
A Joint Analysis of RNA-DNA and DNA-DNA Interactomes Reveals Their Strong Association

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Contacts of Many RNAs Are Associated with the Chromatin Structure
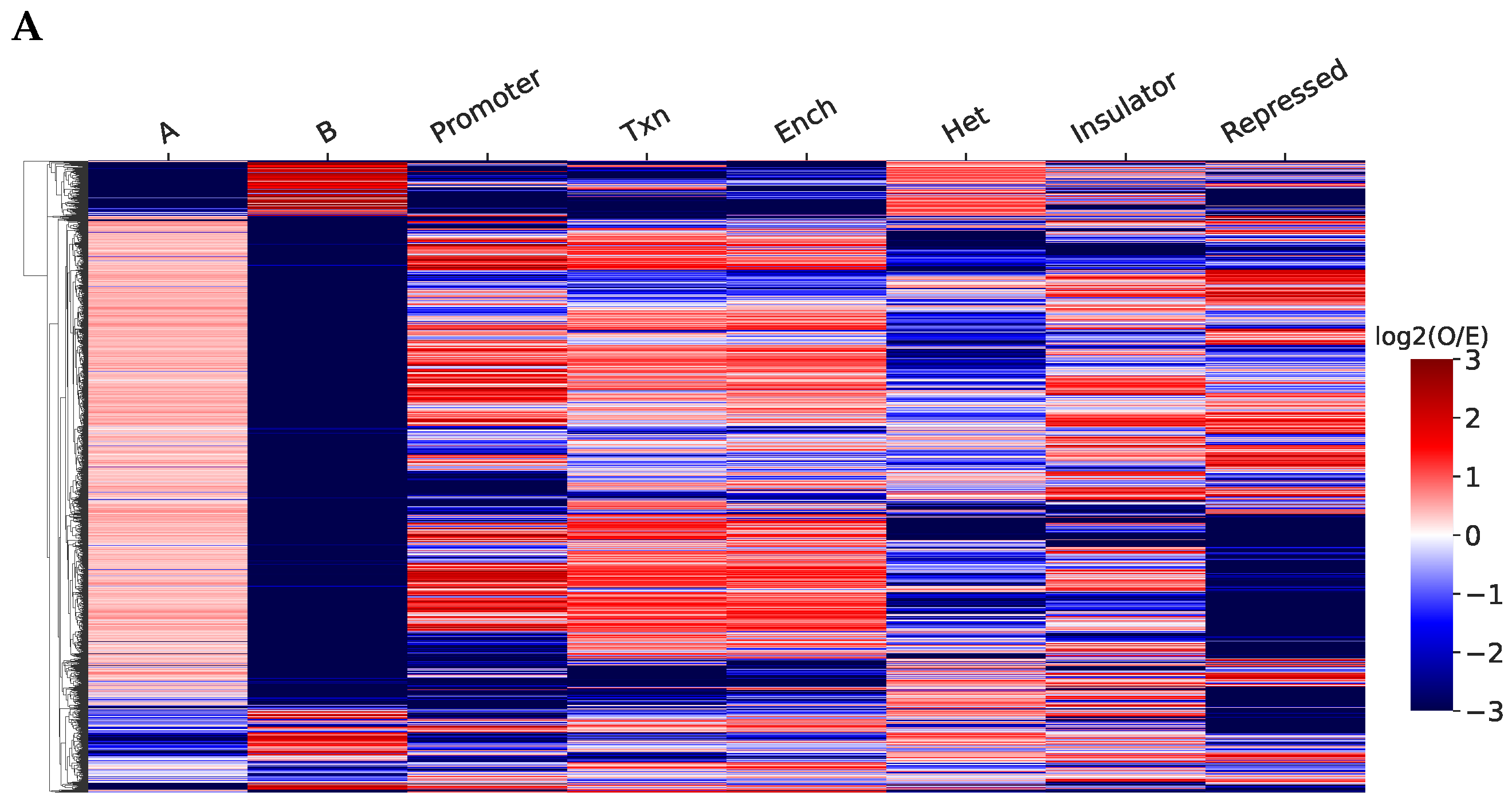
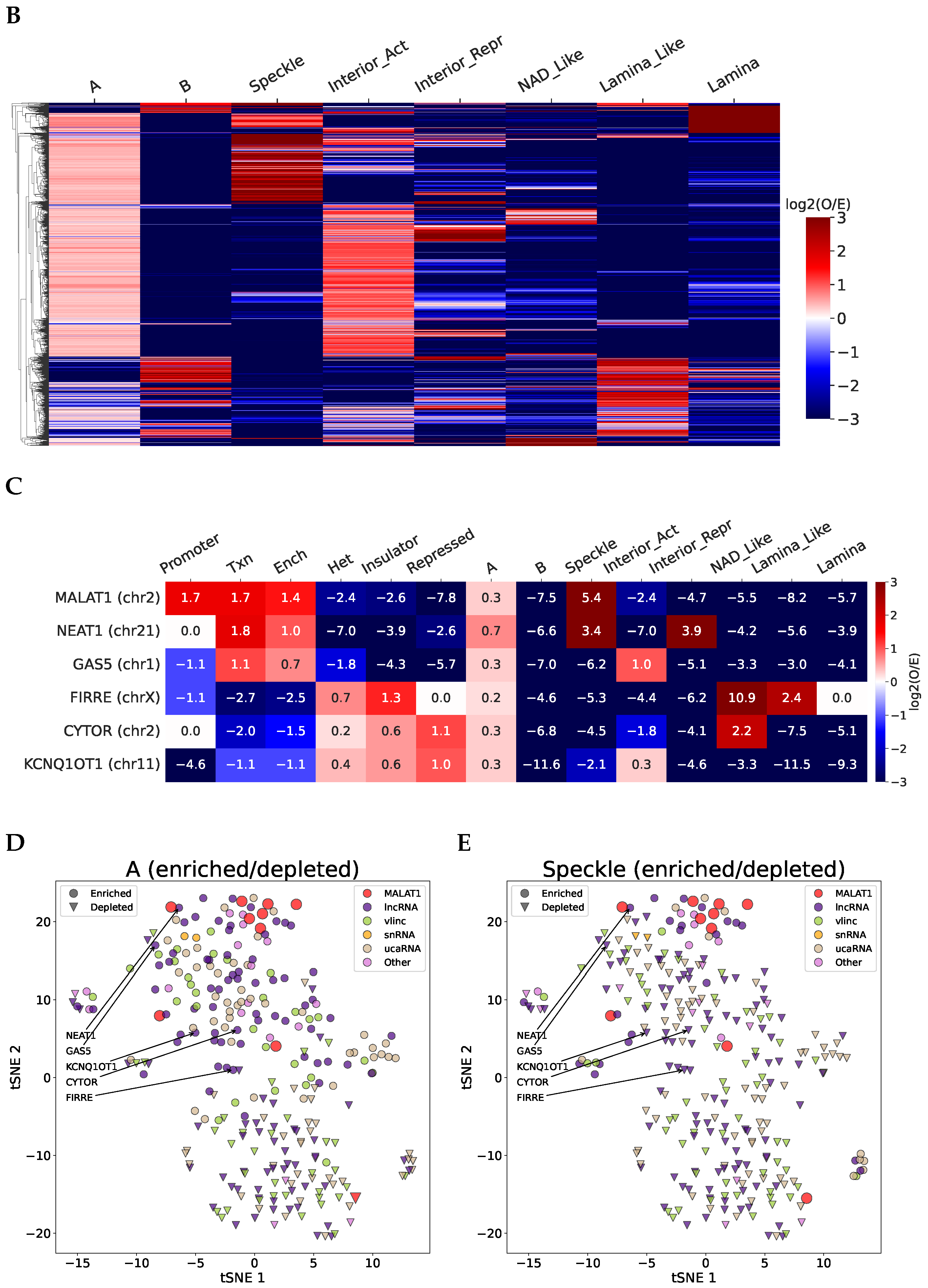
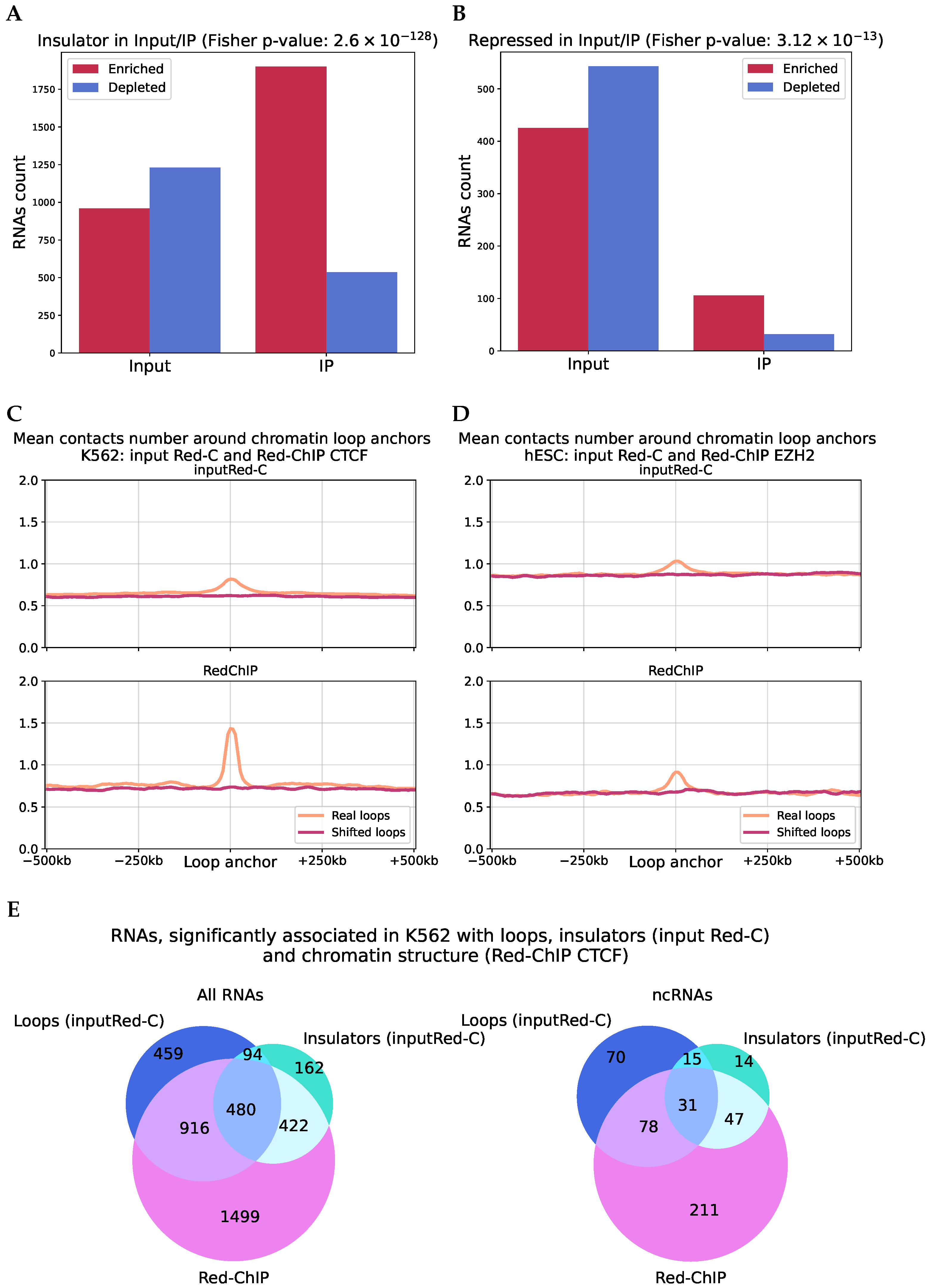
2.2. Structurally Associated Contacts Are Connected with Functional Chromatin Annotations
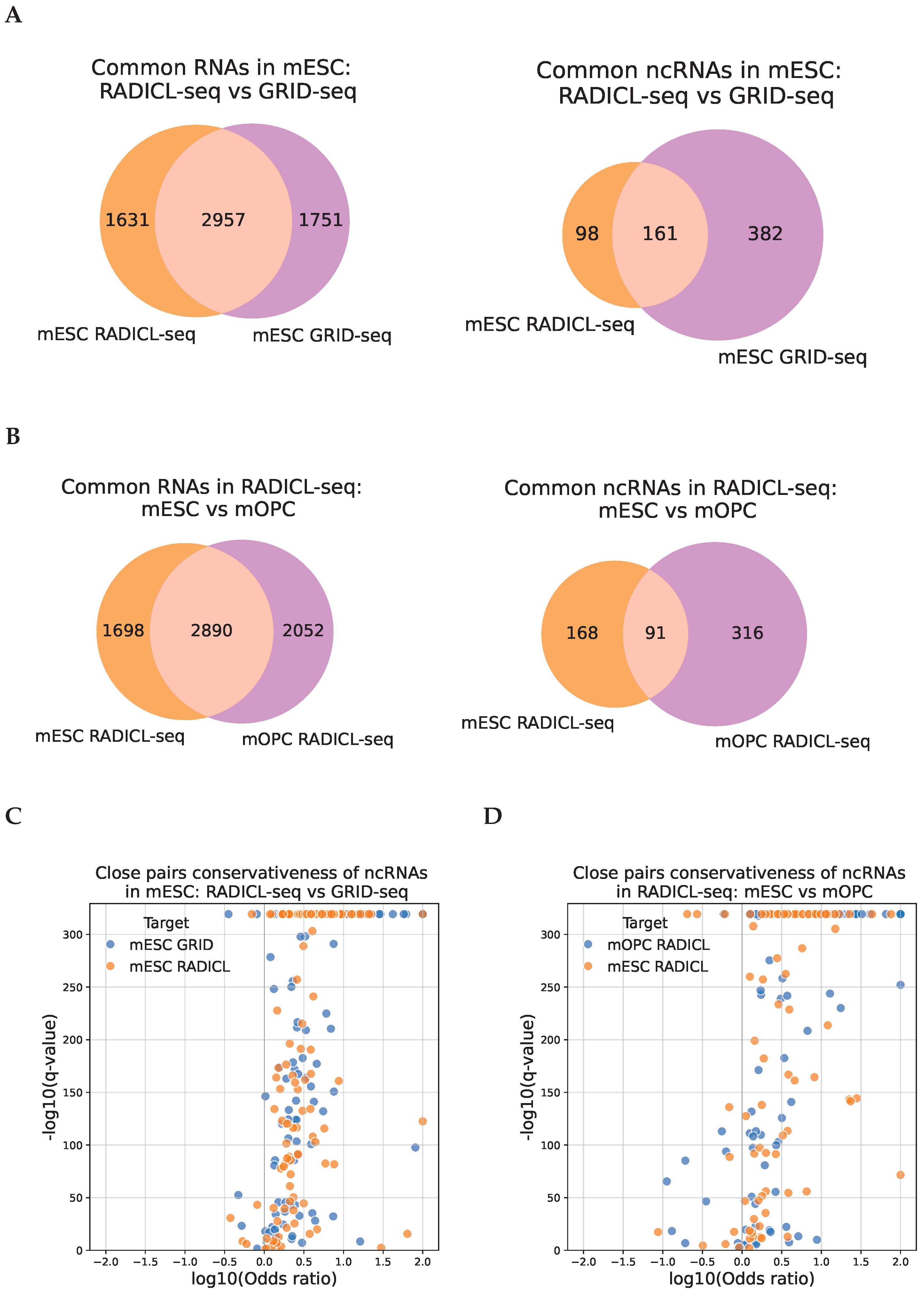
2.3. The Association of the RNA-DNA Interactome and Chromatin Structure Is Conserved Across Experiments
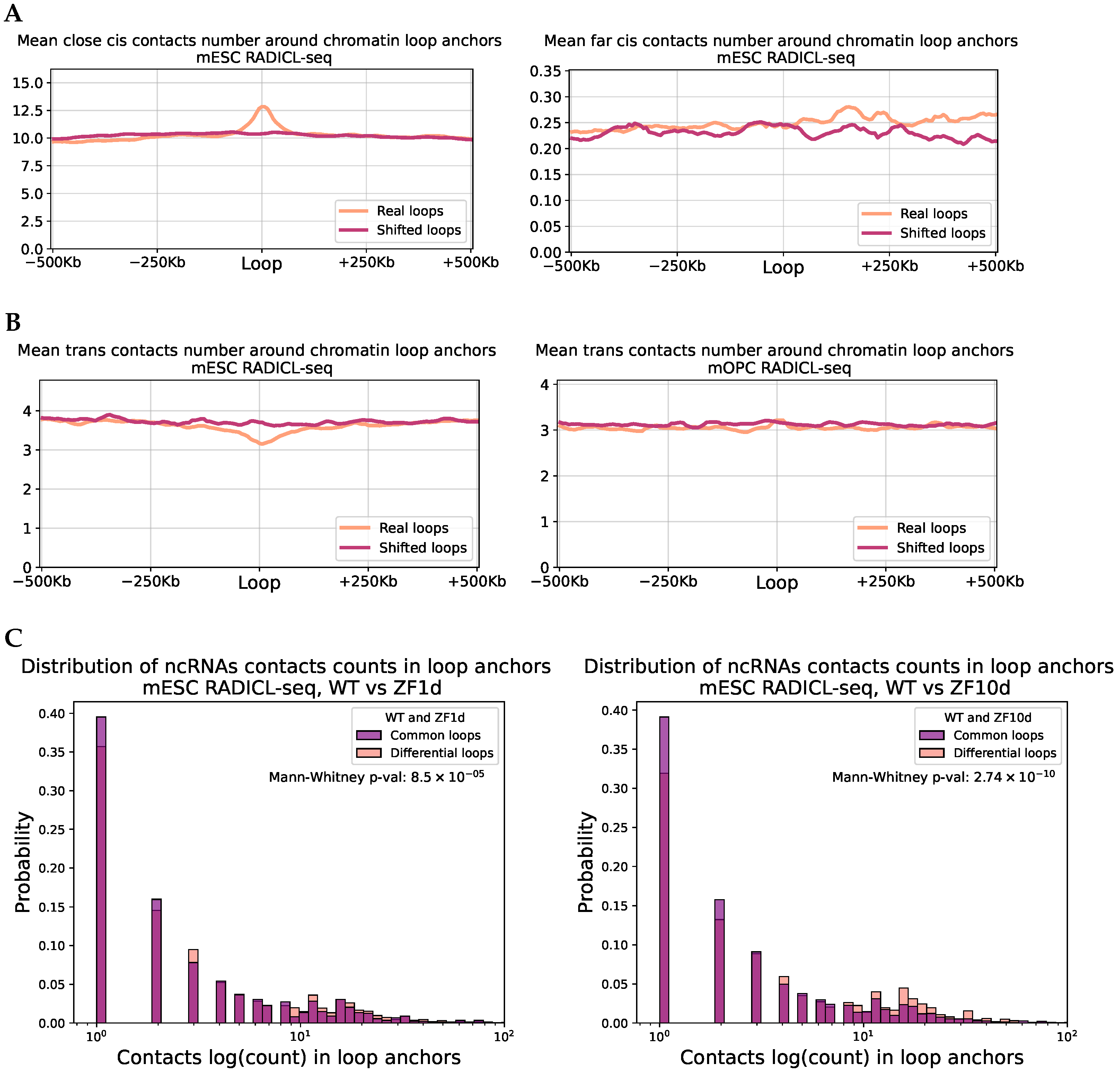
2.4. RNA Contacts Are Associated with Chromatin Loops
2.5. RNA-DNA Interactome and TADs Association
2.6. Analysis of RNA–DNA–Protein Interactions
3. Discussion
4. Materials and Methods
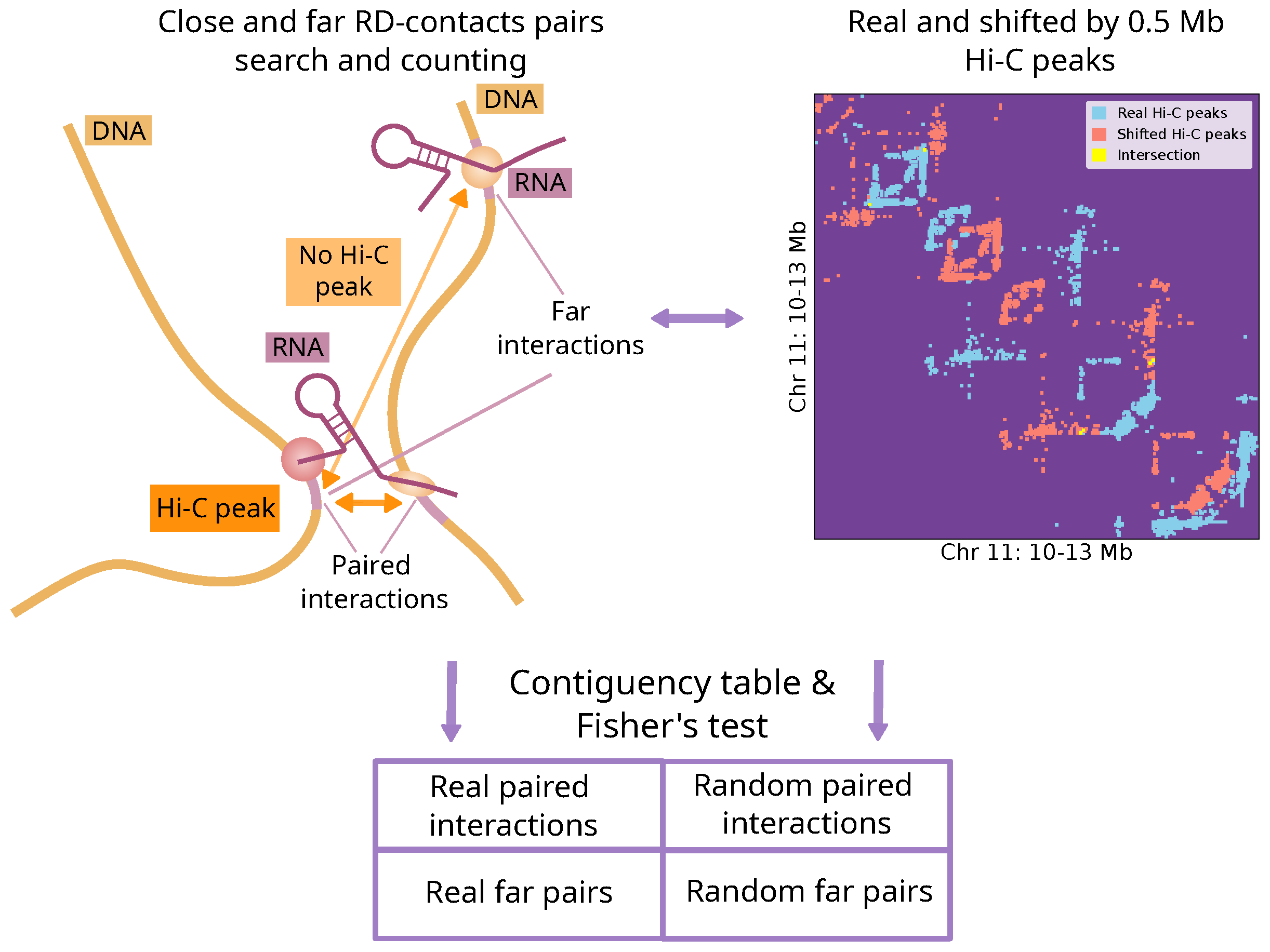
4.1. Procedure for Identifying Associations Between RNA-DNA and DNA-DNA Interactions
4.2. Data Sources
4.3. Data Preprocessing
4.4. Analysis of Functional Chromatin Annotations
4.5. Comparative Analysis of Protocols
4.6. Chromatin Loops
4.7. TADs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RD | RNA-DNA |
| lncRNA | Long non-coding RNA (human) |
| vlincRNA | Very long intergenic non-coding RNA (human) |
| lincRNA | Long intergenic non-coding RNA (mouse) |
| ucaRNA | Unannotated chromatin-associated RNA |
| snRNA | Small nuclear RNA |
| snoRNA | Small nucleolar RNA |
| TAD | Topologically associated domain |
| mESC | Mouse embryonic stem cells |
