
Mapping Inflammatory Markers in Cerebrospinal Fluid Following Aneurysmal Subarachnoid Hemorrhage: An Age- and Sex-Matched Analysis †
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Demographic Characteristics
2.2. Inflammatory Protein Profiles in CSF of SAH Patients vs. Healthy Controls
2.2.1. Comparative Analysis of the Number of Measurable Values
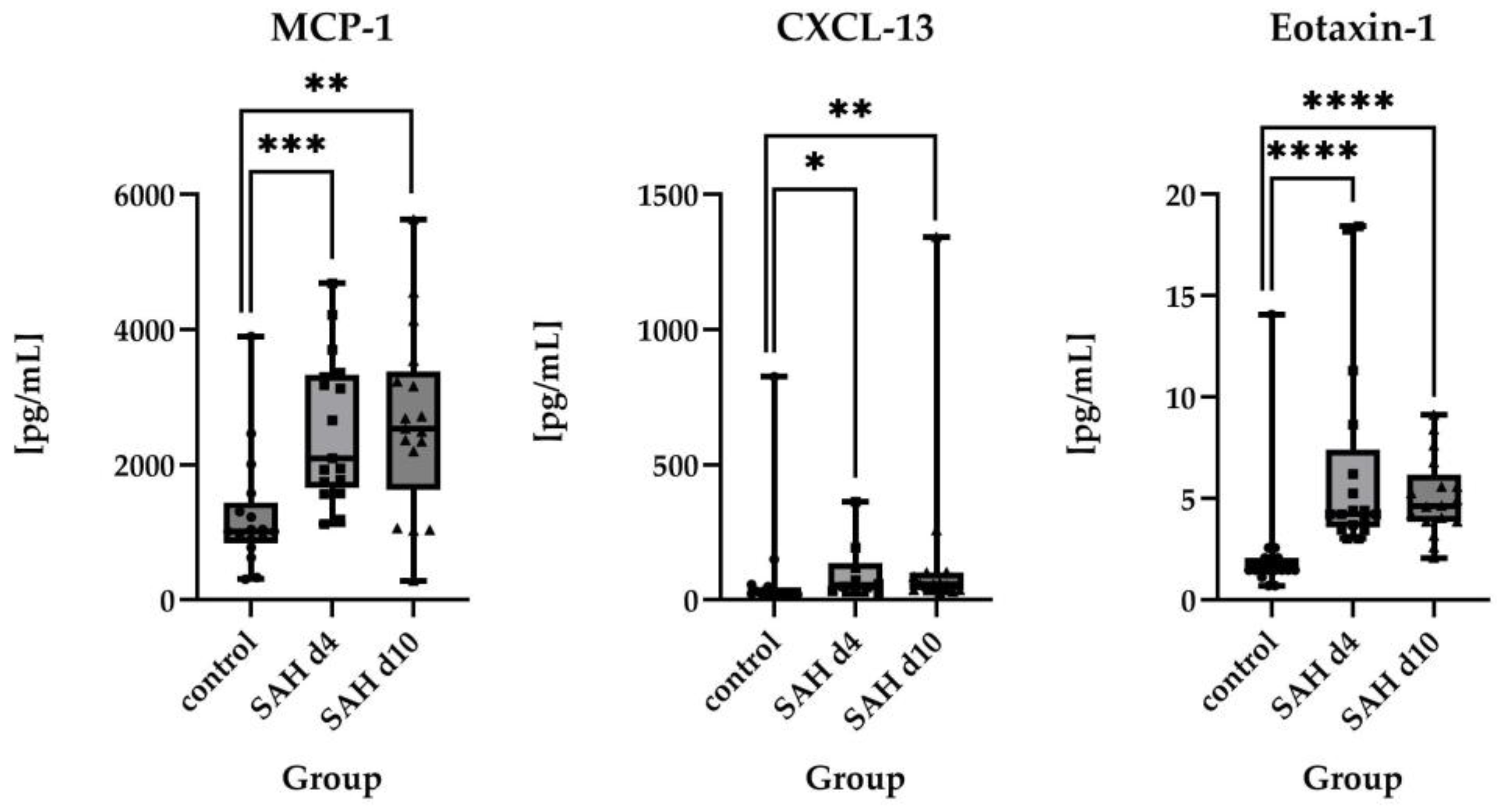
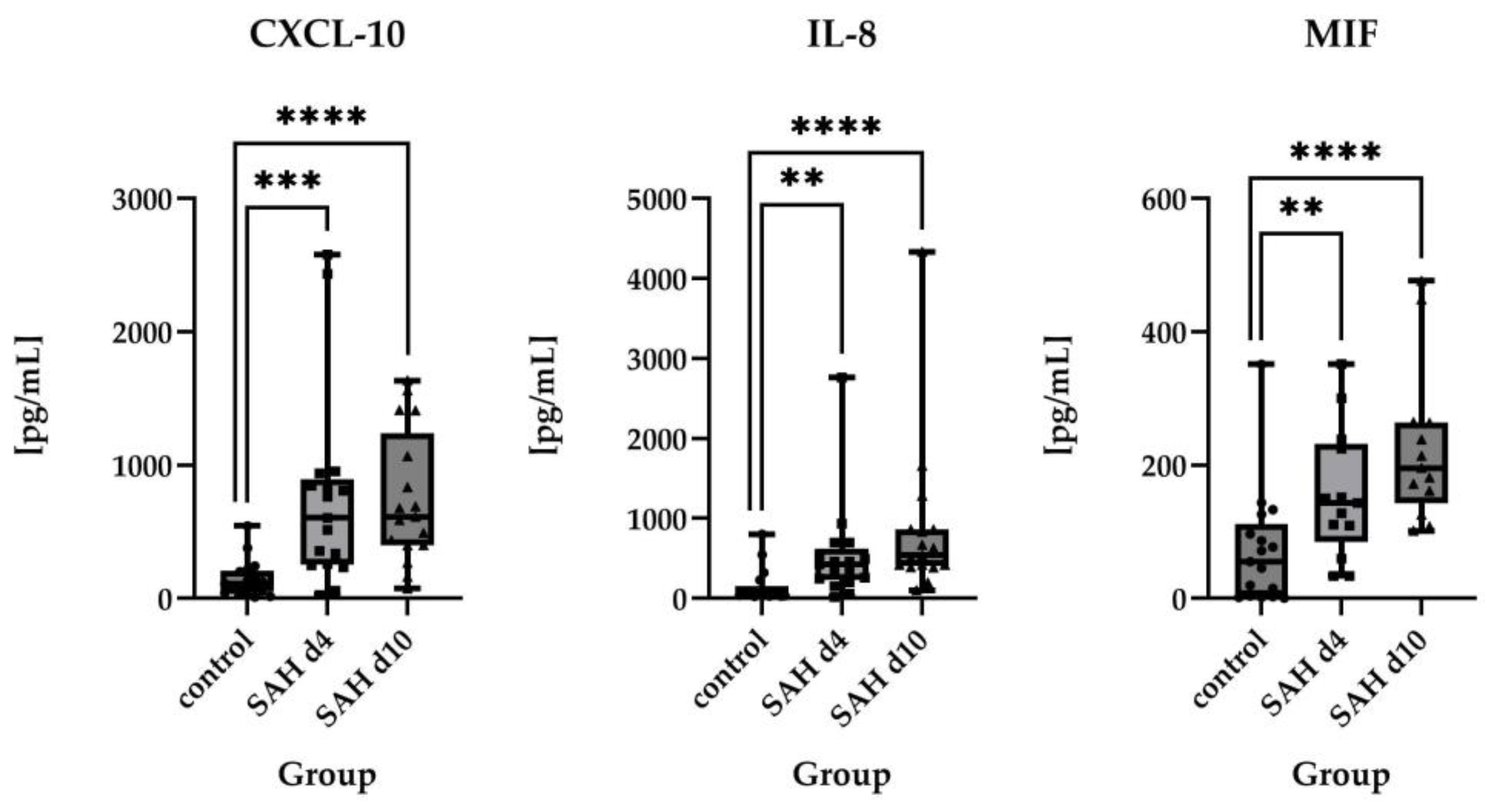
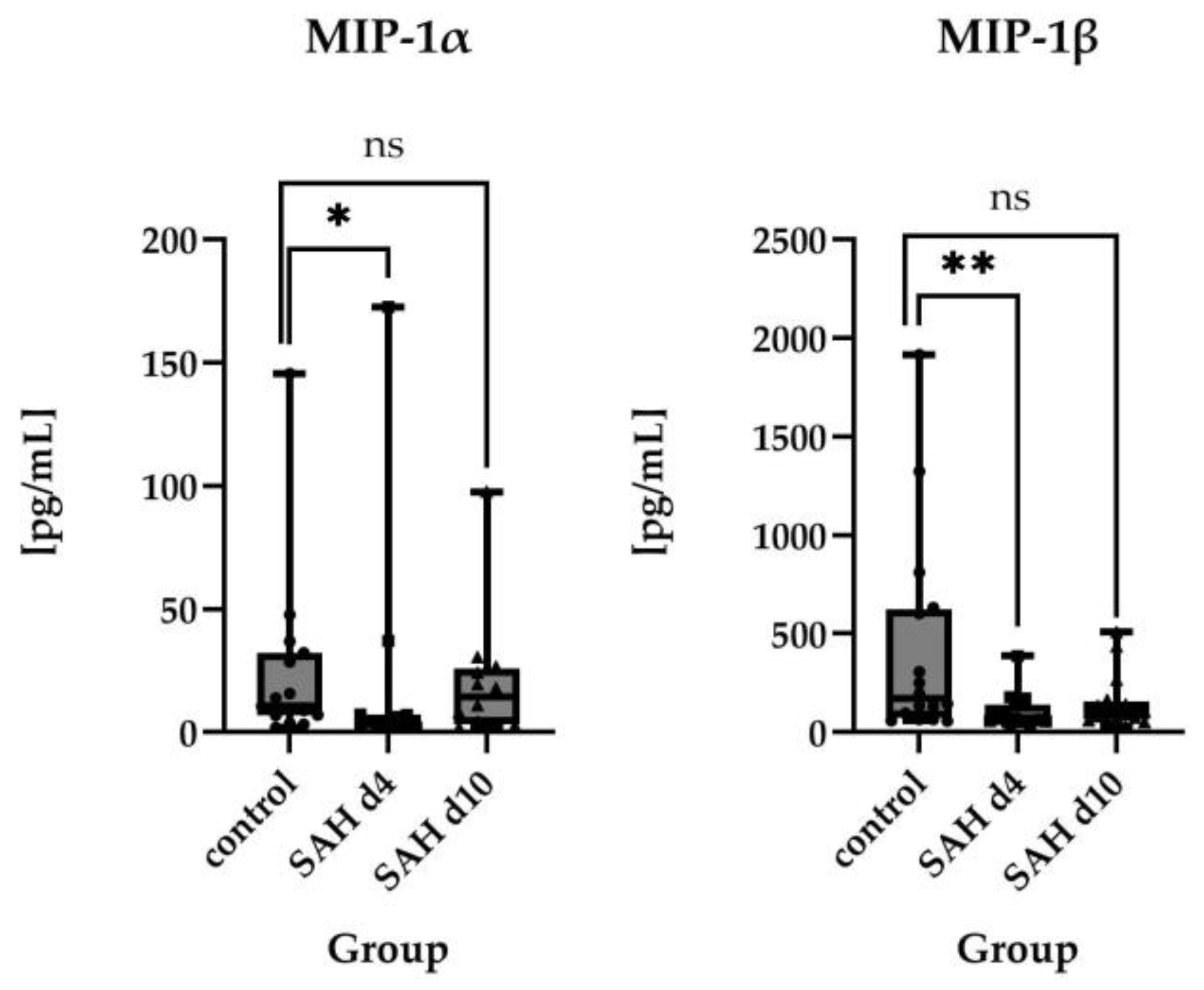
2.2.2. Direct Comparisons of Absolute Protein Levels
2.3. Time Course of Inflammatory Proteins in CSF After SAH
2.4. Subgroup Analysis
3. Discussion
3.1. Inflammatory Protein Profiles and Their Temporal Pattern in CSF of SAH Patients vs. Healthy Controls
3.2. Limitations of the Study
3.3. Therapeutic Implications
4. Materials and Methods
4.1. Inclusion and Exclusion Criteria
4.2. Clinical Management
4.3. Sample Collection and Processing
4.4. Laboratory Analysis
4.5. Choice of Proteins Included in the Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aneurysmal subarachnoid hemorrhage | SAH |
| Early brain injury | EBI |
| Delayed cerebral ischemia | DCI |
| Cerebrospinal fluid | CSF |
| Central nervous system | CNS |
| World Federation of Neurosurgical Societies score | WFNS |
| Glasgow Outcome Scale | GOS |
| Interleukin-1-receptor antagonist | IL-1-RA |
| Interleukin-6 | IL-6 |
| Interleukin-17a | IL-17a |
| Growth-regulated protein alpha | GRO-α |
| C-X-C motif chemokine ligand 1 | CXCL1 |
| Interleukin-1α | IL-1α |
| Interleukin-10 | IL-10 |
| Interleukin-21 | IL-21 |
| Interleukin-9 | IL-9 |
| Monocyte Chemoattractant Protein-1 | MCP-1 |
| C–C motif chemokine ligand 2 | CCL2 |
| C-X-C motif ligand 13 | CXCL-13 |
| Eotaxin-1 (CC-chemokine ligand 11) | CCL11 |
| IFN-gamma-inducible protein 10 | IP-10 |
| C-X-C motif chemokine ligand 10 | CXCL-10 |
| Interleukin-8 | IL-8 |
| C-X-C motif chemokine ligand 8 | CXCL8 |
| Macrophage migration inhibitory factor | MIF |
| Macrophage inflammatory protein-1α | MIP-1α |
| C–C motif chemokine ligand 3 | CCL3 |
| Macrophage inflammatory protein-1β | MIP-1β |
| C–C motif chemokine ligand 4 | CCL4 |
| Stromal cell-derived factor 1α | SDF-1 α |
| C-X-C motif chemokine ligand 12 | CXCL12 |
| Interleukin-2 | IL-2) |
| Triggering receptor expressed on myeloid cells-2 | TREM-2 |
| Traumatic brain injury | TBI |
| External ventricular drain | EVD |
