
mRNA Transcript Variants Expressed in Mammalian Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
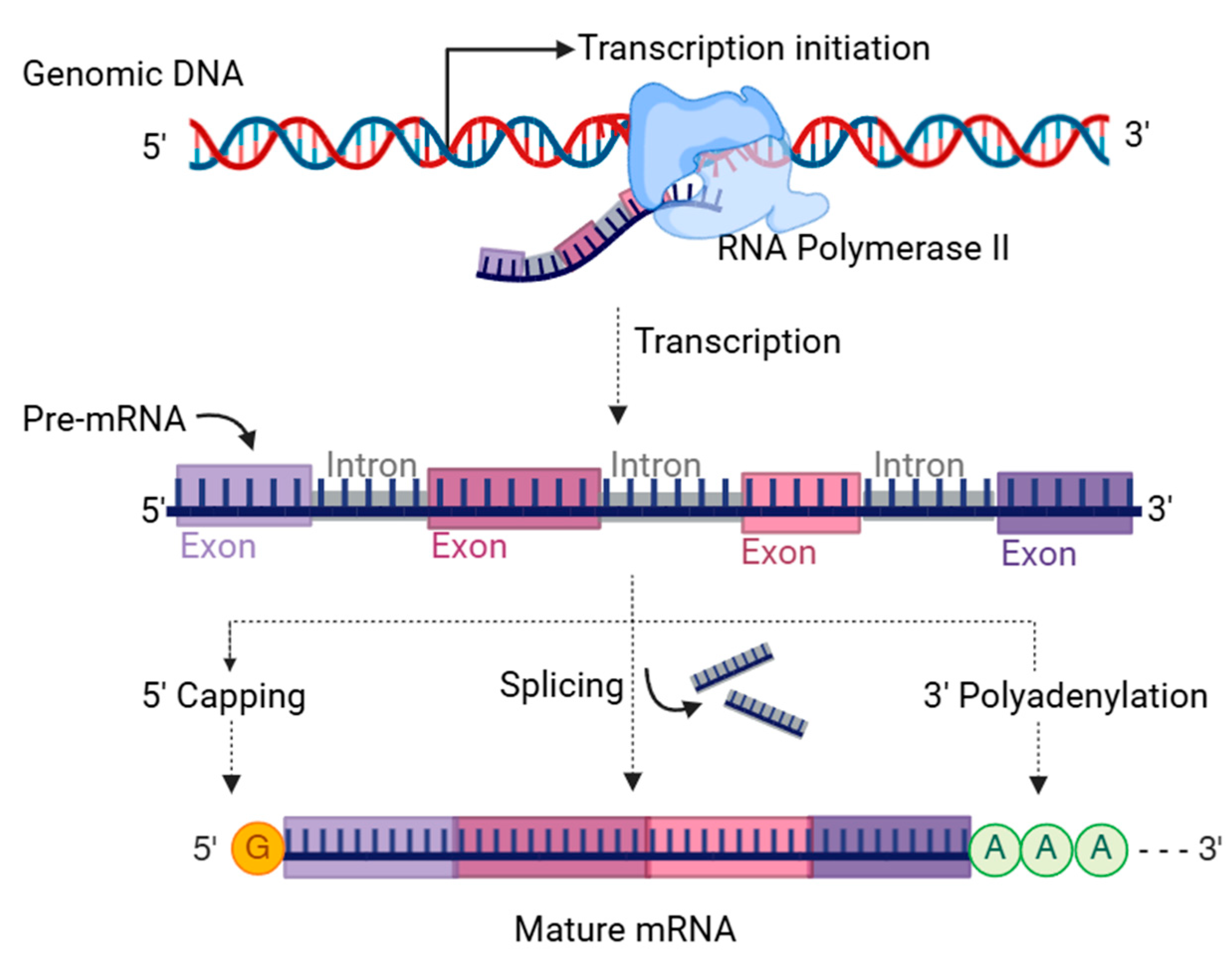
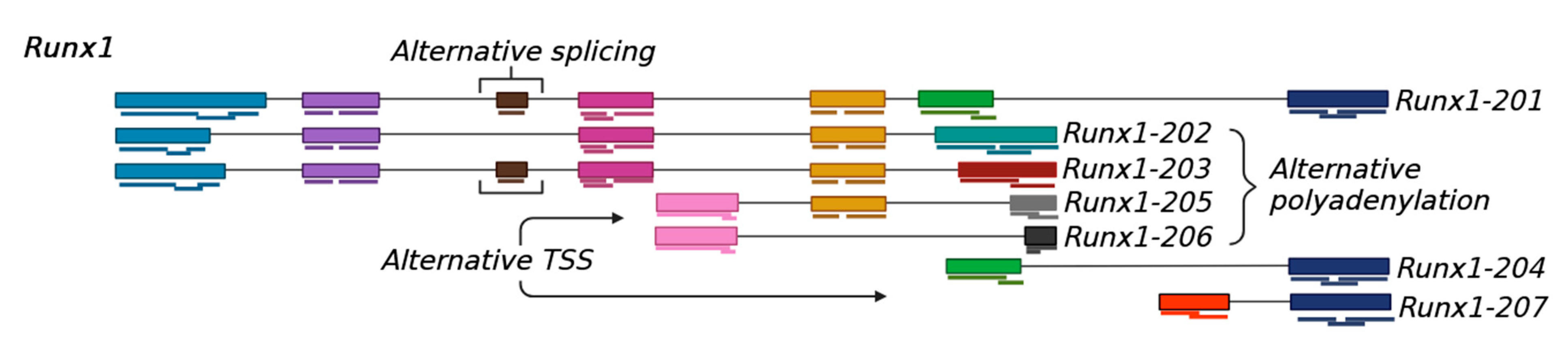
2. Formation of Transcript Variants
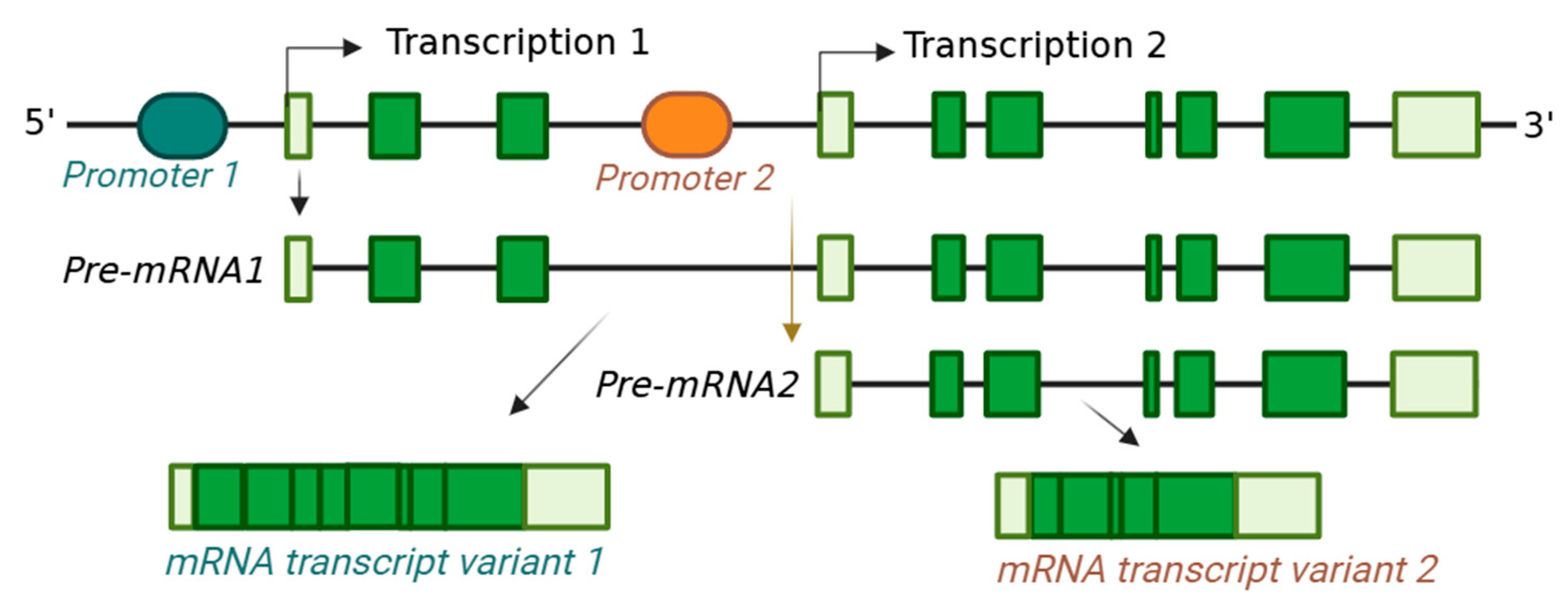
2.1. Transcription from Alternative Start Sites
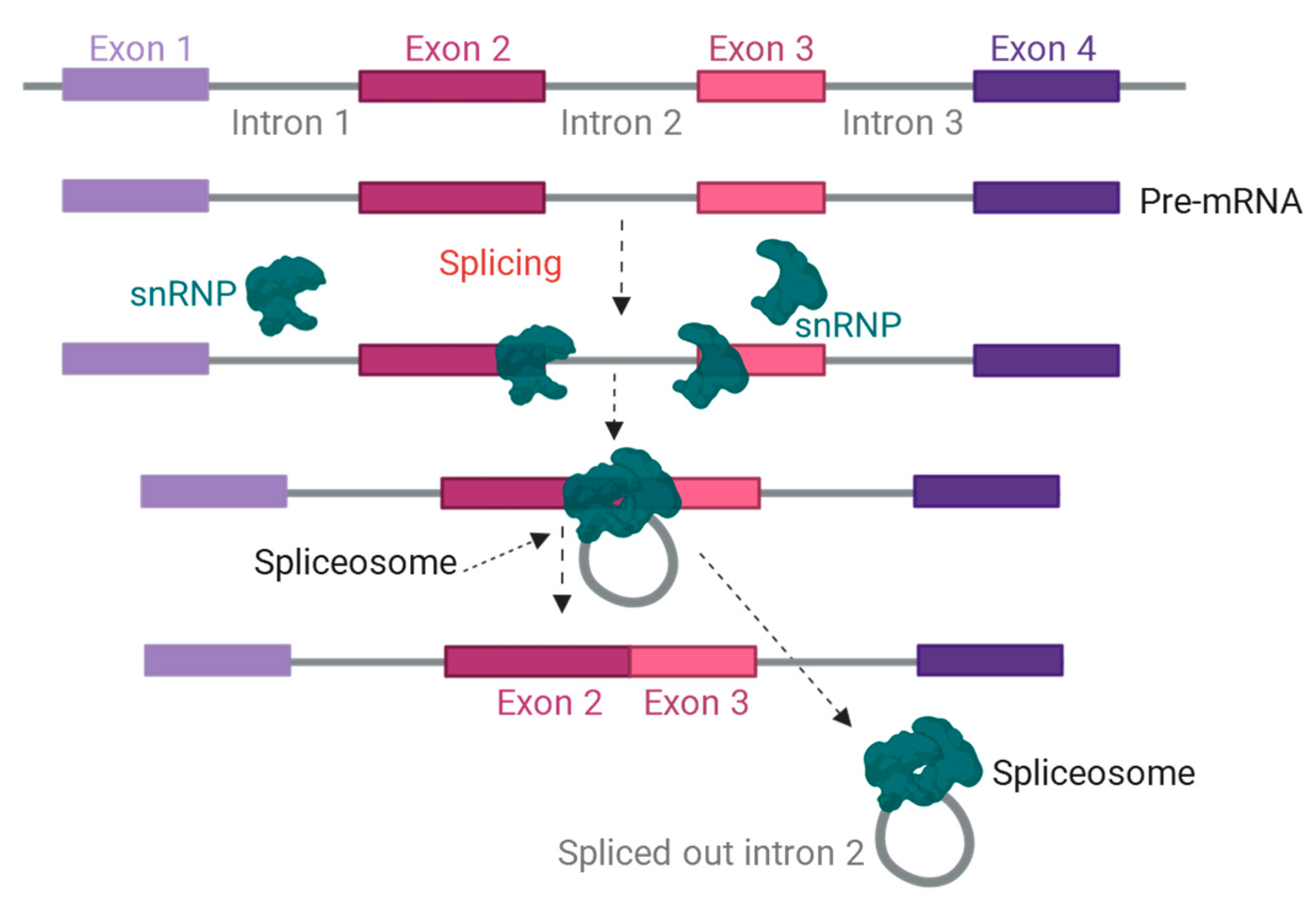
2.2. Alternative Splicing of Pre-mRNA
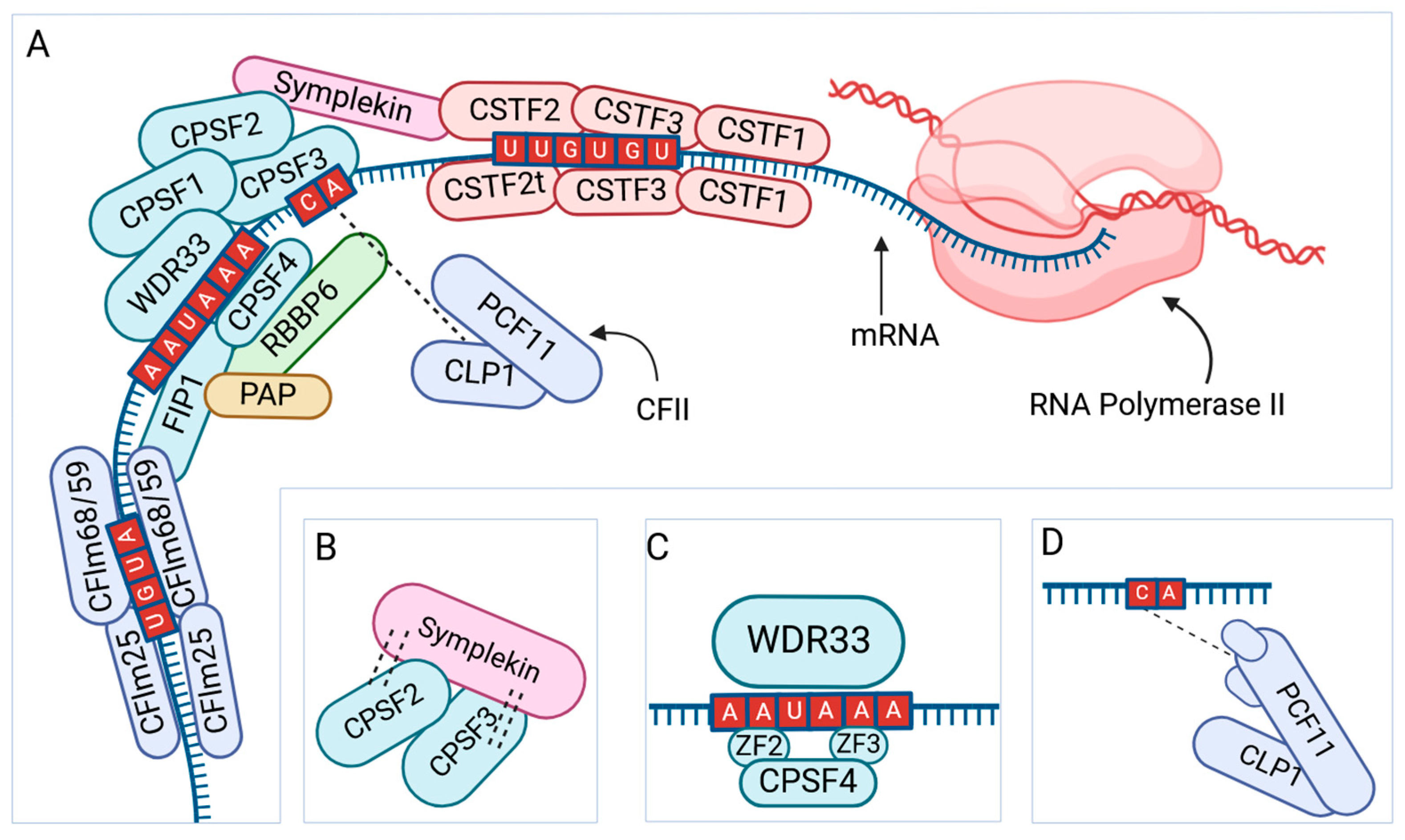
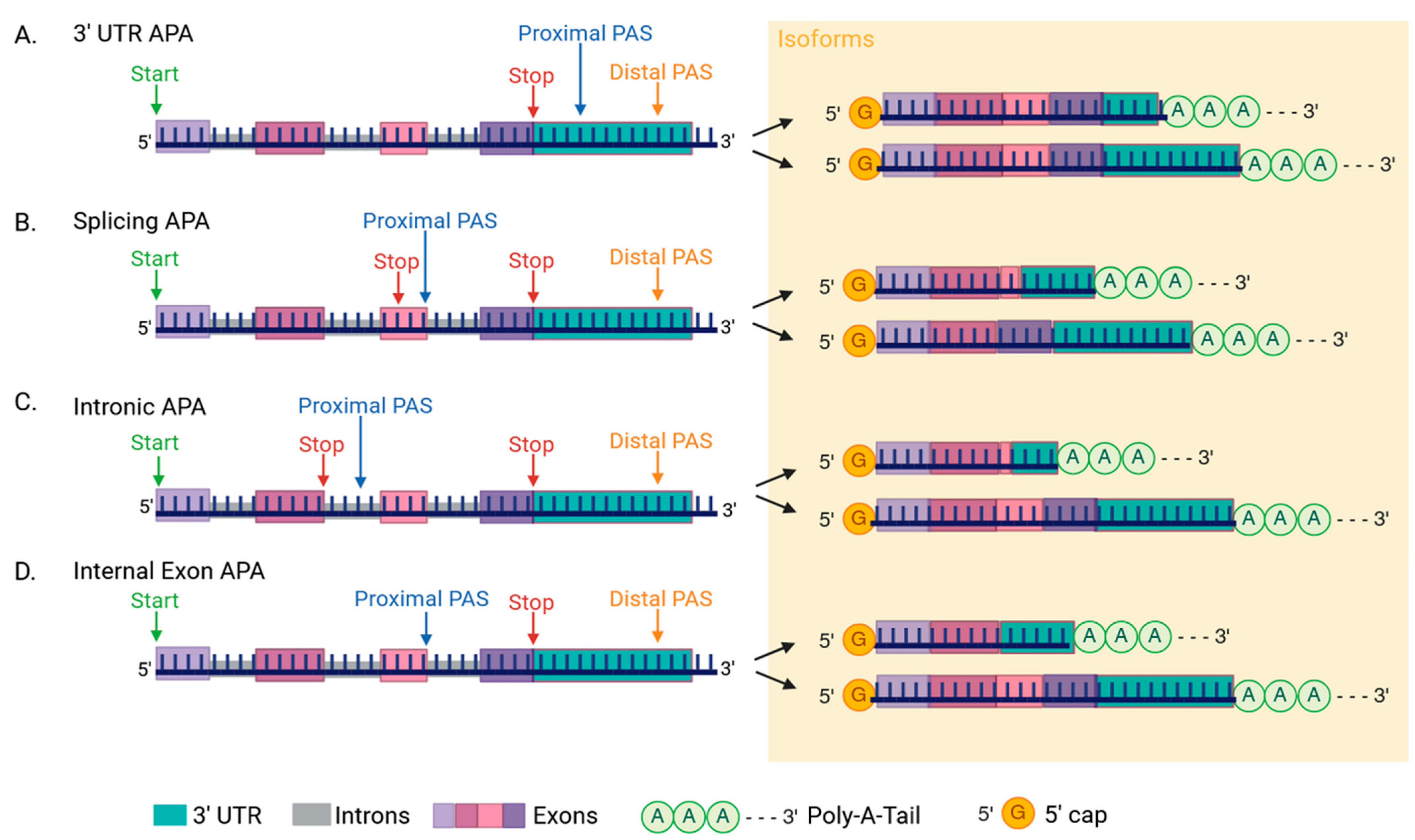
2.3. Alternative Polyadenylation of mRNA
2.4. RNA Editing
2.5. Genetic Mutations
3. Regulation of Transcript Variant Formation
3.1. Regulatory Steps
3.2. Regulation by RNA Modifications
4. Role of the mRNA Transcript Variants
4.1. Coding Proteins with Different Domains
4.2. Non-Coding RNAs
4.3. Decay of mRNAs
5. Transcript Variants in Physiology and Pathological Conditions