| mOPC | Mouse oligodendrocytes progenitor cells |
| hESC | Human embryonic stem cells |
| OutInDR | Contacts densities ratio outside versus inside the TAD |
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fu, X.D. Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat. Rev. Genet. 2019, 20, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hamblin, M.H.; Yin, K.J. The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 2017, 14, 1705–1714. [Google Scholar] [CrossRef]
- Fox, A.H.; Nakagawa, S.; Hirose, T.; Bond, C.S. Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 2018, 43, 124–135. [Google Scholar] [CrossRef]
- Razin, S.V.; Gavrilov, A.A. Non-coding RNAs in chromatin folding and nuclear organization. Cell. Mol. Life Sci. 2021, 78, 5489–5504. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Engreitz, J.; Lander, E.S.; Guttman, M. RNA Antisense Purification (RAP) for Mapping RNA Interactions with Chromatin. In Nuclear Bodies and Noncoding RNAs; Humana Press: New York, NY, USA, 2015; pp. 183–197. [Google Scholar] [CrossRef]
- Chu, C.; Quinn, J.; Chang, H.Y. Chromatin Isolation by RNA Purification (ChIRP). J. Vis. Exp. 2012, e3912. [Google Scholar] [CrossRef]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Zharikova, A.A.; Galitsyna, A.A.; Luzhin, A.V.; Rubanova, N.M.; Golov, A.K.; Petrova, N.V.; Logacheva, M.D.; Kantidze, O.L.; Ulianov, S.V.; et al. Studying RNA–DNA interactome by Red-C identifies noncoding RNAs associated with various chromatin types and reveals transcription