| Computed tomography | CT |
| Intensive care unit | ICU |
| Glasgow Coma Scale | GCS |
| Transcranial doppler | TCD |
| Confidence intervals | CI |
References
- Hoh, B.L.; Ko, N.U.; Amin-Hanjani, S.; Chou, S.H.-Y.; Cruz-Flores, S.; Dangayach, N.S.; Derdeyn, C.P.; Du, R.; Hänggi, D.; Hetts, S.W.; et al. 2023 Guideline for the Management of Patients With Aneurysmal Subarachnoid Hemorrhage: A Guideline From the American Heart Association/American Stroke Association. Stroke 2023, 54, e314–e370. [Google Scholar] [CrossRef] [PubMed]
- Thilak, S.; Brown, P.; Whitehouse, T.; Gautam, N.; Lawrence, E.; Ahmed, Z.; Veenith, T. Diagnosis and Management of Subarachnoid Haemorrhage. Nat. Commun. 2024, 15, 1850. [Google Scholar] [CrossRef]
- Geraghty, J.R.; Lara-Angulo, M.N.; Spegar, M.; Reeh, J.; Testai, F.D. Severe Cognitive Impairment in Aneurysmal Subarachnoid Hemorrhage: Predictors and Relationship to Functional Outcome. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2020, 29, 105027. [Google Scholar] [CrossRef] [PubMed]
- Al-Khindi, T.; Macdonald, R.L.; Schweizer, T.A. Cognitive and Functional Outcome After Aneurysmal Subarachnoid Hemorrhage. Stroke 2010, 41, e519–e536. [Google Scholar] [CrossRef]
- Corraini, P.; Henderson, V.W.; Ording, A.G.; Pedersen, L.; Horváth-Puhó, E.; Toft Sørensen, H. Long-Term Risk of Dementia Among Survivors of Ischemic or Hemorrhagic Stroke. Stroke 2017, 48, 180–186. [Google Scholar] [CrossRef]
- Rost, N.S.; Brodtmann, A.; Pase, M.P.; van Veluw, S.J.; Biffi, A.; Duering, M.; Hinman, J.D.; Dichgans, M. Post-Stroke Cognitive Impairment and Dementia. Circ. Res. 2022, 130, 1252–1271. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Schweizer, T.A. Spontaneous Subarachnoid Haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Vergouwen, M.D.; Vermeulen, M.; van Gijn, J.; Rinkel, G.J.; Wijdicks, E.F.; Muizelaar, J.P.; Mendelow, A.D.; Juvela, S.; Yonas, H.; Terbrugge, K.G.; et al. Definition of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage as an Outcome Event in Clinical Trials and Observational Studies: Proposal of a Multidisciplinary Research Group. Stroke 2010, 41, 2391–2395. [Google Scholar] [CrossRef]
