
Gender Differences in Adenine Diet-Induced Kidney Toxicity: The Impact of 17β-Estradiol on Renal Inflammation and Fibrosis
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
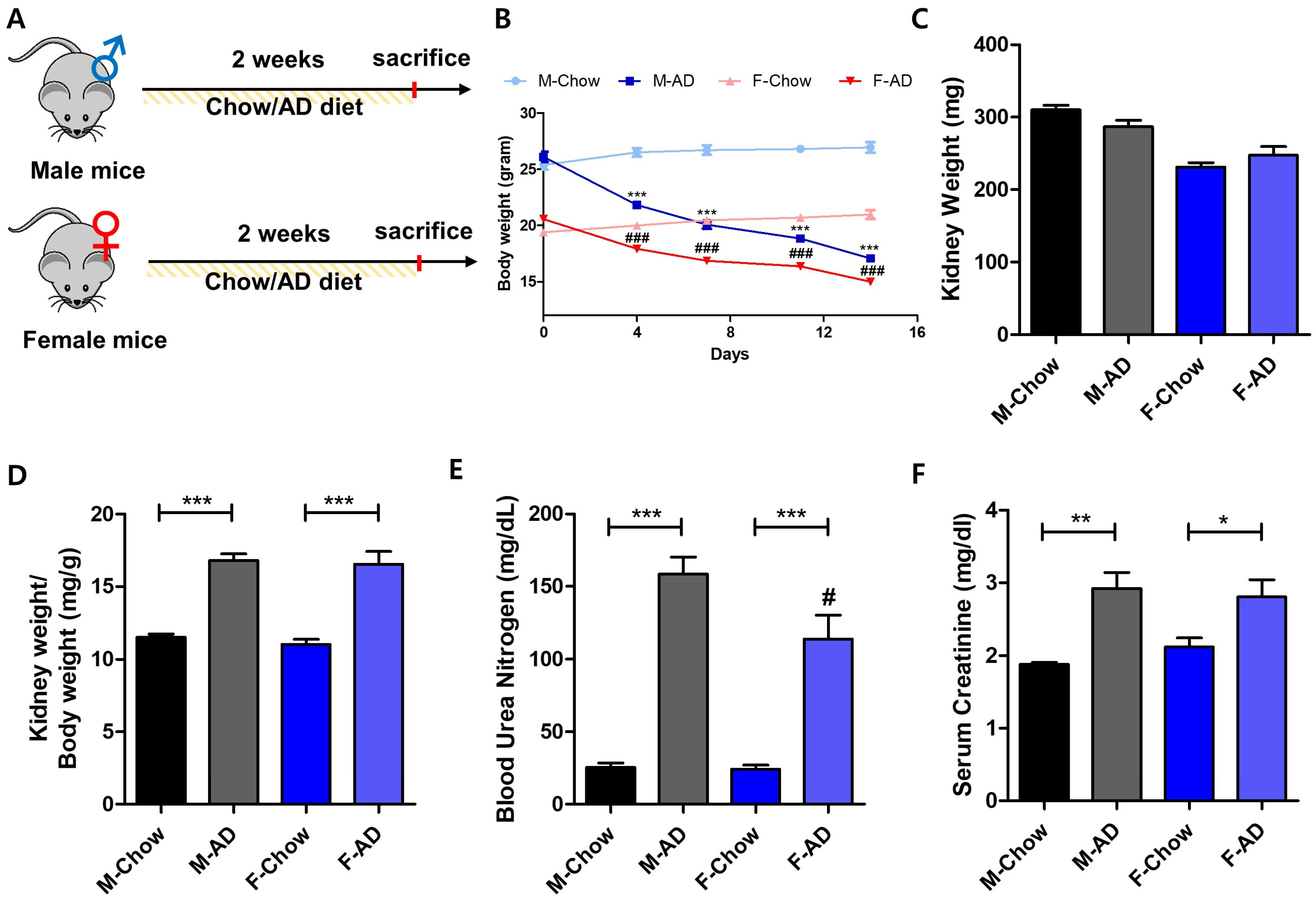
2.1. Male Mice Exhibit Heightened Vulnerability to AD-Induced Kidney Injury
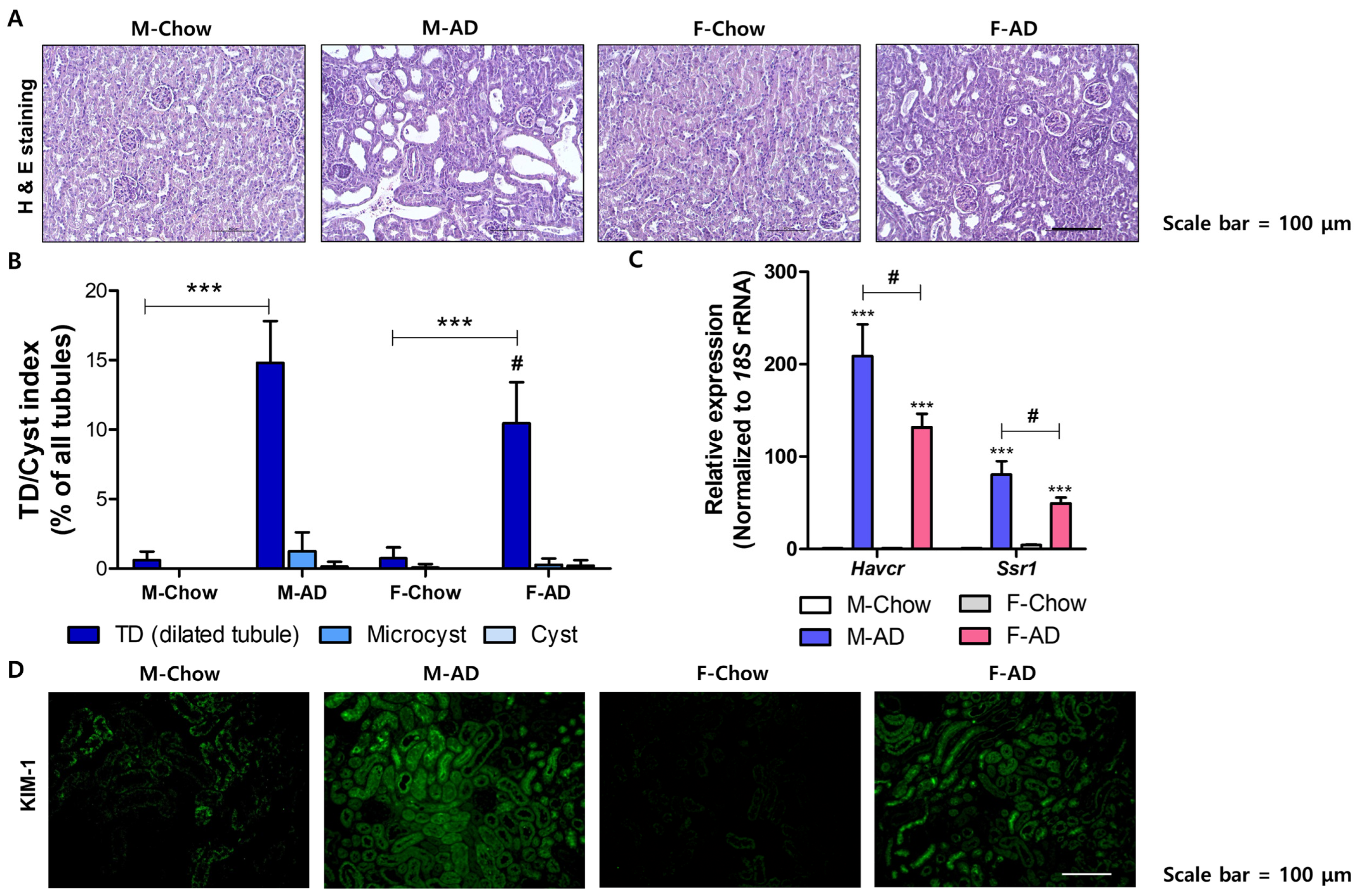
2.2. Male Mice Exhibited Severe Renal Fibrosis Development in the AD Model
2.3. Male Mice Are Susceptive to Inflammatory Responses in the Kidney Under AD Conditions
2.4. E2 Shows Protective Effects on LPS-Induced Inflammatory Responses in Renal Tubular Epithelial Cells
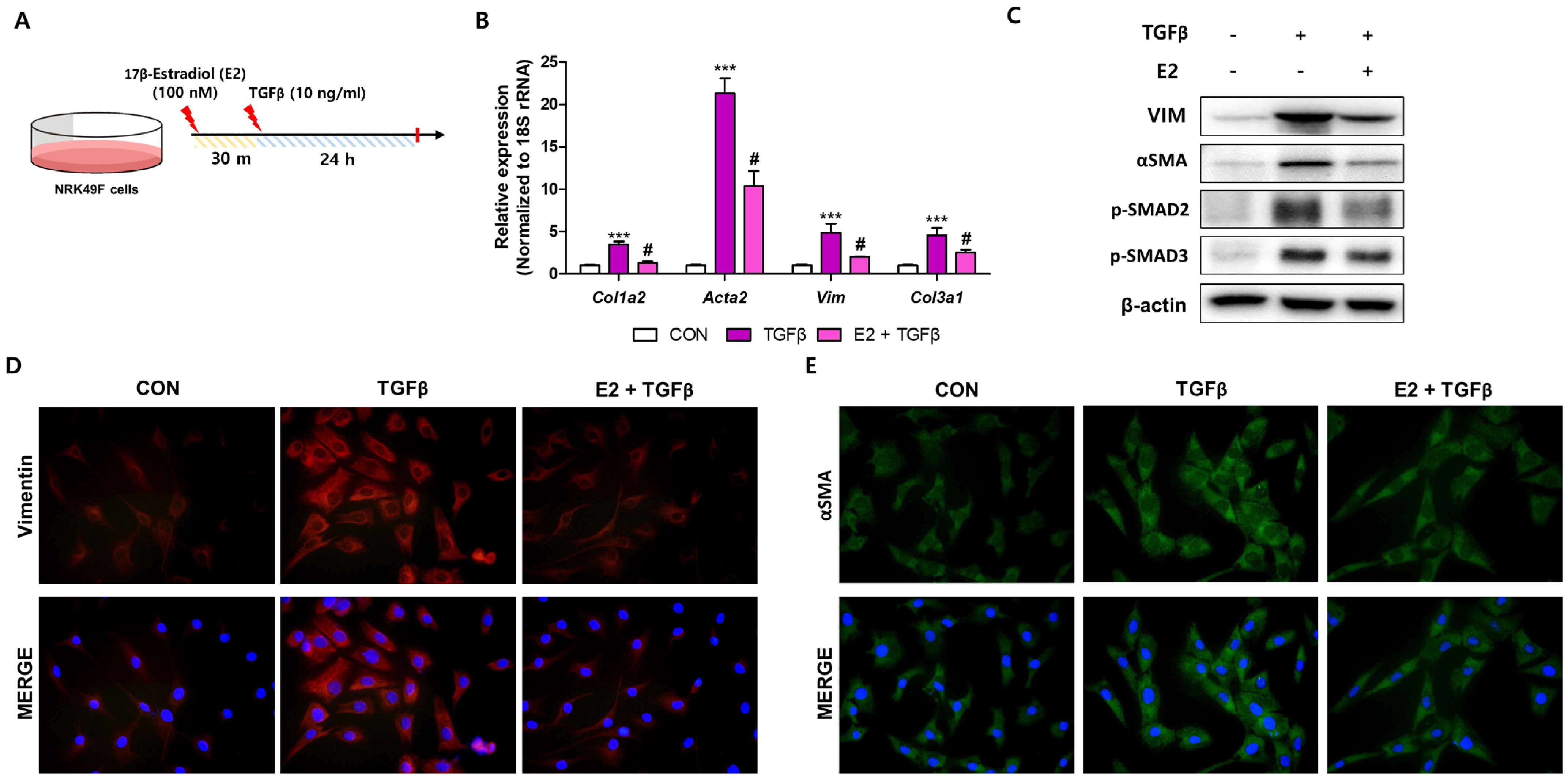
2.5. E2 Decreases TGFβ-Induced Fibroblast Activation in NRK49F Cells
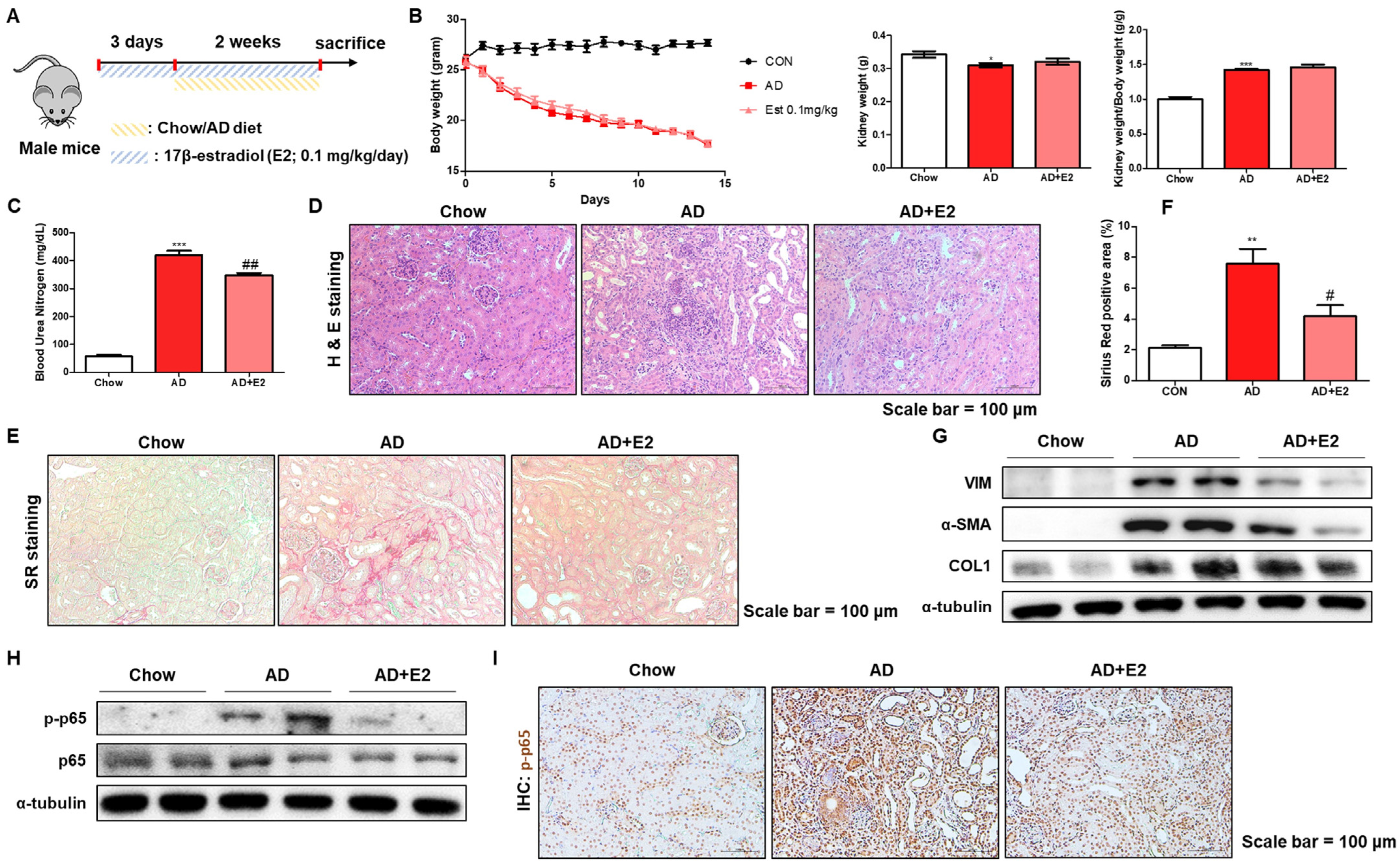
2.6. E2 Treatment Ameliorates AD-Induced Kidney Inflammation and Fibrosis
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Cell Culture and Treatments
4.3. Biochemical Assays for BUN and Creatinine
4.4. Protein Extraction and Western Blot Analysis
4.5. RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR)
4.6. Histological Analysis
4.7. Quantification of Dilated Tubules
4.8. In Situ Hybridization (ISH) Staining
4.9. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Farahmand, M.; Ramezani Tehrani, F.; Khalili, D.; Cheraghi, L.; Azizi, F. Endogenous estrogen exposure and chronic kidney disease; a 15-year prospective cohort study. BMC Endocr. Disord. 2021, 21, 155. [Google Scholar] [CrossRef]
- Carrero, J.J.; Hecking, M.; Chesnaye, N.C.; Jager, K.J. Sex and gender disparities in the epidemiology and outcomes of chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 151–164. [Google Scholar] [CrossRef]