dynamics. Nucleic Acids Res. 2020, 48, 6699–6714. [Google Scholar] [CrossRef]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.T.; Li, H.; Fu, X.D. GRID-seq reveals the global RNA–chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.F.; et al. RADICL-seq identifies general and cell type–specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 2020, 11, 1018. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yan, Z.; Nguyen, T.C.; Bouman Chen, Z.; Chien, S.; Zhong, S. Mapping RNA–chromatin interactions by sequencing with iMARGI. Nat. Protoc. 2019, 14, 3243–3272. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Rao, S.; Huntley, M.; Durand, N.; Stamenova, E.; Bochkov, I.; Robinson, J.; Sanborn, A.; Machol, I.; Omer, A.; Lander, E.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Calandrelli, R.; Wen, X.; Charles Richard, J.L.; Luo, Z.; Nguyen, T.C.; Chen, C.J.; Qi, Z.; Xue, S.; Chen, W.; Yan, Z.; et al. Genome-wide analysis of the interplay between chromatin-associated RNA and 3D genome organization in human cells. Nat. Commun. 2023, 14, 6519. [Google Scholar] [CrossRef]
- Kuang, S.; Pollard, K.S. Exploring the roles of RNAs in chromatin architecture using deep learning. Nat. Commun. 2024, 15, 6373. [Google Scholar] [CrossRef]
- Mumbach, M.R.; Granja, J.M.; Flynn, R.A.; Roake, C.M.; Satpathy, A.T.; Rubin, A.J.; Qi, Y.; Jiang, Z.; Shams, S.; Louie, B.H.; et al. HiChIRP reveals RNA-associated chromosome conformation. Nat. Methods 2019, 16, 489–492. [Google Scholar] [CrossRef]
