
Rubiadin Mediates the Upregulation of Hepatic Hepcidin and Alleviates Iron Overload via BMP6/SMAD1/5/9-Signaling Pathway
 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
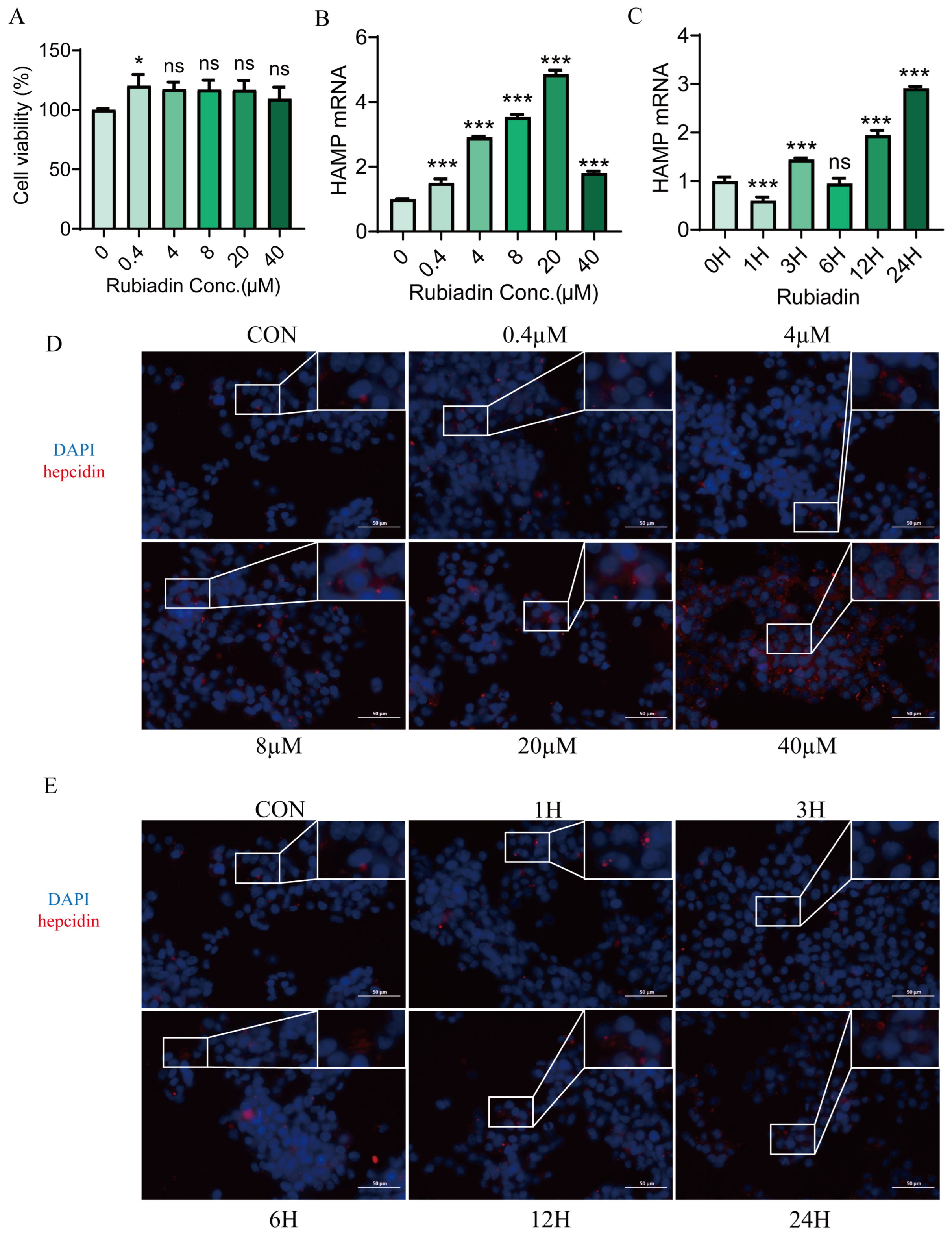
2.1. Rubiadin Significantly Upregulates Hepcidin Expression in HepG2 Cells
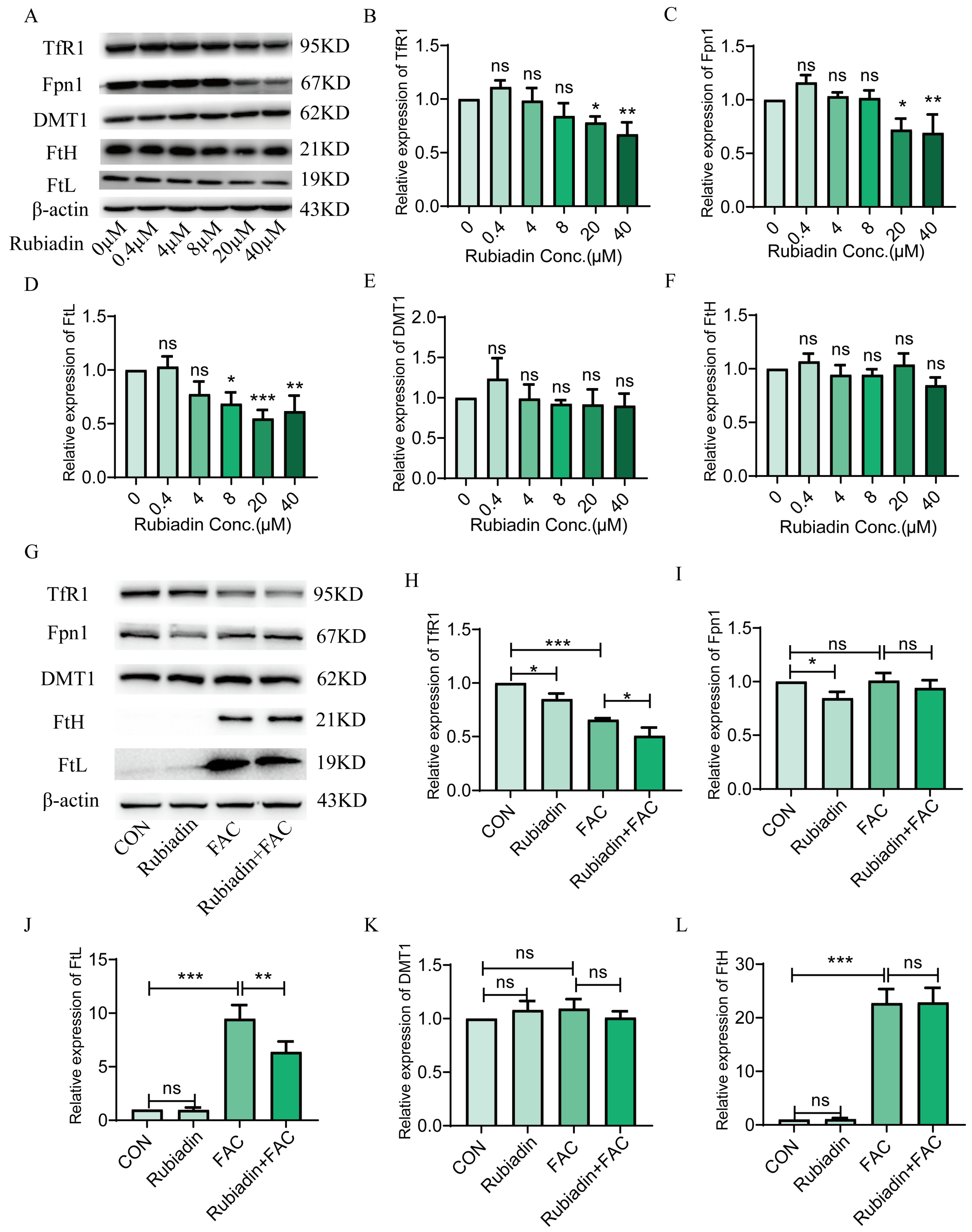
2.2. Rubiadin Decreases the Protein Expression of TfR1, Fpn1 and FtL in FAC Treated or Untreated HepG2 Cells
2.3. Rubiadin Enhances the Phosphorylation of STAT3 and SMAD1/5/9 in a Dose-Dependent Manner
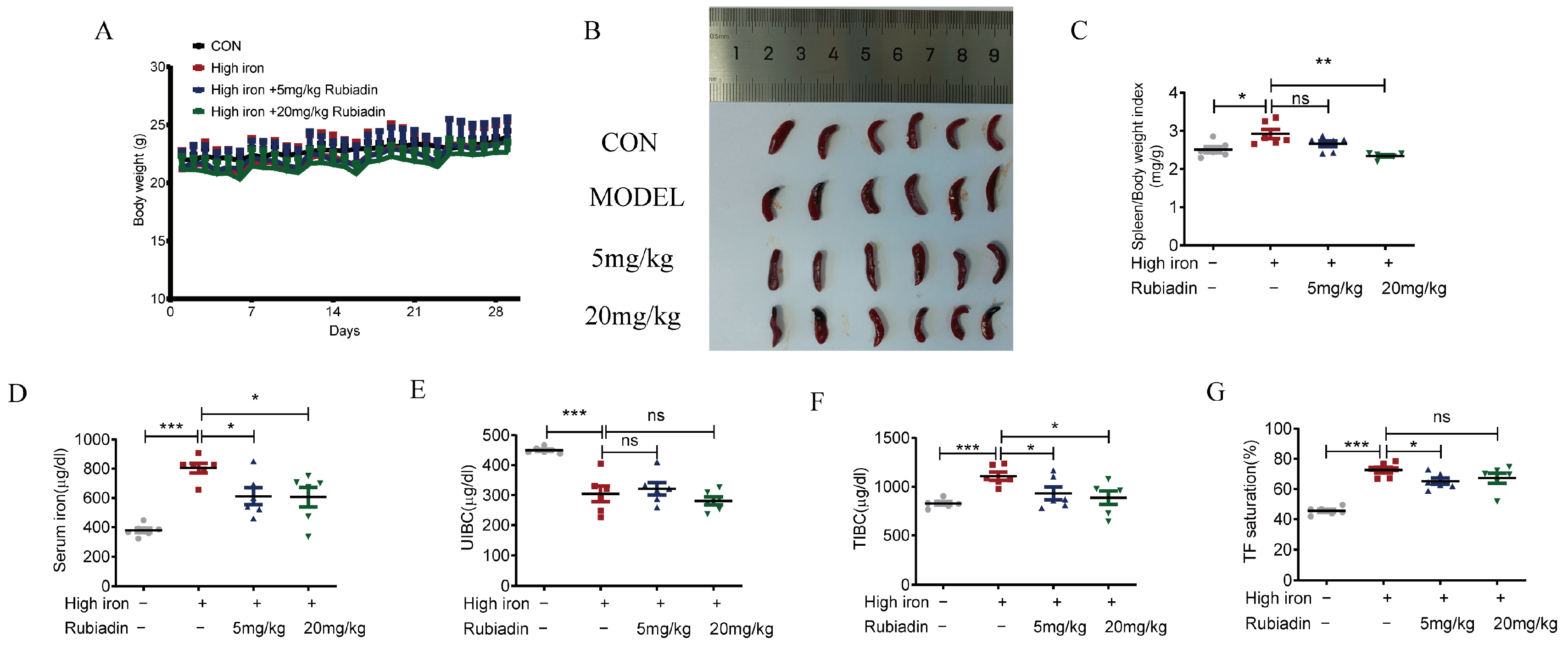
2.4. Rubiadin Remarkably Reverses the Abnormal Elevation of Serum Iron and Alleviates Splenomegaly Caused by Iron Overload
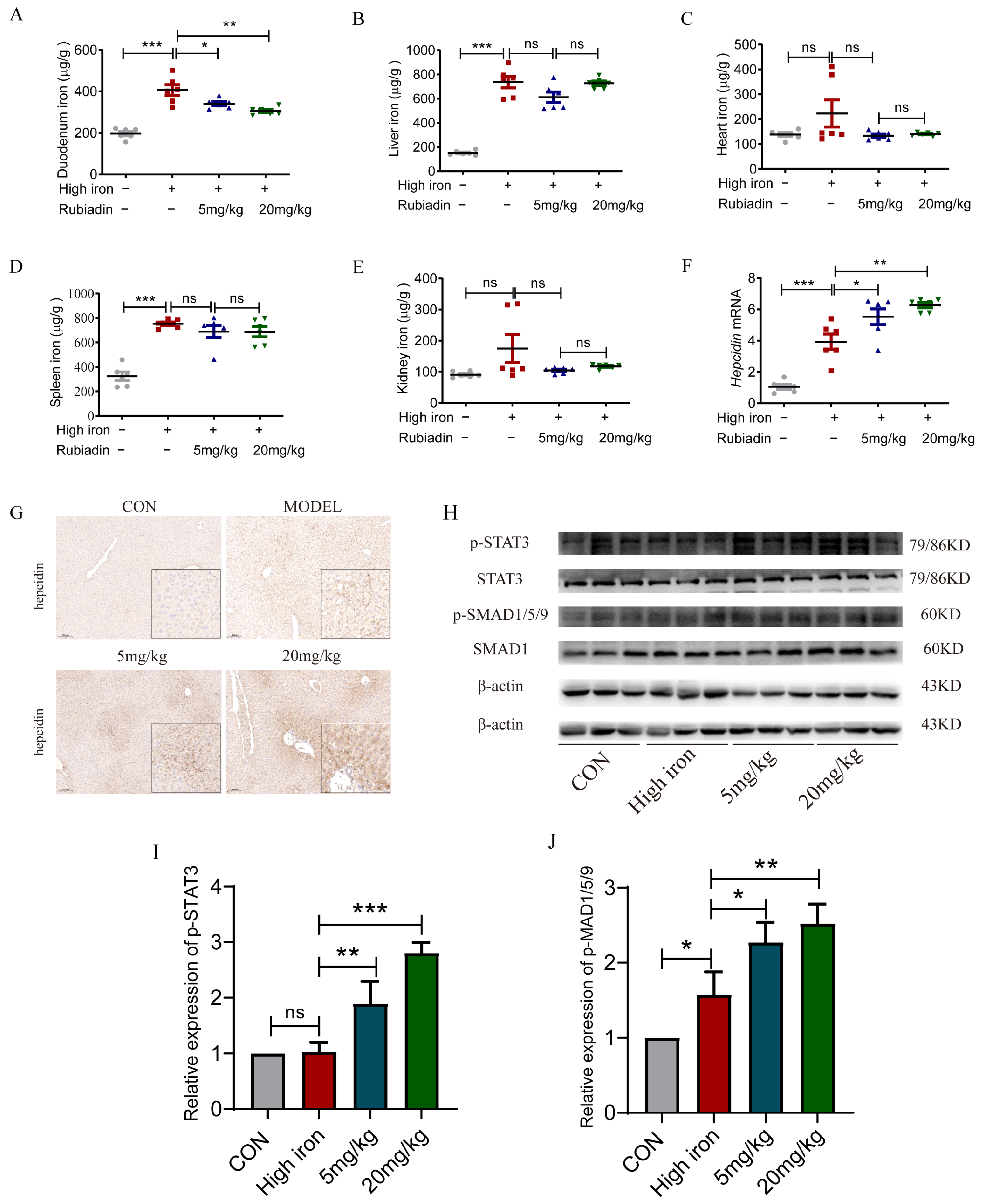
2.5. Rubiadin Remarkably Reverses the Elevation of Duodenal Iron Content and Further Enhances the Hepcidin mRNA and the Phosphorylation of STAT3 and SMAD1/5/9 Caused by Iron Overload
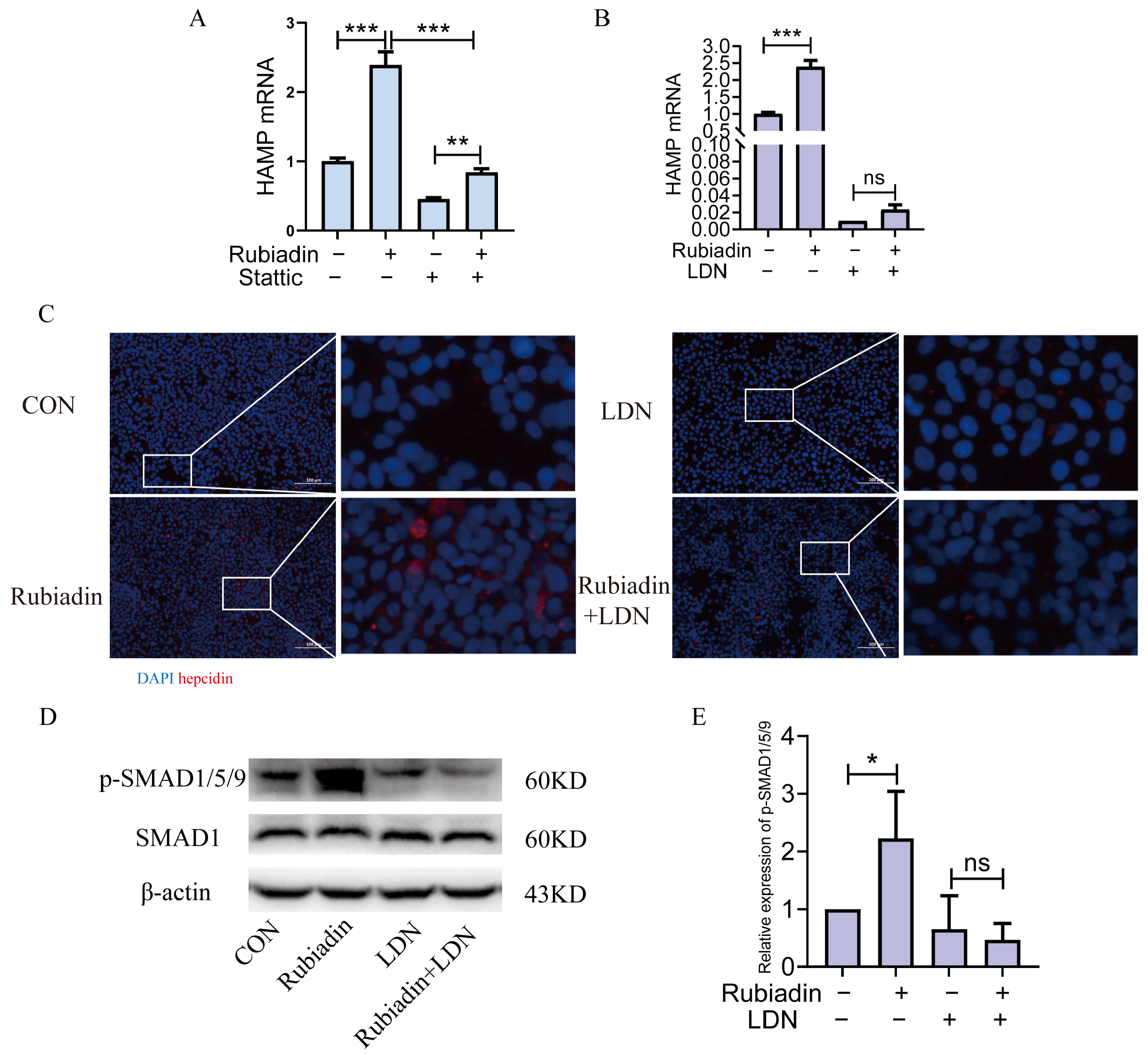
2.6. Rubiadin Upregulates Hepcidin via BMP6/SMAD1/5/9-Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Animals