- Neugarten, J. Gender and the progression of renal disease. J. Am. Soc. Nephrol. 2002, 13, 2807–2809. [Google Scholar] [CrossRef]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2020, 21, 5203. [Google Scholar] [CrossRef]
- Cantaluppi, V.; Quercia, A.D.; Dellepiane, S.; Ferrario, S.; Camussi, G.; Biancone, L. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Chung, K.W.; Lee, J.; Chung, H.Y.; Moon, H.R. Renal tubular PAR2 promotes interstitial fibrosis by increasing inflammatory responses and EMT process. Arch. Pharmacal Res. 2022, 45, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Jiang, H.; Pan, J.; Huang, X.R.; Wang, Y.C.; Huang, H.F.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y.; Chen, J.H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.L.; Adamczak, M.; Rabe, T.; Harbi, N.A.; Krtil, J.; Koch, A.; Hamar, P.; Amann, K.; Ritz, E. Beneficial Effects of Estrogens on Indices of Renal Damage in Uninephrectomized SHRsp Rats. J. Am. Soc. Nephrol. 2004, 15, 348–358. [Google Scholar] [CrossRef]
- Inada, A.; Inada, O.; Fujii, N.L.; Nagafuchi, S.; Katsuta, H.; Yasunami, Y.; Matsubara, T.; Arai, H.; Fukatsu, A.; Nabeshima, Y.I. Adjusting the 17β-Estradiol-to-Androgen Ratio Ameliorates Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 3035–3050. [Google Scholar] [CrossRef]
- Wu, C.C.; Chang, C.Y.; Chang, S.T.; Chen, S.H. 17β-Estradiol Accelerated Renal Tubule Regeneration in Male Rats After Ischemia/Reperfusion-Induced Acute Kidney Injury. Shock 2016, 46, 158–163. [Google Scholar] [CrossRef]
