ZEB2 Gene Pathogenic Variants Across Protein-Coding Regions and Impact on Clinical Manifestations: A Review

 , and
, and 
Abstract
1. Introduction
2. Subjects and Methods
2.1. Literature Search
2.2. Demographics
3. Results and Discussion
3.1. ZEB2 Gene and Protein Description
ZEB2 Gene
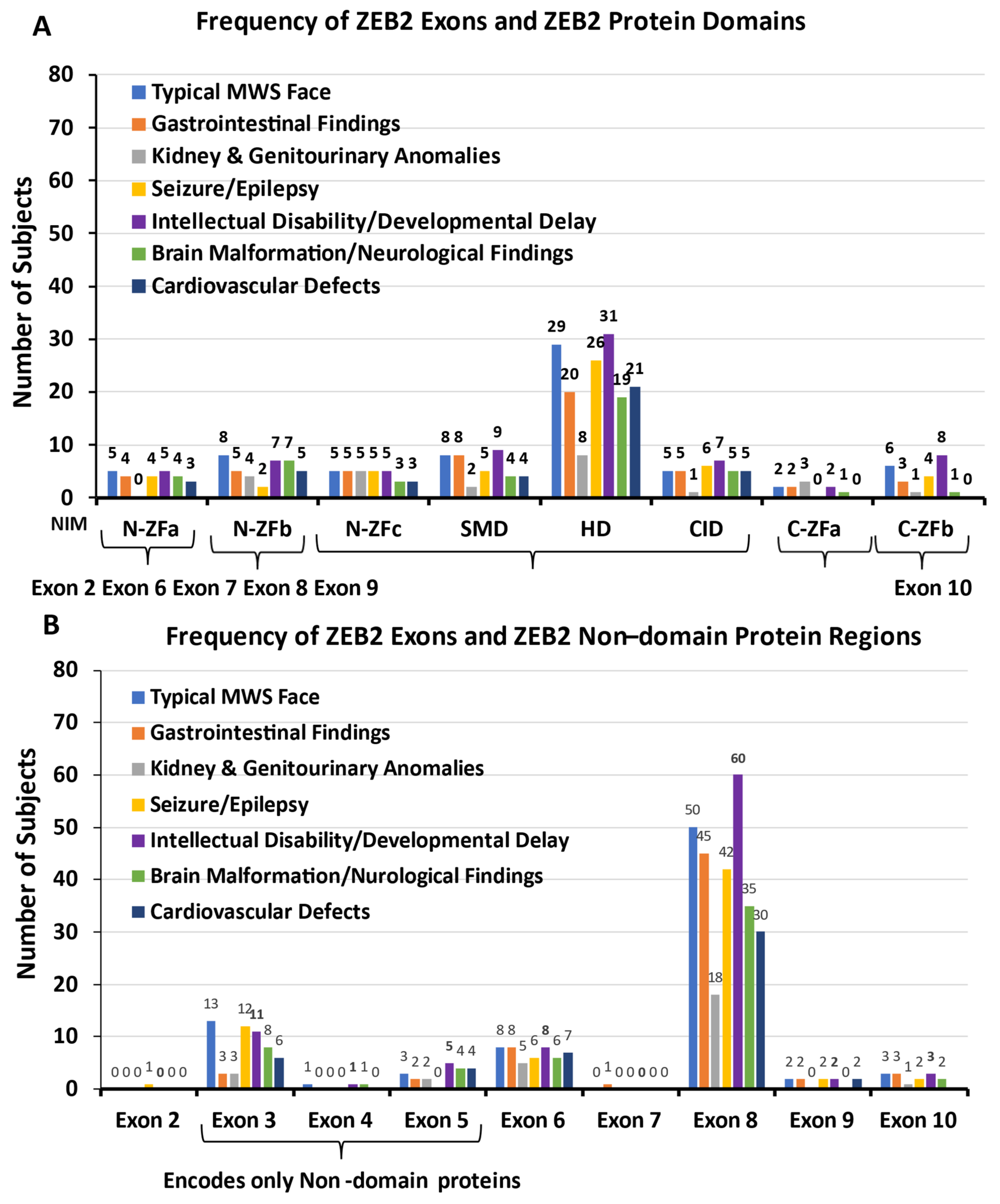
3.2. ZEB2 Protein Domains and Non-Domains
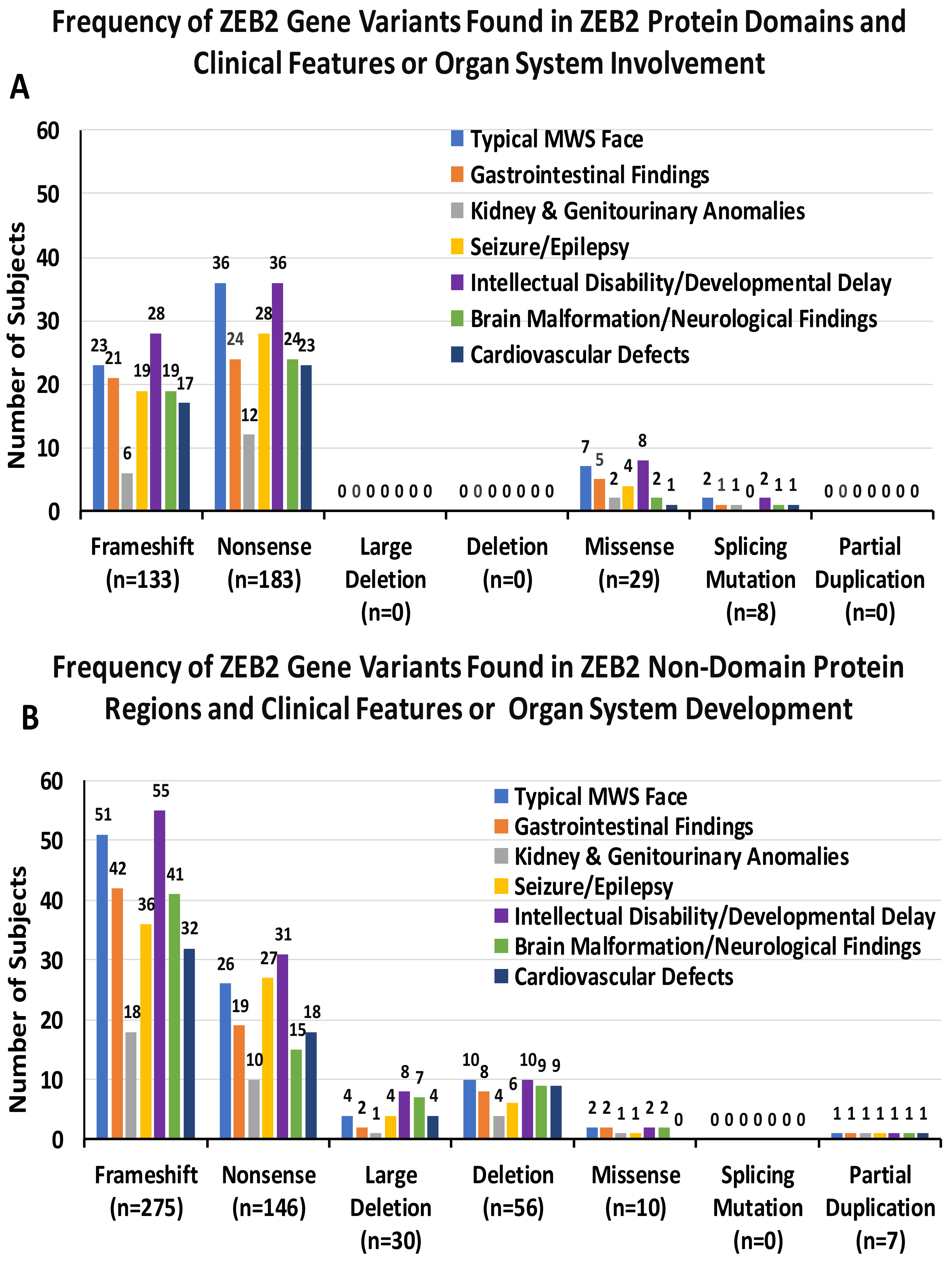
3.3. ZEB2 Gene Variants and Organ System Involvement
3.4. Clinical Manifestations and Organ System Development
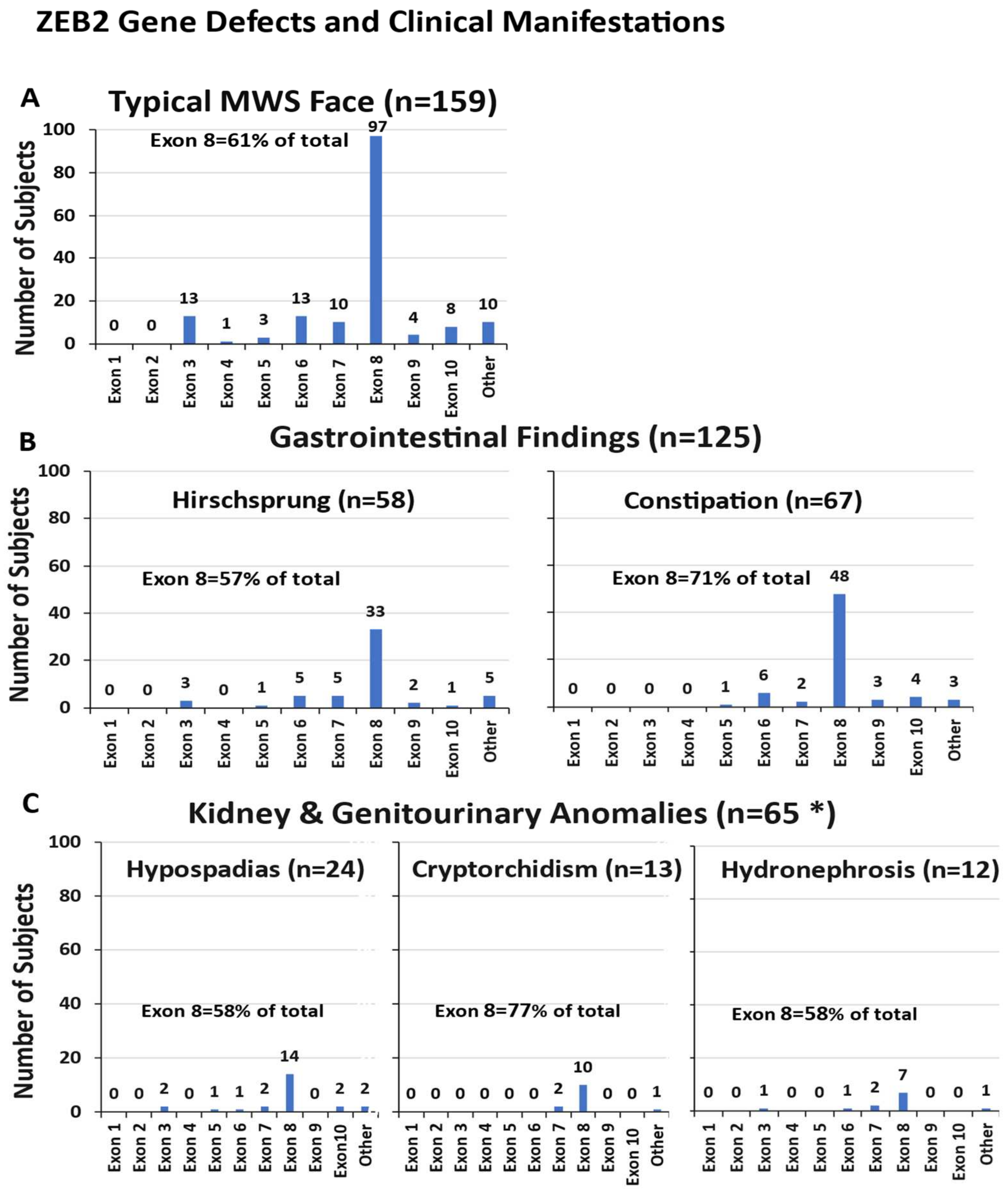
3.4.1. Typical MWS Face Description
3.4.2. Gastrointestinal Involvement
3.4.3. Kidney and Genitourinary Anomalies
3.4.4. Developmental Delays and Intellectual Disabilities
3.4.5. Description of Neurologic Manifestations and Seizures
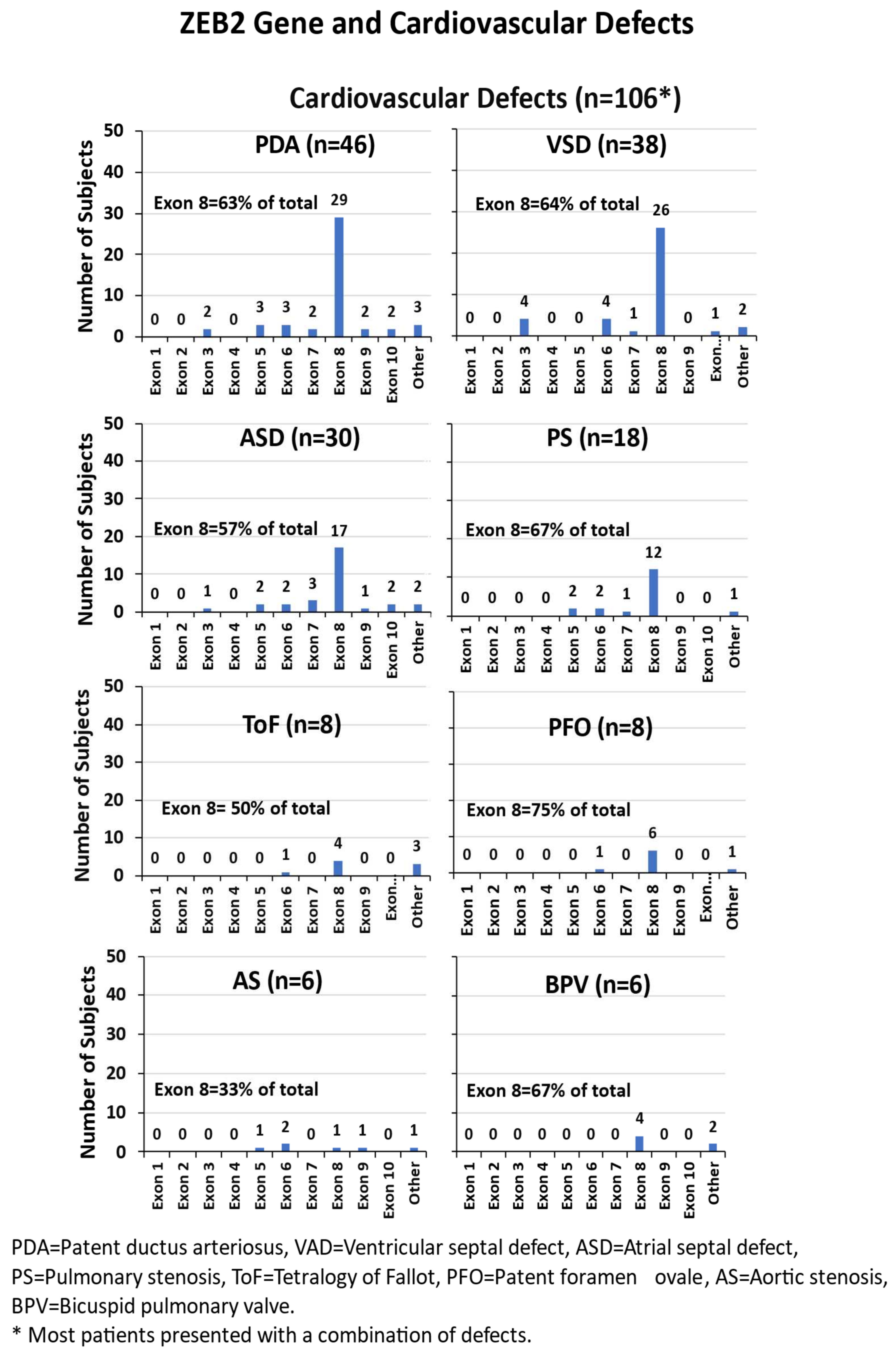
3.4.6. Cardiovascular Defects
3.4.7. Other Phenotypic Findings
3.4.8. ZEB2 Exons and ZEB2 Protein Domains and Non-Domain Regions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mowat, D.R.; Croaker, G.D.; Cass, D.T.; Kerr, B.A.; Chaitow, J.; Adès, L.C.; Chia, N.L.; Wilson, M.J. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J. Med. Genet. 1998, 8, 617–623. [Google Scholar] [CrossRef]
- Garavelli, L.; Mainardi, P.C. Mowat-Wilson syndrome. Orphanet J. Rare Dis. 2007, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Mowat, D.R.; Wilson, M.J.; Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 2003, 40, 305–310. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jones, K.L.; Jones, M.C.; del Campo, M. Smith’s Recognizable Patterns of Human Malformation, 7th ed.; Elsevier-Sanders: Philadelphia, PA, USA, 2013. [Google Scholar]