- Dodd, W.S.; Laurent, D.; Dumont, A.S.; Hasan, D.M.; Jabbour, P.M.; Starke, R.M.; Hosaka, K.; Polifka, A.J.; Hoh, B.L.; Chalouhi, N. Pathophysiology of Delayed Cerebral Ischemia After Subarachnoid Hemorrhage: A Review. J. Am. Heart Assoc. 2021, 10, e021845. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. Int. J. Mol. Sci. 2016, 17, 497. [Google Scholar] [CrossRef]
- Miller, B.A.; Turan, N.; Chau, M.; Pradilla, G. Inflammation, Vasospasm, and Brain Injury after Subarachnoid Hemorrhage. BioMed Res. Int. 2014, 2014, e384342. [Google Scholar] [CrossRef] [PubMed]
- Muroi, C.; Hugelshofer, M.; Seule, M.; Tastan, I.; Fujioka, M.; Mishima, K.; Keller, E. Correlation among Systemic Inflammatory Parameter, Occurrence of Delayed Neurological Deficits, and Outcome after Aneurysmal Subarachnoid Hemorrhage. Neurosurgery 2013, 72, 367–375. [Google Scholar] [CrossRef]
- Vlachogiannis, P.; Hillered, L.; Enblad, P.; Ronne-Engström, E. Elevated Levels of Several Chemokines in the Cerebrospinal Fluid of Patients with Subarachnoid Hemorrhage Are Associated with Worse Clinical Outcome. PLoS ONE 2023, 18, e0282424. [Google Scholar] [CrossRef]
- Devlin, P.; Ishrat, T.; Stanfill, A.G. A Systematic Review of Inflammatory Cytokine Changes Following Aneurysmal Subarachnoid Hemorrhage in Animal Models and Humans. Transl. Stroke Res. 2022, 13, 881–897. [Google Scholar] [CrossRef]
- Liu, Q.; Li, K.; He, H.; Miao, Z.; Cui, H.; Wu, J.; Ding, S.; Wen, Z.; Chen, J.; Lu, X.; et al. The Markers and Risk Stratification Model of Intracranial Aneurysm Instability in a Large Chinese Cohort. Sci. Bull. 2023, 68, 1162–1175. [Google Scholar] [CrossRef]
- Vlachogiannis, P.; Hillered, L.; Enblad, P.; Ronne-Engström, E. Temporal Patterns of Inflammation-Related Proteins Measured in the Cerebrospinal Fluid of Patients with Aneurysmal Sub-arachnoid Hemorrhage Using Multiplex Proximity Extension Assay Technology. PLoS ONE 2022, 17, e0263460. [Google Scholar] [CrossRef]
- Al-Tamimi, Y.Z.; Bhargava, D.; Orsi, N.M.; Teraifi, A.; Cummings, M.; Ekbote, U.V.; Quinn, A.C.; Homer-Vanniasinkam, S.; Ross, S. Compartmentalisation of the Inflammatory Response Following Aneurysmal Subarachnoid Haemorrhage. Cytokine 2019, 123, 154778. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, X.; Lu, J.; Shi, H.; Huang, L.; Shao, A.; Zhang, A.; Liu, Y.; Ren, R.; Lenahan, C.; et al. Inhibition of Caspase-1-Mediated Inflammasome Activation Reduced Blood Coagulation in Cerebrospinal Fluid after Sub-arachnoid Haemorrhage. eBioMedicine 2022, 76, 103843. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, Y.; Chen, L.; Wang, J.; Zhang, J.; Zhang, H.; Tian, S.; Zhang, A.; Zhang, J.; Zhang, J.H.; et al. Cerebrospinal Fluid Markers of Neuroinflammation and Coagulation in Severe Cerebral Edema and Chronic Hydrocephalus after Subarachnoid Hemorrhage: A Prospective Study. J. Neuroinflamm. 2024, 21, 237. [Google Scholar] [CrossRef]