- Mylarshchikov, D.E.; Nikolskaya, A.I.; Bogomaz, O.D.; Zharikova, A.A.; Mironov, A.A. BaRDIC: Robust peak calling for RNA–DNA interaction data. NAR Genom. Bioinform. 2024, 6, lqae054. [Google Scholar] [CrossRef]
- Quinodoz, S.A.; Jachowicz, J.W.; Bhat, P.; Ollikainen, N.; Banerjee, A.K.; Goronzy, I.N.; Blanco, M.R.; Chovanec, P.; Chow, A.; Markaki, Y.; et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 2021, 184, 5775–5790.e30. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, A.; Hua, N.; Boninsegna, L.; Zhan, Y.; Polles, G.; Gong, K.; Hao, S.; Li, W.; Zhou, X.J.; Alber, F. Evaluating the role of the nuclear microenvironment in gene function by population-based modeling. Nat. Struct. Mol. Biol. 2023, 30, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, E.M.; Dekker, J. Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem. Sci. 2020, 45, 385–396. [Google Scholar] [CrossRef]
- Sexton, C.E.; Han, M.V. Genome wide clustering on integrated chromatin states and Micro-C contacts reveals chromatin interaction signatures. bioRxiv 2023. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Zhang, R.; van Schaik, T.; Zhang, L.; Sasaki, T.; Peric-Hupkes, D.; Chen, Y.; Gilbert, D.M.; van Steensel, B.; et al. SPIN reveals genome-wide landscape of nuclear compartmentalization. Genome Biol. 2021, 22, 36. [Google Scholar] [CrossRef]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Lin, G.; Wu, T.; Gao, X.; He, Z.; Nong, W. Research Progress of Long Non-Coding RNA GAS5 in Malignant Tumors. Front. Oncol. 2022, 12, 846497. [Google Scholar] [CrossRef]