- Adam, M.P.; Conta, J.; Bean, L.J.H. Mowat-Wilson Syndrome. 2007 March 28 [Updated 2019 July 25]. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallacw, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1412/ (accessed on 4 March 2024). [PubMed]
- Ivanovski, I.; Djuric, O.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Rosato, S.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet. Med. 2018, 20, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Cordelli, D.M.; Di Pisa, V.; Fetta, A.; Garavelli, L.; Maltoni, L.; Soliani, L.; Ricci, E. Neurological Phenotype of Mowat-Wilson Syndrome. Genes 2021, 12, 982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- St Peter, C.; Hossain, W.A.; Lovell, S.; Rafi, S.K.; Butler, M.G. Mowat-Wilson Syndrome: Case Report and Review of ZEB2 Gene Variant Types, Protein Defects and Molecular Interactions. Int. J. Mol. Sci. 2024, 25, 2838. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; De Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J. Biol. Chem. 1999, 274, 20489–20498. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Farkas, C.; Benyouce, A.; Carmichael, C.; Haigh, K.; Wong, N.; Huylebroeck, D.; Stemmler, M.P.; Brabletz, S.; Brabletz, T.; et al. Interplay between the EMT transcription factors ZEB1 and ZEB2 regulates hematopoietic stem and progenitor cell differentiation and hematopoietic lineage fidelity. PLoS Biol. 2021, 19, e3001394. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.; van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Souopgui, J.; Van Damme, J.; Bellefroid, E.; Vandekerckhove, J.; Huylebroeck, D. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet. 2008, 17, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, G.L.; Lindtner, S.; Trzcinski, B.; Visel, A.; Pennacchio, L.A.; Huylebroeck, D.; Higashi, Y.; Rubenstein, J.L. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 2013, 77, 83–98. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, H.; Xiao, Z.; Zheng, J.; Wu, J.; Hu, X.L.; Yang, X.; Shen, Q. ZEB1 Represses Neural Differentiation and Cooperates with CTBP2 to Dynamically Regulate Cell Migration during Neocortex Development. Cell Rep. 2019, 27, 2335–2353.e6. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Alvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes. Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Iwafuchi-Doi, M.; Matsuda, K.; Murakami, K.; Niwa, H.; Tesar, P.; Aruga, J.; Matsuo, I.; Kondoh, H. Transcriptional regulatory networks in epiblast cells and during anterior neural plate development as modeled in epiblast stem cells. Development 2012, 139, 3926–3937, Erratum in: Development 2012, 139, 4675. [Google Scholar] [CrossRef] [PubMed]
- Bendoraite, A.; Knouf, E.C.; Garg, K.S.; Parkin, R.K.; Kroh, E.M.; O’Briant, K.C.; Ventura, A.P.; Godwin, A.K.; Karlan, B.Y.; Drescher, C.W.; et al. Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: Evidence supporting a mesothelial-to-epithelial transition. Gynecol. Oncol. 2010, 116, 117–125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van de Putte, T.; Francis, A.; Nelles, L.; van Grunsven, L.A.; Huylebroeck, D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum. Mol. Genet. 2007, 16, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte, A.F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur. J. Hum. Genet. 2015, 23, 252–255. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jády, B.E.; Darzacq, X.; Tucker, K.E.; Matera, A.G.; Bertrand, E.; Kiss, T. Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J. 2003, 22, 1878–1888. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Han, J.H.; Batey, S.; Nickson, A.A.; Teichmann, S.A.; Clarke, J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 2007, 8, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, M.; Lai, L.; Liu, Z. Allostery of multidomain proteins with disordered linkers. Curr. Opin. Struct. Biol. 2020, 62, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cao, H.; Lai, L.; Liu, Z. Disordered linkers in multidomain allosteric proteins: Entropic effect to favor the open state or enhanced local concentration to favor the closed state? Protein Sci. 2018, 27, 1600–1610. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, M.; Wang, B.; Wang, B. Classification of protein domains based on their three-dimensional shapes (CPD3DS). Synth. Syst. Biotechnol. 2021, 6, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Kulesha, E.; Gough, J.; Murzin, A.G. The SCOP database in 2020: Expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res 2020, 48, D376–D382. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.; Sillitoe, I.; Marsden, R.L.; Orengo, C.A. The Classification of Protein Domains. Methods Mol. Biol. 2017, 1525, 137–164. [Google Scholar] [PubMed]
- Sillitoe, I.; Dawson, N.; Thornton, J.; Orengo, C. The history of the CATH structural classification of protein domains. Biochimie 2015, 119, 209–217. [Google Scholar] [CrossRef]
- Gemovic, B.; Perovic, V.; Glisic, S.; Veljkovic, N. Feature-based classification of amino acid substitutions outside conserved functional protein domains. ScientificWorldJournal 2013, 2013, 948617. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Güleray Lafcı, N.; Karaosmanoglu, B.; Taskiran, E.Z.; Simsek-Kiper, P.O.; Utine, G.E. Mutated Transcripts of ZEB2 Do Not Undergo Nonsense-Mediated Decay in Mowat-Wilson Syndrome. Mol. Syndromol. 2023, 14, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. CtBP Family Proteins: Unique Transcriptional Regulators in the Nucleus with Diverse Cytosolic Functions. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6557/ (accessed on 1 November 2023).