- Ohashi, S.N.; DeLong, J.H.; Kozberg, M.G.; Mazur-Hart, D.J.; van Veluw, S.J.; Alkayed, N.J.; Sansing, L.H. Role of Inflammatory Processes in Hemorrhagic Stroke. Stroke 2023, 54, 605–619. [Google Scholar] [CrossRef]
- Chai, C.-Z.; Ho, U.-C.; Kuo, L.-T. Systemic Inflammation after Aneurysmal Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2023, 24, 10943. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef] [PubMed]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; Khoury, J.E. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2021, 91, 498. [Google Scholar] [CrossRef]
- Simon, M.; Grote, A. Interleukin 6 and Aneurysmal Subarachnoid Hemorrhage. A Narrative Review. Int. J. Mol. Sci. 2021, 22, 4133. [Google Scholar] [CrossRef]
- Vlachogiannis, P.; Hillered, L.; Khalil, F.; Enblad, P.; Ronne-Engström, E. Interleukin-6 Levels in Cerebrospinal Fluid and Plasma in Patients with Severe Spontaneous Subarachnoid Hemorrhage. World Neurosurg. 2019, 122, e612–e618. [Google Scholar] [CrossRef]
- Niwa, A.; Osuka, K.; Nakura, T.; Matsuo, N.; Watabe, T.; Takayasu, M. Interleukin-6, MCP-1, IP-10, and MIG Are Sequentially Expressed in Cerebrospinal Fluid after Subarachnoid Hemorrhage. J. Neuroinflamm. 2016, 13, 217. [Google Scholar] [CrossRef]
- Sobowale, O.A.; Parry-Jones, A.R.; Smith, C.J.; Tyrrell, P.J.; Rothwell, N.J.; Allan, S.M. Interleukin-1 in Stroke. Stroke 2016, 47, 2160–2167. [Google Scholar] [CrossRef]
- Gruber, A.; Rössler, K.; Graninger, W.; Donner, A.; Illievich, U.M.; Czech, T. Ventricular Cerebrospinal Fluid and Serum Concentrations of sTNFR-I, IL-1ra, and IL-6 After Aneurysmal Subarachnoid Hemorrhage. J. Neurosurg. Anesthesiol. 2000, 12, 297. [Google Scholar] [CrossRef]
- Moraes, L.; Trias, N.; Brugnini, A.; Grille, P.; Lens, D.; Biestro, A.; Grille, S. TH17/Treg Imbalance and IL-17A Increase after Severe Aneurysmal Subarachnoid Hemorrhage. J. Neuroimmunol. 2020, 346, 577310. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Z.; Zhou, M.; Ling, S.; Zhou, X.; Yuan, B.; Zhao, X.; Qi, M.; Han, Y.; Qin, F.; et al. TREM2 Alleviates Subarachnoid Hemorrhage-Induced Brain Injury through Attenuating Neuroinflammation and Programmed Cell Death in Vivo and in Vitro. Front. Biosci.-Landmark 2024, 29, 50. [Google Scholar] [CrossRef] [PubMed]