4.4. Cell Proliferation Detection
4.5. Quantitative Real-Time PCR
4.6. Western Blot Analysis
4.7. Cellular Immunofluorescence
4.8. Serum Iron and Transferrin Saturation
4.9. Tissue Iron Measurement
4.10. Immunohistochemistry
4.11. Enzyme-Linked Immunosorbent Assay (ELISA) Measurements
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Macrophages and systemic iron homeostasis. J. Innate Immun. 2012, 4, 446–453. [Google Scholar] [CrossRef]
- Green, R.; Charlton, R.; Seftel, H.; Bothwell, T.; Mayet, F.; Adams, B.; Finch, C.; Layrisse, M. Body iron excretion in man: A collaborative study. Am. J. Med. 1968, 45, 336–353. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Kämmerer, L.; Mohammad, G.; Wolna, M.; Robbins, P.A.; Lakhal-Littleton, S. Fetal liver hepcidin secures iron stores in utero. Blood 2020, 136, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Vujic Spasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef]
- Rao, G.M.; Rao, C.V.; Pushpangadan, P.; Shirwaikar, A. Hepatoprotective effects of rubiadin, a major constituent of Rubia cordifolia Linn. J. Ethnopharmacol. 2006, 103, 484–490. [Google Scholar] [CrossRef]