- Li, X.; Han, M.; Zhang, H.; Liu, F.; Pan, Y.; Zhu, J.; Liao, Z.; Chen, X.; Zhang, B. Structures and biological functions of zinc finger proteins and their roles in hepatocellular carcinoma. Biomark. Res. 2022, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Birkhoff, J.C.; Huylebroeck, D.; Conidi, A. ZEB2, the Mowat-Wilson Syndrome Transcription Factor: Confirmations, Novel Functions, and Continuing Surprises. Genes 2021, 12, 1037. [Google Scholar] [CrossRef]
- Conidi, A.; van den Berghe, V.; Leslie, K.; Stryjewska, A.; Xue, H.; Chen, Y.G.; Seuntjens, E.; Huylebroeck, D. Four amino acids within a tandem QxVx repeat in a predicted extended α-helix of the Smad-binding domain of Sip1 are necessary for binding to activated Smad proteins. PLoS ONE 2013, 8, e76733. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef]
- Bürglin, T.R. Homeodomain subtypes and functional diversity. Subcell. Biochem. 2011, 52, 95–122. [Google Scholar]
- Berezovsky, I.N.; Guarnera, E.; Zheng, Z. Basic units of protein structure, folding, and function. Prog. Biophys. Mol. Biol. 2017, 128, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Van Hateren, N.; Shenton, T.; Borycki, A.G. Expression of avian C-terminal binding proteins (Ctbp1 and Ctbp2) during embryonic development. Dev. Dyn. 2006, 235, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.D.; Soriano, P. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol. Cell Biol. 2002, 22, 5296–5307. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Trainor, P.A.; Melton, K.R.; Manzanares, M. Origins and plasticity of neural crest cells and their roles in jaw and evolution. Int. J. Dev. Biol. 2003, 47, 541–553. [Google Scholar] [PubMed]
- Wakamatsu, N.; Yamada, Y.; Yamada, K.; Ono, T.; Nomura, N.; Taniguchi, H.; Kitoh, H.; Mutoh, N.; Yamanaka, T.; Mushiake, K.; et al. Mutations in SIP1, encoding Smad interacting protein-1, causes a form of Hirschsprung disease. Nat. Genet. 2001, 27, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Srivatsa, S.; Parthasarathy, S.; Molnár, Z.; Tarabykin, V. Sip1 downstream Effector ninein controls neocortical axonal growth, ipsilateral branching, and microtubule growth and stability. Neuron 2015, 85, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Fetta, A.; Garavelli, L.; Caraffi, S.; Ivanovski, I.; Bonanni, P.; Accorsi, P.; Giordano, L.; Pantaleoni, C.; Romeo, A.; et al. Further delineation and long-term evolution of electroclinical phenotype in Mowat Wilson Syndrome. A longitudinal study in 40 individuals. Epilepsy Behav. 2021, 124, 108315. [Google Scholar] [CrossRef] [PubMed]
- Cordelli, D.M.; Garavelli, L.; Savasta, S.; Guerra, A.; Pellicciari, A.; Giordano, L.; Bonetti, S.; Cecconi, I.; Wischmeijer, A.; Seri, M.; et al. Epilepsy in Mowat-Wilson syndrome: Delineation of the electroclinical phenotype. Am. J. Med. Genet. A 2013, 161A, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Cordelli, D.M.; Pellicciari, A.; Kiriazopulos, D.; Franzoni, E.; Garavelli, L. Epilepsy in Mowat-Wilson syndrome: Is it a matter of GABA? Epilepsia 2013, 54, 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Epifanova, E.; Babaev, A.; Newman, A.G.; Tarabykin, V. Role of Zeb2/Sip1 in neuronal development. Brain Res. 2019, 1705, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Dagorno, C.; Pio, L.; Capri, Y.; Ali, L.; Giurgea, I.; Qoshe, L.; Morcrette, G.; Julien-Marsollier, F.; Sommet, J.; Chomton, M.; et al. Mowat Wilson syndrome and Hirschsprung disease: A retrospective study on functional outcomes. Pediatr. Surg. Int. 2020, 36, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Espinosa-Parrilla, Y.; Steffann, J.; Gosset, P.; Pelet, A.; Prieur, M.; Boute, O.; Choise, A.; Lacombe, D.; Philip, N.; et al. Large-scale deletions and SMADIP1 truncating mutations in syndromic Hirschsprung disease with involvement of midline structures. Am. J. Hum. Genet. 2001, 69, 1370–1377. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nagaya, M.; Kato, J.; Niimi, N.; Tanaka, S.; Wakamatsu, N. Clinical features of a form of Hirschsprung’s disease caused by a novel genetic abnormality. J. Pediatr. Surg. 2002, 37, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C.; Thiel, C.T.; Dufke, A.; Crow, Y.J.; Meinecke, P.; Suri, M.; Ala-Mello, S.; Beemer, F.; Bernasconi, S.; Bianchi, P.; et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur. J. Med. Genet. 