- Thakur, N.; Tiwari, V.K.; Thomassin, H.; Pandey, R.R.; Kanduri, M.; Göndör, A.; Grange, T.; Ohlsson, R.; Kanduri, C. An Antisense RNA Regulates the Bidirectional Silencing Property of the Kcnq1 Imprinting Control Region. Mol. Cell. Biol. 2004, 24, 7855–7862. [Google Scholar] [CrossRef]
- Yang, F.; Deng, X.; Ma, W.; Berletch, J.B.; Rabaia, N.; Wei, G.; Moore, J.M.; Filippova, G.N.; Xu, J.; Liu, Y.; et al. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015, 16, 52. [Google Scholar] [CrossRef]
- Wang, X.; Yu, H.; Sun, W.; Kong, J.; Zhang, L.; Tang, J.; Wang, J.; Xu, E.; Lai, M.; Zhang, H. The long non-coding RNA CYTOR drives colorectal cancer progression by interacting with NCL and Sam68. Mol. Cancer 2018, 17, 110. [Google Scholar] [CrossRef]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 Is Dispensable for Mouse Development but Its Transcription Plays a cis-Regulatory Role in the Adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Saldaña-Meyer, R.; Rodriguez-Hernaez, J.; Escobar, T.; Nishana, M.; Jácome-López, K.; Nora, E.P.; Bruneau, B.G.; Tsirigos, A.; Furlan-Magaril, M.; Skok, J.; et al. RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol. Cell 2019, 76, 412–422.e5. [Google Scholar] [CrossRef]
- Ulianov, S.V.; Khrameeva, E.E.; Gavrilov, A.A.; Flyamer, I.M.; Kos, P.; Mikhaleva, E.A.; Penin, A.A.; Logacheva, M.D.; Imakaev, M.V.; Chertovich, A.; et al. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 2016, 26, 70–84. [Google Scholar] [CrossRef]