- Wach, J.; Vychopen, M.; Güresir, A.; Güresir, E. Anti-Inflammatory Drug Therapy in Aneurysmal Subarachnoid Hemorrhage: A Systematic Review and Meta-Analysis of Prospective Randomized and Placebo-Controlled Trials. J. Clin. Med. 2023, 12, 4165. [Google Scholar] [CrossRef] [PubMed]
- Güresir, E.; Lampmann, T.; Bele, S.; Czabanka, M.; Czorlich, P.; Gempt, J.; Goldbrunner, R.; Hurth, H.; Hermann, E.; Jabbarli, R.; et al. Fight INflammation to Improve Outcome after Aneurysmal Subarachnoid HEmorRhage (FINISHER) Trial: Study Protocol for a Randomized Controlled Trial. Int. J. Stroke Off. J. Int. Stroke Soc. 2023, 18, 242–247. [Google Scholar] [CrossRef]
- Hunt, W.E.; Hess, R.M. Surgical Risk as Related to Time of Intervention in the Repair of Intracranial Aneurysms. J. Neurosurg. 1968, 28, 14–20. [Google Scholar] [CrossRef]
- Teasdale, G.M.; Drake, C.G.; Hunt, W.; Kassell, N.; Sano, K.; Pertuiset, B.; De Villiers, J.C. A Universal Subarachnoid Hemorrhage Scale: Report of a Committee of the World Federation of Neurosurgical Societies. J. Neurol. Neurosurg. Psychiatry 1988, 51, 1457. [Google Scholar] [CrossRef]
- Frontera, J.A.; Claassen, J.; Schmidt, J.M.; Wartenberg, K.E.; Temes, R.; Connolly, E.S.; MacDonald, R.L.; Mayer, S.A. Prediction of Symptomatic Vasospasm after Subarachnoid Hemorrhage: The Modified Fisher Scale. Neurosurgery 2006, 59, 21–27. [Google Scholar] [CrossRef]
- Jennett, B.; Bond, M. Assessment of Outcome after Severe Brain Damage. Lancet Lond. Engl. 1975, 1, 480–484. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Migliorini, P.; Bossù, P. Cause or Consequence? The Role of IL-1 Family Cytokines and Receptors in Neuroinflammatory and Neurodegenerative Diseases. Front. Immunol. 2023, 14. [Google Scholar] [CrossRef]
- Burmeister, A.R.; Marriott, I. The Interleukin-10 Family of Cytokines and Their Role in the CNS. Front. Cell Neurosci. 2018, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Hauptmann, J.; Regen, T. The Role of IL-17 in CNS Diseases. Acta Neuropathol. 