- Watroly, M.N.; Sekar, M.; Fuloria, S.; Gan, S.H.; Jeyabalan, S.; Wu, Y.S.; Subramaniyan, V.; Sathasivam, K.V.; Ravi, S.; Mat Rani, N.N.I.; et al. Chemistry, Biosynthesis, Physicochemical and Biological Properties of Rubiadin: A Promising Natural Anthraquinone for New Drug Discovery and Development. Drug Des. Devel. Ther. 2021, 15, 4527–4549. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.C.; Barton, J.C. How I treat hemochromatosis. Blood 2010, 116, 317–325. [Google Scholar] [CrossRef]
- Pennell, D.J.; Porter, J.B.; Cappellini, M.D.; El-Beshlawy, A.; Chan, L.L.; Aydinok, Y.; Elalfy, M.S.; Sutcharitchan, P.; Li, C.K.; Ibrahim, H.; et al. Efficacy of deferasirox in reducing and preventing cardiac iron overload in beta-thalassemia. Blood 2010, 115, 2364–2371. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. Hepcidin and Iron in Health and Disease. Annu. Rev. Med. 2023, 74, 261–277. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Ruchala, P.; Goodnough, J.B.; Kautz, L.; Preza, G.C.; Nemeth, E.; Ganz, T. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012, 120, 3829–3836. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Wu, Q.; Wang, H.; Zhao, L.; Wang, X.; Mu, M.; Xie, E.; He, X.; Shao, D.; et al. Adenine alleviates iron overload by cAMP/PKA mediated hepatic hepcidin in mice. J. Cell. Physiol. 2018, 233, 7268–7278. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Genetics, Genetic Testing, and Management of Hemochromatosis: 15 Years Since Hepcidin. Gastroenterology 2015, 149, 1240–1251.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.W.; Nguyen, N.H.; Gibert, Y.; Motola, S.; Buckett, P.; Wessling-Resnick, M.; Fraenkel, E.; Fraenkel, P.G. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology 2013, 58, 1315–1325. [Google Scholar] [CrossRef]
- Akhtar, M.N.; Zareen, S.; Yeap, S.K.; Ho, W.Y.; Lo, K.M.; Hasan, A.; Alitheen, N.B. Total synthesis, cytotoxic effects of damnacanthal, nordamnacanthal and related anthraquinone analogues. Molecules 2013, 18, 10042–10055. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.W.; Yang, G.; Zhou, Y.F.; Qian, C.; Mu, M.D.; Ke, Y.; Qian, Z.M. Regulating ferroportin-1 and transferrin receptor-1 expression: A novel function of hydrogen sulfide. J. Cell. Physiol. 2019, 234, 3158–3169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.F.; Wu, X.M.; Zhou, G.; Mu, M.D.; Zhang, F.L.; Li, F.M.; Qian, C.; Du, F.; Yung, W.H.; Qian, Z.M.; et al. Cystathionine β-synthase is required for body iron homeostasis. Hepatology 2018, 67, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Qian, Z.M.; Luo, Q.; Yung, W.H.; Ke, Y. Hepcidin Suppresses Brain Iron Accumulation by Downregulating Iron Transport Proteins in Iron-Overloaded Rats. Mol. Neurobiol. 2015, 52, 101–114. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, X.; Chang, L.; Zhu, X.; Gong, F.; Che, L.; Zhang, R.; Wang, L.; Gong, C.; Fang, C.; Yao, C.; et al. Rubiadin Mediates the Upregulation of Hepatic Hepcidin and Alleviates Iron Overload via BMP6/SMAD1/5/9-Signaling Pathway. Int. J. Mol. Sci. 2025, 26, 1385. https://doi.org/10.3390/ijms26031385