- Dogan, E.; Erkoc, R.; Demir, C.; Sayarlioglu, H.; Dilek, I.; Sayarlioglu, M. Effect of hormone replacement therapy on CD4+ and CD8+ numbers, CD4+/CD8+ ratio, and immunoglobulin levels in hemodialysis patients. Ren. Fail. 2005, 27, 421–424. [Google Scholar]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Kang, S.C.; Jhee, J.H.; Joo, Y.S.; Lee, S.M.; Nam, K.H.; Yun, H.R.; Han, S.H.; Yoo, T.H.; Kang, S.W.; Park, J.T. Association of Reproductive Lifespan Duration and Chronic Kidney Disease in Postmenopausal Women. Mayo Clin. Proc. 2020, 95, 2621–2632. [Google Scholar] [CrossRef]
- Ren, L.; Li, F.; Di, Z.; Xiong, Y.; Zhang, S.; Ma, Q.; Bian, X.; Lang, Z.; Ye, Q.; Wang, Y. Estradiol Ameliorates Acute Kidney Ischemia-Reperfusion Injury by Inhibiting the TGF-betaRI-SMAD Pathway. Front. Immunol. 2022, 13, 822604. [Google Scholar] [CrossRef]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-edged Sword. J. Histochem. Cytochem. 2019, 67, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Oe, Y.; Fushima, T.; Sato, E.; Sato, H.; Ito, S.; Takahashi, N. Protease-activated receptor 2 exacerbates adenine-induced renal tubulointerstitial injury in mice. Biochem. Biophys. Res. Commun. 2017, 483, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhang, T.; Xiao, X.; Shi, Y.; Duan, H.; Ren, Y. Protease-activated receptor-2 promotes kidney tubular epithelial inflammation by inhibiting autophagy via the PI3K/Akt/mTOR signalling pathway. Biochem. J. 2017, 474, 2733–2747. [Google Scholar] [CrossRef]
- Kattah, A.G.; Smith, C.Y.; Gazzuola Rocca, L.; Grossardt, B.R.; Garovic, V.D.; Rocca, W.A. CKD in Patients with Bilateral Oophorectomy. Clin. J. Am. Soc. Nephrol. 2018, 13, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Pesce, C.; Zheng, W.; Kim, J.; Zhang, Y.; Menini, S.; Haywood, J.R.; Sandberg, K. Sex differences in renal injury and nitric oxide production in renal wrap hypertension. Am. J. Physiol. -Heart Circ. Physiol. 2005, 288, H43–H47. [Google Scholar] [CrossRef]