2015, 129, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Lykhopiy, V.; Malviya, V.; Humblet-Baron, S.; Schlenner, S.M. IL-2 Immunotherapy for Targeting Regulatory T Cells in Autoimmunity. Genes. Immun. 2023, 24, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Yshii, L.; Pasciuto, E.; Bielefeld, P.; Mascali, L.; Lemaitre, P.; Marino, M.; Dooley, J.; Kouser, L.; Verschoren, S.; Lagou, V.; et al. Astrocyte-Targeted Gene Delivery of Interleukin 2 Specifically Increases Brain-Resident Regulatory T Cell Numbers and Protects against Pathological Neuroinflammation. Nat. Immunol. 2022, 23, 878–891. [Google Scholar] [CrossRef]
- Shbeer, A.M.; Ahmed Robadi, I. The Role of Interleukin-21 in Autoimmune Diseases: Mechanisms, Therapeutic Implications, and Future Directions. Cytokine 2024, 173, 156437. [Google Scholar] [CrossRef]
- Rothaug, M.; Becker-Pauly, C.; Rose-John, S. The Role of Interleukin-6 Signaling in Nervous Tissue. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1218–1227. [Google Scholar] [CrossRef]
- Pajulas, A.; Zhang, J.; Kaplan, M.H. The World According to IL-9. J. Immunol. 2023, 211, 7–14. [Google Scholar] [CrossRef]
- Li, H.; Nourbakhsh, B.; Ciric, B.; Zhang, G.-X.; Rostami, A. Neutralization of IL-9 Ameliorates Experimental Autoimmune Encephalomyelitis by Decreasing the Effector T Cell Population. J. Immunol. 2010, 185, 4095–4100. [Google Scholar] [CrossRef]
- Ding, X.; Cao, F.; Cui, L.; Ciric, B.; Zhang, G.-X.; Rostami, A. IL-9 Signaling Affects Central Nervous System Resident Cells during Inflammatory Stimuli. Exp. Mol. Pathol. 2015, 99, 570–574. [Google Scholar] [CrossRef]
- Painter, M.M.; Atagi, Y.; Liu, C.-C.; Rademakers, R.; Xu, H.; Fryer, J.D.; Bu, G. TREM2 in CNS Homeostasis and Neurodegenerative Disease. Mol. Neurodegener. 2015, 10, 43. [Google Scholar] [CrossRef]
- Zhong, J.; Xing, X.; Gao, Y.; Pei, L.; Lu, C.; Sun, H.; Lai, Y.; Du, K.; Xiao, F.; Yang, Y.; et al. Distinct Roles of TREM2 in Central Nervous System Cancers and Peripheral Cancers. Cancer Cell 2024, 42, 968–984.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Z.; Ye, N.; Zhen, X. Macrophage Migration Inhibitory Factor (MIF) in CNS Diseases: Functional Regulation and Potential Therapeutic Indication. Fundam. Res. 2024, 4, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, M.; Abdi, Z.; Murdjeva, M.; Macedo, D.; Maes, A.; Maes, M. CCL-11 or Eotaxin-1: An Immune Marker for Ageing and Accelerated Ageing in Neuro-Psychiatric Disorders. Pharmaceuticals 2020, 13, 230. [Google Scholar] [CrossRef]