5.1. Transcript Variants in Physiological Conditions
5.2. Transcript Variants in Pathological Conditions
5.3. Translational Relevance of Studying mRNA Transcript Variants
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, J.; Hyman, L.; Moore, C. Formation of mRNA 3′ ends in eukaryotes: Mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev. 1999, 63, 405–445. [Google Scholar] [CrossRef] [PubMed]
- Ruth, E.B. Replication, Transcription, and Translation. Am. Biol. Teach. 1984, 46, 470–472. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Pimentel, H.; Bray, N.L.; Pachter, L. Gene-level differential analysis at transcript-level resolution. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. BioSystems 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Kochetov, A.V. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays 2008, 30, 683–691. [Google Scholar] [CrossRef]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef]
- Sharma, M.G. RNA Transcription. In Genetics Fundamentals Notes; Springer: Berlin/Heidelberg, Germany, 2022; pp. 491–535. [Google Scholar]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Reyes, A.; Huber, W. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018, 46, 582–592. [Google Scholar] [CrossRef]
- Arzumanian, V.A.; Dolgalev, G.V.; Kurbatov, I.Y.; Kiseleva, O.I.; Poverennaya, E.V. Epitranscriptome: Review of Top 25 Most-Studied RNA Modifications. Int. J. Mol. Sci. 2022, 23, 13851. [Google Scholar] [CrossRef]
- Chanfreau, G.F. Impact of RNA modifications and RNA-modifying enzymes on eukaryotic ribonucleases. Enzymes 2017, 41, 299–329. [Google Scholar] [PubMed]
