic analysis. Cells 2020, 9, 1517. [Google Scholar] [CrossRef]
- Scandola, S.; Mehta, D.; Rodriguez, M.; Li, Q.; Uhrig, R.J. Multi-omic analysis reveals the biochemical changes underpinning the varied phenotypes of the Arabidopsis long-period mutant rve 4 6 8. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gallo, M.R.; Li, Q.; Talasila, M.; Uhrig, R.J.M.; Proteomics, C. Quantitative Time-Course Analysis of Osmotic and Salt Stress in Arabidopsis thaliana Using Short Gradient Multi-CV FAIMSpro BoxCar DIA. Mol. Cell. Proteom. 2023, 22, 100638. [Google Scholar] [CrossRef]
- Grubb, L.E.; Scandola, S.; Mehta, D.; Khodabocus, I.; Uhrig, R.G. Quantitative Proteomic Analysis of Brassica Napus Reveals Intersections Between Nutrient Deficiency Responses. Plant Cell Environ. 2024, 48, 1409–1428. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, B.; Lee, Y.J.; You, S.; Freeman, M.R.; Yang, W. BoxCar and shotgun proteomic analyses reveal molecular networks regulated by UBR5 in prostate cancer. Proteomics 2022, 22, 2100172. [Google Scholar] [CrossRef]
- Gasparri, R.; Noberini, R.; Cuomo, A.; Yadav, A.; Tricarico, D.; Salvetto, C.; Maisonneuve, P.; Caminiti, V.; Sedda, G.; Sabalic, A. Serum proteomics profiling identifies a preliminary signature for the diagnosis of early-stage lung cancer. Proteom. Clin. Appl. 2023, 17, 2200093. [Google Scholar] [CrossRef]
- Cortés-Ríos, J.; Zárate, A.M.; Figueroa, J.D.; Medina, J.; Fuentes-Lemus, E.; Rodríguez-Fernández, M.; Aliaga, M.; López-Alarcón, C. Protein quantification by bicinchoninic acid (BCA) assay follows complex kinetics and can be performed at short incubation times. Anal. Biochem. 2020, 608, 113904. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.R.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.; Xiao, N.; Lu, Y.; Fu, Y.; Yang, C.; Li, M.; Wu, S.; et al. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2022, 50, D1522–D1527. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, B.; Huang, Y.; Lu, A.; Liao, H.; Zhai, R.; Gong, X.; Dong, L.; Jiang, Y.; Dai, X.; Fang, X.; et al. Enhanced Analysis of Low-Abundance Proteins in Soybean Seeds Using Advanced Mass Spectrometry. Int. J. Mol. Sci. 2025, 26, 949. https://doi.org/10.3390/ijms26030949
Meng B, Huang Y, Lu A, Liao H, Zhai R, Gong X, Dong L, Jiang Y, Dai X, Fang X, et al. Enhanced Analysis of Low-Abundance Proteins in Soybean Seeds Using Advanced Mass Spectrometry. International Journal of Molecular Sciences. 2025; 26(3):949. https://doi.org/10.3390/ijms26030949
Chicago/Turabian StyleMeng, Bo, Yuanyuan Huang, Ao Lu, Huanyue Liao, Rui Zhai, Xiaoyun Gong, Lianhua Dong, You Jiang, Xinhua Dai, Xiang Fang, and et al. 2025. "Enhanced Analysis of Low-Abundance Proteins in Soybean Seeds Using Advanced Mass Spectrometry" International Journal of Molecular Sciences 26, no. 3: 949. https://doi.org/10.3390/ijms26030949
APA StyleMeng, B., Huang, Y., Lu, A., Liao, H., Zhai, R., Gong, X., Dong, L., Jiang, Y., Dai, X., Fang, X., & Zhao, Y. (2025). Enhanced Analysis of Low-Abundance Proteins in Soybean Seeds Using Advanced Mass Spectrometry. International Journal of Molecular Sciences, 26(3), 949. https://doi.org/10.3390/ijms26030949