2005, 48, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Ghoumid, J.; Drevillon, L.; Alavi-Naini, S.M.; Bondurand, N.; Rio, M.; Briand-Suleau, A.; Nasser, M.; Goodwin, L.; Raymond, P.; Yanicostas, C.; et al. ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum. Mol. Genet. 2013, 22, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Dastot-Le Moal, F.; Wilson, M.; Mowat, D.; Collot, N.; Niel, F.; Goossens, M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum. Mutat. 2007, 28, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Heinritz, W.; Zweier, C.; Froster, U.G.; Strenge, S.; Kuja, A.; Syrbe, S.; Rauch, A.; Schuster, V. A missense mutation in the ZFHX1B gene associated with an atypical Mowat-Wilson syndrome phenotype. Am. J. Med. Genet. A 2006, 140, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Wang, L.; Wen, F.; Xiao, H.; Duan, J.; Zhang, T.; Yin, Z.; Dong, Q.; Guo, J.; Liao, J. Genotype-phenotype analysis in Mowat-Wilson syndrome associated with two novel and two recurrent ZEB2 variants. Exp. Ther. Med. 2020, 6, 263. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, Y.; Peng, Q.; Ma, K.; Li, S.; Rao, C.; Zhong, B.; Lu, X. A novel nonsense mutation of ZEB2 gene in a Chinese patient with Mowat-Wilson syndrome. J. Clin. Lab. Anal. 2020, 9, e23413. [Google Scholar] [CrossRef]
- Ho, S.; Luk, H.M.; Chung, B.H.; Fung, J.L.; Mak, H.H.; Lo, I.F.M. Mowat-Wilson syndrome in a Chinese population: A case series. Am. J. Med. Genet. A. 2020, 182, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Wenger, T.L.; Harr, M.; Ricciardi, S.; Bhoj, E.; Santani, A.; Adam, M.P.; Barnett, S.S.; Ganetzky, R.; McDonald-McGinn, D.M.; Battaglia, D. CHARGE-like presentation, craniosynostosis and mild Mowat-Wilson Syndrome diagnosed by recognition of the distinctive facial gestalt in a cohort of 28 new cases. Am. J. Med. Genet. A 2014, 164A, 2557–2566, Erratum in: Am. J. Med. Genet. A 2015, 167, 1682–1683. [Google Scholar] [CrossRef] [PubMed]
- Mundhofir, F.E.; Yntema, H.G.; van der Burgt, I.; Hame, B.C.; Faradz, S.M.; van Bon, B.W. Mowat-Wilson syndrome: The first clinical and molecular report of an indonesian patient. Case Rep. Genet. 2012, 2012, 949507. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murray, S.B.; Spangler, B.B.; Helm, B.M.; Vergano, S.S. Polymicrogyria in a 10-month-old boy with Mowat-Wilson syndrome. Am. J. Med. Genet. A 2015, 167A, 2402–2405. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, Y.C.; Li, Q.; Zhang, Z.; Xiao, P.; Yuan, X.Y.; Li, L.; Jiang, Q. Clinical and genetic features of Mowat-Wilson syndrome: An analysis of 3 cases. Zhongguo Dang Dai Er Ke Za Zhi 2019, 21, 468–473. (In Chinese) [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yamada, Y.; Nomura, N.; Yamada, K.; Matsuo, M.; Suzuki, Y.; Sameshima, K.; Kimura, R.; Yamamoto, Y.; Fukushi, D.; Fukuhara, Y.; et al. The spectrum of ZEB2 mutations causing the Mowat-Wilson syndrome in Japanese populations. Am. J. Med. Genet. A 2014, 164A, 1899–1908, Erratum in: Am. J. Med. Genet. A 2015, 167, 1428. [Google Scholar] [CrossRef] [PubMed]
- Tronina, A.; Świerczyńska, M.; Filipek, E. First Case Report of Developmental Bilateral Cataract with a Novel Mutation in the ZEB2 Gene Observed in Mowat-Wilson Syndrome. Medicina 2023, 59, 101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jakubiak, A.; Szczałuba, K.; Badura-Stronka, M.; Kutkowska-Kaźmierczak, A.; Jakubiuk-Tomaszuk, A.; Chilarska, T.; Pilch, J.; Braun-Walicka, N.; Castaneda, J.; Wołyńska, K.; et al. Clinical characteristics of Polish patients with molecularly confirmed Mowat-Wilson syndrome. J. Appl. Genet. 2021, 62, 477–485. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Refaat, K.; Helmy, N.; Elawady, M.; El Ruby, M.; Kamel, A.; Mekkawy, M.; Ashaat, E.; Eid, O.; Mohamed, A.; Rady, M. Interstitial Deletion of 2q22.2q22.3 Involving the Entire ZEB2 Gene in a Case of Mowat-Wilson Syndrome. Mol. Syndromol. 2021, 12, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Musaad, W.; Lyons, A.; Allen, N.; Letshwiti, J. Mowat-Wilson syndrome presenting with Shone’s complex cardiac anomaly. BMJ Case Rep. 2022, 15, e246913. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pachajoa, H.; Gomez-Pineda, E.; Giraldo-Ocampo, S.; Lores, J. Mowat-Wilson Syndrome as a Differential Diagnosis in Patients with Congenital Heart Defects and Dysmorphic Facies. Pharmgenomics Pers. Med. 2022, 15, 913–918. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, L.; Wang, J.; Wang, L.; Xu, Q.; Zhou, B.; Zhang, Z.; Li, Q.; Wang, H.; Han, L.; Jiang, Q.; et al. Physical, language, neurodevelopment and phenotype-genotype correlation of Chinese patients with Mowat-Wilson syndrome. Front. Genet. 2022, 13, 1016677. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fu, Y.; Xu, W.; Wang, Q.; Lin, Y.; He, P.; Liu, Y.; Yuan, H. Three Novel De Novo ZEB2 Variants Identified in Three Unrelated Chinese Patients with Mowat-Wilson Syndrome and A Systematic Review. Front. Genet. 2022, 13, 853183. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, L.; Han, X.; Li, X.; Han, B.; Nie, W. A Chinese Boy with Mowat-Wilson Syndrome Caused by a 10 bp Deletion in the ZEB2 Gene. Pharmgenomics Pers. Med. 2021, 14, 1041–1045. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Şenbil, N.; Arslan, Z.; Sayın Kocakap, D.B.; Bilgili, Y. A Case Report of a Prenatally Missed Mowat-Wilson Syndrome with Isolated Corpus Callosum Agenesis. Child Neurol. Open 2021, 8, 2329048X211006511. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van de Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Higashi, Y.; Huylebroeck, D. Mice lacking Zfhx1b, the gene that encodes Smad-interacting protein-1, reveal a role for multiple TGFβ and BMP receptor-regulated Smads in neural crest formation, neuronal patterning, and TGFβ-mediated axon growth inhibition. Development 2003, 130, 5213–5226. [Google Scholar]
- Zweier, C.; Thiel, C.T.; Degenhardt, F.; Sticht, H.; Rossier, E.; Gillessen-Kaesbach, G.; Rauch, A. Mowat-Wilson syndrome. Clin. Genet. 2005, 67, 71–80. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frameshift | Nonsense | Missense | Large Deletion | Small Deletion | Others | |
|---|---|---|---|---|---|---|
| Typical MWS face | 46% (n = 74) | 38% (n = 62) | 6% (n = 9) | 2% (n = 4) | 6% (n = 10) | 2% (n = 3) |
| Gastrointestinal findings | 50% (n = 63) | 34% (n = 43) | 6% (n = 7) | 2% (n = 2) | 6% (n = 8) | 2% (n = 2) |
| Kidney and genitourinary anomalies | 43% (n = 24) | 39% (n = 22) | 5% (n = 3) | 2% (n = 1) | 7% (n = 4) | 4% (n = 2) |
| Seizure/epilepsy | 43% (n = 55) | 43% (n = 55) | 4% (n = 5) | 3% (n = 4) | 5% (n = 6) | 2% (n = 2) |
| Intellectual disability/developmental delay | 46% (n = 83) | 37% (n = 67) | 6% (n = 10) | 4% (n = 8) | 6% (n = 10) | 1% (n = 3) |
| Seizure/epilepsy | 43% (n = 55) | 43% (n = 55) | 4% (n = 5) | 3% (n = 4) | 5% (n = 6) | 2% (n = 2) |
| Brain malformation/ neurological findings | 50% (n = 60) | 32% (n = 39) | 3% (n = 4) | 6% (n = 7) | 7% (n = 9) | 2% (n = 2) |
| Cardiovascular defects | 46% (n = 49) | 39% (n = 41) | 1% (n = 1) | 4% (n = 4) | 8% (n = 9) | 2% (n = 2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, W.A.; St. Peter, C.; Lovell, S.; Rafi, S.K.; Butler, M.G. ZEB2 Gene Pathogenic Variants Across Protein-Coding Regions and Impact on Clinical Manifestations: A Review. Int. J. Mol. Sci. 2025, 26, 1307. https://doi.org/10.3390/ijms26031307
Hossain WA, St. Peter C, Lovell S, Rafi SK, Butler MG. ZEB2 Gene Pathogenic Variants Across Protein-Coding Regions and Impact on Clinical Manifestations: A Review. International Journal of Molecular Sciences. 2025; 26(3):1307. https://doi.org/10.3390/ijms26031307
Chicago/Turabian StyleHossain, Waheeda A., Caroline St. Peter, Scott Lovell, Syed K. Rafi, and Merlin G. Butler. 2025. "ZEB2 Gene Pathogenic Variants Across Protein-Coding Regions and Impact on Clinical Manifestations: A Review" International Journal of Molecular Sciences 26, no. 3: 1307. https://doi.org/10.3390/ijms26031307
APA StyleHossain, W. A., St. Peter, C., Lovell, S., Rafi, S. K., & Butler, M. G. (2025). ZEB2 Gene Pathogenic Variants Across Protein-Coding Regions and Impact on Clinical Manifestations: A Review. International Journal of Molecular Sciences, 26(3), 1307. https://doi.org/10.3390/ijms26031307