- Silbiger, S.R.; Neugarten, J. The role of gender in the progression of renal disease. Adv. Ren. Replace. Ther. 2003, 10, 3–14. [Google Scholar] [CrossRef]
- Yu, M.; Ryu, D.R.; Kim, S.J.; Choi, K.B.; Kang, D.H. Clinical implication of metabolic syndrome on chronic kidney disease depends on gender and menopausal status: Results from the Korean National Health and Nutrition Examination Survey. Nephrol. Dial. Transplant. 2010, 25, 469–477. [Google Scholar] [CrossRef]
- Miao, J.; Huang, J.; Liang, Y.; Zhang, Y.; Li, J.; Meng, P.; Shen, W.; Li, X.; Wu, Q.; Wang, X.; et al. Sirtuin 6 is a key contributor to gender differences in acute kidney injury. Cell Death Discov. 2023, 9, 134. [Google Scholar] [CrossRef]
- Kim, N.Y.; Lee, H.S.; Park, J.H.; Jeon, S.; Oh, C.; Kim, S.Y. Influence of age on gender-related differences in acute kidney injury after minimally invasive radical or partial nephrectomy. Surg. Endosc. 2022, 36, 2962–2972. [Google Scholar] [CrossRef]
- Darvishzadeh Mahani, F.; Khaksari, M.; Raji-Amirhasani, A. Renoprotective effects of estrogen on acute kidney injury: The role of SIRT1. Int. Urol. Nephrol. 2021, 53, 2299–2310. [Google Scholar] [CrossRef]
- Ma, H.Y.; Chen, S.; Du, Y. Estrogen and estrogen receptors in kidney diseases. Ren. Fail. 2021, 43, 619–642. [Google Scholar] [CrossRef]