Xie X, Chang L, Zhu X, Gong F, Che L, Zhang R, Wang L, Gong C, Fang C, Yao C, et al. Rubiadin Mediates the Upregulation of Hepatic Hepcidin and Alleviates Iron Overload via BMP6/SMAD1/5/9-Signaling Pathway. International Journal of Molecular Sciences. 2025; 26(3):1385. https://doi.org/10.3390/ijms26031385
Chicago/Turabian StyleXie, Xueting, Linyue Chang, Xinyue Zhu, Fengbei Gong, Linlin Che, Rujun Zhang, Lixin Wang, Chenyuan Gong, Cheng Fang, Chao Yao, and et al. 2025. "Rubiadin Mediates the Upregulation of Hepatic Hepcidin and Alleviates Iron Overload via BMP6/SMAD1/5/9-Signaling Pathway" International Journal of Molecular Sciences 26, no. 3: 1385. https://doi.org/10.3390/ijms26031385
APA StyleXie, X., Chang, L., Zhu, X., Gong, F., Che, L., Zhang, R., Wang, L., Gong, C., Fang, C., Yao, C., Hu, D., Zhao, W., Zhou, Y., & Zhu, S. (2025). Rubiadin Mediates the Upregulation of Hepatic Hepcidin and Alleviates Iron Overload via BMP6/SMAD1/5/9-Signaling Pathway. International Journal of Molecular Sciences, 26(3), 1385. https://doi.org/10.3390/ijms26031385