- Khani-Habibabadi, F.; Zare, L.; Sahraian, M.A.; Javan, M.; Behmanesh, M. Hotair and Malat1 Long Noncoding RNAs Regulate Bdnf Expression and Oligodendrocyte Precursor Cell Differentiation. Mol. Neurobiol. 2022, 59, 4209–4222. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA Interactions Enable Specific Targeting of Noncoding RNAs to Nascent Pre-mRNAs and Chromatin Sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Sultanov, R.I.; Magnitov, M.D.; Galitsyna, A.A.; Dashinimaev, E.B.; Aiden, E.L.; Razin, S.V. Brief Report: RedChIP identifies noncoding RNAs associated with genomic sites occupied by Polycomb and CTCF proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2116222119. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Nassar, L.R.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.; Lee, B.; et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2023, 51, D1188–D1195. [Google Scholar] [CrossRef]
- Laurent, G., St.; Shtokalo, D.; Dong, B.; Tackett, M.R.; Fan, X.; Lazorthes, S.; Nicolas, E.; Sang, N.; Triche, T.J.; McCaffrey, T.A.; et al. VlincRNAs controlled by retroviral elements are a hallmark of pluripotency and cancer. Genome Biol. 2013, 14, R73. [Google Scholar] [CrossRef]
- Ryabykh, G.K.; Kuznetsov, S.V.; Korostelev, Y.D.; Sigorskikh, A.I.; Zharikova, A.A.; Mironov, A.A. RNA-Chrom: A manually curated analytical database of RNA–chromatin interactome. Database 2023, 2023, baad025. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Open2C; Abdennur, N.; Fudenberg, G.; Flyamer, I.M.; Galitsyna, A.A.; Goloborodko, A.; Imakaev, M.; Venev, S. Bioframe: Operations on genomic intervals in Pandas Dataframes. Bioinformatics 2024, 40, btae088. [Google Scholar] [CrossRef]
- Bailey, T.; Krajewski, P.; Ladunga, I.; Lefebvre, C.; Li, Q.; Liu, T.; Madrigal, P.; Taslim, C.; Zhang, J. Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLoS Comput. Biol. 2013, 9, e1003326. [Google Scholar] [CrossRef]
- Abdennur, N.; Mirny, L.A. Cooler: Scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 2020, 36, 311–316. [Google Scholar] [CrossRef]
- Abdennur, N.; Abraham, S.; Fudenberg, G.; Flyamer, I.M.; Galitsyna, A.A.; Goloborodko, A.; Imakaev, M.; Oksuz, B.A.; Venev, S.V. Cooltools: Enabling high-resolution Hi-C analysis in Python. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kaul, A.; Bhattacharyya, S.; Ay, F. Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat. Protoc. 2020, 15, 991–1012. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Beagrie, R.A.; Thieme, C.J.; Annunziatella, C.; Baugher, C.; Zhang, Y.; Schueler, M.; Kukalev, A.; Kempfer, R.; Chiariello, A.M.; Bianco, S.; et al. Multiplex-GAM: Genome-wide identification of chromatin contacts yields insights overlooked by Hi-C. Nat. Methods 2023, 20, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Shi, Y.; Dai, C.; Tjong, H.; Gong, K.; Alber, F.; Zhou, X.J. TopDom: An efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016, 44, e70. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, M.; Tavernari, D.; Oricchio, E.; Ciriello, G. Comparison of computational methods for the identification of topologically associating domains. Genome Biol. 2018, 19, 217. [Google Scholar] [CrossRef]
- Mylarshchikov, D.E.; Mironov, A.A. ortho2align: A sensitive approach for searching for orthologues of novel lncRNAs. BMC Bioinform. 2022, 23, 384. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zvezdin, D.S.; Tyukaev, A.A.; Zharikova, A.A.; Mironov, A.A. A Joint Analysis of RNA-DNA and DNA-DNA Interactomes Reveals Their Strong Association. Int. J. Mol. Sci. 2025, 26, 1137. https://doi.org/10.3390/ijms26031137
Zvezdin DS, Tyukaev AA, Zharikova AA, Mironov AA. A Joint Analysis of RNA-DNA and DNA-DNA Interactomes Reveals Their Strong Association. International Journal of Molecular Sciences. 2025; 26(3):1137. https://doi.org/10.3390/ijms26031137
Chicago/Turabian StyleZvezdin, Dmitry S., Artyom A. Tyukaev, Anastasia A. Zharikova, and Andrey A. Mironov. 2025. "A Joint Analysis of RNA-DNA and DNA-DNA Interactomes Reveals Their Strong Association" International Journal of Molecular Sciences 26, no. 3: 1137. https://doi.org/10.3390/ijms26031137
APA StyleZvezdin, D. S., Tyukaev, A. A., Zharikova, A. A., & Mironov, A. A. (2025). A Joint Analysis of RNA-DNA and DNA-DNA Interactomes Reveals Their Strong Association. International Journal of Molecular Sciences, 26(3), 1137. https://doi.org/10.3390/ijms26031137