- Callewaert, F.; Boonen, S.; Vanderschueren, D. Sex steroids and the male skeleton: A tale of two hormones. Trends Endocrinol. Metab. 2010, 21, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, Y.H.; Jung, S.W.; Kim, D.J.; Park, S.H.; Song, S.J.; Jeong, K.H.; Moon, J.Y.; Ihm, C.G.; Lee, T.W.; et al. Sex-related differences in the intratubular renin-angiotensin system in two-kidney, one-clip hypertensive rats. Am. J. Physiol.-Ren. Physiol. 2019, 317, F670–F682. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Ma, L.; Zhou, L.; Fu, P. Renal Protective Effects of 17β-Estradiol on Mice with Acute Aristolochic Acid Nephropathy. Molecules 2016, 21, 1391. [Google Scholar] [CrossRef] [PubMed]
- Maric, C.; Sandberg, K.; Hinojosa-Laborde, C. Glomerulosclerosis and tubulointerstitial fibrosis are attenuated with 17β-estradiol in the aging Dahl salt sensitive rat. J. Am. Soc. Nephrol. 2004, 15, 1546–1556. [Google Scholar] [CrossRef]
- Pezeshki, Z.; Nematbakhsh, M.; Nasri, H.; Talebi, A.; Pilehvarian, A.A.; Safari, T.; Eshraghi-Jazi, F.; Haghighi, M.; Ashrafi, F. Evidence against protective role of sex hormone estrogen in Cisplatin-induced nephrotoxicity in ovarectomized rat model. Toxicol. Int. 2013, 20, 43–47. [Google Scholar]
- Ha, S.; Yang, Y.; Kim, B.M.; Kim, J.; Son, M.; Kim, D.; Yu, H.S.; Im, D.S.; Chung, H.Y.; Chung, K.W. Activation of PAR2 promotes high-fat diet-induced renal injury by inducing oxidative stress and inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166474. [Google Scholar] [CrossRef]
- Marko, L.; Vigolo, E.; Hinze, C.; Park, J.K.; Roel, G.; Balogh, A.; Choi, M.; Wubken, A.; Cording, J.; Blasig, I.E.; et al. Tubular Epithelial NF-kappaB Activity Regulates Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 2658–2669. [Google Scholar] [CrossRef]