- Korbecki, J.; Gąssowska-Dobrowolska, M.; Wójcik, J.; Szatkowska, I.; Barczak, K.; Chlubek, M.; Baranowska-Bosiacka, I. The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System. Int. J. Mol. Sci. 2022, 23, 4205. [Google Scholar] [CrossRef]
- Irani, D.N. Regulated Production of CXCL13 within the Central Nervous System. J. Clin. Cell Immunol. 2016, 7, 460. [Google Scholar] [CrossRef]
- Righi, D.; Manco, C.; Pardini, M.; Stufano, A.; Schino, V.; Pelagotti, V.; Massa, F.; Stefano, N.D.; Plantone, D. Investigating Interleukin-8 in Alzheimer’s Disease: A Comprehensive Review. J. Alzheimer’s Dis. 2024, 2024, 13872877241298973. [Google Scholar] [CrossRef]
- Matsushima, K.; Yang, D.; Oppenheim, J.J. Interleukin-8: An Evolving Chemokine. Cytokine 2022, 153, 155828. [Google Scholar] [CrossRef]
- Michlmayr, D.; McKimmie, C.S. Role of CXCL10 in Central Nervous System Inflammation. Int. J. Interferon Cytokine Mediat. Res. 2014, 6, 1–18. [Google Scholar] [CrossRef]
- Conductier, G.; Blondeau, N.; Guyon, A.; Nahon, J.-L.; Rovère, C. The Role of Monocyte Chemoattractant Protein MCP1/CCL2 in Neuroinflammatory Diseases. J. Neuroimmunol. 2010, 224, 93–100. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Bhavsar, I.; Miller, C.S.; Al-Sabbagh, M. Macrophage Inflammatory Protein-1 Alpha (MIP-1 Alpha)/CCL3: As a Biomarker. General. Methods Biomark. Res. Their Appl. 2015, 223–249. [Google Scholar] [CrossRef]
- Puthenparampil, M.; Stropparo, E.; Zywicki, S.; Bovis, F.; Cazzola, C.; Federle, L.; Grassivaro, F.; Rinaldi, F.; Perini, P.; Sormani, M.P.; et al. Wide Cytokine Analysis in Cerebrospinal Fluid at Diagnosis Identified CCL-3 as a Possible Prognostic Factor for Multiple Sclerosis. Front. Immunol. 2020, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Szczuciński, A.; Losy, J. Chemokines and Chemokine Receptors in Multiple Sclerosis. Potential Targets for New Therapies. Acta Neurol. Scand. 2007, 115, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Menten, P.; Wuyts, A.; Van Damme, J. Macrophage Inflammatory Protein-1. Cytokine Growth Factor. Rev. 2002, 13, 455–481. [Google Scholar] [CrossRef]
- Li, M.; Ransohoff, R.M. Multiple Roles of Chemokine CXCL12 in the Central Nervous System: A Migration from Immunology to Neurobiology. Prog. Neurobiol. 2008, 84, 116–131. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | SAH (n = 17) |