- Kumar, D.; Singla, S.K.; Puri, V.; Puri, S. The restrained expression of NF-kB in renal tissue ameliorates folic acid induced acute kidney injury in mice. PLoS ONE 2015, 10, e115947. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef]
- Elliot, S.J.; Karl, M.; Berho, M.; Potier, M.; Zheng, F.; Leclercq, B.; Striker, G.E.; Striker, L.J. Estrogen deficiency accelerates progression of glomerulosclerosis in susceptible mice. Am. J. Pathol. 2003, 162, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. -Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Vindevoghel, L.; Lechleider, R.J.; Uitto, J.; Roberts, A.B.; Mauviel, A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene 2001, 20, 3332–3340. [Google Scholar] [CrossRef] [PubMed]
- Tingskov, S.J.; Jensen, M.S.; Pedersen, C.T.; de Araujo, I.; Mutsaers, H.A.M.; Norregaard, R. Tamoxifen attenuates renal fibrosis in human kidney slices and rats subjected to unilateral ureteral obstruction. Biomed. Pharmacother. 2021, 133, 111003. [Google Scholar] [CrossRef]
- Kim, D.; Lee, A.S.; Jung, Y.J.; Yang, K.H.; Lee, S.; Park, S.K.; Kim, W.; Kang, K.P. Tamoxifen ameliorates renal tubulointerstitial fibrosis by modulation of estrogen receptor α-mediated transforming growth factor-β1/Smad signaling pathway. Nephrol. Dial. Transplant. 2014, 29, 2043–2053. [Google Scholar] [CrossRef]
- Cao, R.; Su, W.; Sheng, J.; Guo, Y.; Su, J.; Zhang, C.; Wang, H.; Tang, Y.; Chen, L.; Qiao, R.; et al. Estrogen receptor beta attenuates renal fibrosis by suppressing the transcriptional activity of Smad3. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166755. [Google Scholar]
- Chung, K.W.; Ha, S.; Kim, S.M.; Kim, D.H.; An, H.J.; Lee, E.K.; Moon, H.R.; Chung, H.Y. PPARalpha/beta Activation Alleviates Age-Associated Renal Fibrosis in Sprague Dawley Rats. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 452–458. [Google Scholar]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef]
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, S.; Son, M.; Kim, J.; Kim, D.; Kim, M.-J.; Yoo, J.; Kim, B.M.; Kim, D.; Chung, H.Y.; Chung, K.W. Gender Differences in Adenine Diet-Induced Kidney Toxicity: The Impact of 17β-Estradiol on Renal Inflammation and Fibrosis. Int. J. Mol. Sci. 2025, 26, 1358. https://doi.org/10.3390/ijms26031358
Ha S, Son M, Kim J, Kim D, Kim M-J, Yoo J, Kim BM, Kim D, Chung HY, Chung KW. Gender Differences in Adenine Diet-Induced Kidney Toxicity: The Impact of 17β-Estradiol on Renal Inflammation and Fibrosis. International Journal of Molecular Sciences. 2025; 26(3):1358. https://doi.org/10.3390/ijms26031358
Chicago/Turabian StyleHa, Sugyeong, Minjung Son, Jeongwon Kim, Doyeon Kim, Mi-Jeong Kim, Jian Yoo, Byeong Moo Kim, Donghwan Kim, Hae Young Chung, and Ki Wung Chung. 2025. "Gender Differences in Adenine Diet-Induced Kidney Toxicity: The Impact of 17β-Estradiol on Renal Inflammation and Fibrosis" International Journal of Molecular Sciences 26, no. 3: 1358. https://doi.org/10.3390/ijms26031358
APA StyleHa, S., Son, M., Kim, J., Kim, D., Kim, M.-J., Yoo, J., Kim, B. M., Kim, D., Chung, H. Y., & Chung, K. W. (2025). Gender Differences in Adenine Diet-Induced Kidney Toxicity: The Impact of 17β-Estradiol on Renal Inflammation and Fibrosis. International Journal of Molecular Sciences, 26(3), 1358. https://doi.org/10.3390/ijms26031358