|---|---|
| Female sex | 14 |
| Median age | 56 (95% CI 49–67 years) |
| Clinical condition at admission | |
| Hunt and Hess 1–3 | 10 |
| Hunt and Hess 4–5 | 7 |
| WFNS I–III | 5 |
| WFNS IV–V | 12 |
| Radiological features (modified Fisher) | |
| Intracerebral or intraventricular hemorrhage | 13 |
| Thick subarachnoid blood clot | 4 |
| Aneurysm location | |
| Anterior circulation | 13 |
| Posterior circulation | 4 |
| Treatment | |
| Clipping | 4 |
| Endovascular | 13 |
| Complications | |
| DCI | 7 |
| Shunt dependency | 6 |
| Outcome at time of discharge | |
| Unfavorable (GOS 1–3) | 7 |
| Favorable (GOS 4–5) | 10 |
| Outcome after six months | |
| Unfavorable (GOS 1–3) | 8 |
| Favorable (GOS 4–5) | 5 |
| Lost to follow-up | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seyfried, K.S.; Kremer, B.; Conzen-Dilger, C.; Veldeman, M.; Bertram, U.; Blume, C.; Mueller, C.A.; Bi, T.; Jütten, K.; Clusmann, H.; et al. Mapping Inflammatory Markers in Cerebrospinal Fluid Following Aneurysmal Subarachnoid Hemorrhage: An Age- and Sex-Matched Analysis. Int. J. Mol. Sci. 2025, 26, 1302. https://doi.org/10.3390/ijms26031302
Seyfried KS, Kremer B, Conzen-Dilger C, Veldeman M, Bertram U, Blume C, Mueller CA, Bi T, Jütten K, Clusmann H, et al. Mapping Inflammatory Markers in Cerebrospinal Fluid Following Aneurysmal Subarachnoid Hemorrhage: An Age- and Sex-Matched Analysis. International Journal of Molecular Sciences. 2025; 26(3):1302. https://doi.org/10.3390/ijms26031302
Chicago/Turabian StyleSeyfried, Katharina Sophie, Benedikt Kremer, Catharina Conzen-Dilger, Michael Veldeman, Ulf Bertram, Christian Blume, Christian Andreas Mueller, Tianshu Bi, Kerstin Jütten, Hans Clusmann, and et al. 2025. "Mapping Inflammatory Markers in Cerebrospinal Fluid Following Aneurysmal Subarachnoid Hemorrhage: An Age- and Sex-Matched Analysis" International Journal of Molecular Sciences 26, no. 3: 1302. https://doi.org/10.3390/ijms26031302
APA StyleSeyfried, K. S., Kremer, B., Conzen-Dilger, C., Veldeman, M., Bertram, U., Blume, C., Mueller, C. A., Bi, T., Jütten, K., Clusmann, H., & Höllig, A. (2025). Mapping Inflammatory Markers in Cerebrospinal Fluid Following Aneurysmal Subarachnoid Hemorrhage: An Age- and Sex-Matched Analysis. International Journal of Molecular Sciences, 26(3), 1302. https://doi.org/10.3390/ijms26031302