- Vo, K.; Sharma, Y.; Paul, A.; Mohamadi, R.; Mohamadi, A.; Fields, P.E.; Rumi, M.A.K. Importance of Transcript Variants in Transcriptome Analyses. Cells 2024, 13, 1502. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Wright, C.J.; Smith, C.W.; Jiggins, C.D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022, 23, 697–710. [Google Scholar] [CrossRef]
- Welle, S.; Brooks, A.I.; Delehanty, J.M.; Needler, N.; Thornton, C.A. Gene expression profile of aging in human muscle. Physiol. Genom. 2003, 14, 149–159. [Google Scholar] [CrossRef]
- Albert, F.W.; Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015, 16, 197–212. [Google Scholar] [CrossRef]
- Steri, M.; Idda, M.L.; Whalen, M.B.; Orrù, V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA 2018, 9, e1474. [Google Scholar] [CrossRef]
- Manning, K.S.; Cooper, T.A. The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell Biol. 2017, 18, 102–114. [Google Scholar] [CrossRef]
- Zhang, G.; Pradhan, S. Mammalian epigenetic mechanisms. IUBMB Life 2014, 66, 240–256. [Google Scholar] [CrossRef]
- Alfonso-Gonzalez, C.; Hilgers, V. (Alternative) transcription start sites as regulators of RNA processing. Trends Cell Biol. 2024, 34, 1018–1028. [Google Scholar] [CrossRef]
- Xu, C.; Park, J.K.; Zhang, J. Evidence that alternative transcriptional initiation is largely nonadaptive. PLoS Biol. 2019, 17, e3000197. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Huang, Q.; Liu, D.; Kong, S.; Kamada, R.; Ozato, K.; Zhang, Y.; Zhu, J. A single-cell genomic strategy for alternative transcript start sites identification. Biotechnol. J. 2024, 19, e2300516. [Google Scholar] [CrossRef]
- Beyer, A.L.; Osheim, Y.N. Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 1988, 2, 754–765. [Google Scholar]
- Cotmore, S.F.; Tattersall, P. Alternate splicing in a parvoviral nonstructural gene links a common amino-terminal sequence to downstream domains which confer radically different localization and turnover characteristics. Virology 1990, 177, 477–487. [Google Scholar]
- Rosikiewicz, W.; Sikora, J.; Skrzypczak, T.; Kubiak, M.R.; Makałowska, I. Promoter switching in response to changing environment and elevated expression of protein-coding genes overlapping at their 5’ends. Sci. Rep. 2021, 11, 8984. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, S.; Lu, H.; Xu, H. Differential regulation of alternative promoters emerges from unified kinetics of enhancer-promoter interaction. Nat. Commun. 2022, 13, 2714. [Google Scholar] [CrossRef]
- Rogalska, M.E.; Vivori, C.; Valcárcel, J. Regulation of pre-mRNA splicing: Roles in physiology and disease, and therapeutic prospects. Nat. Rev. Genet. 2023, 24, 251–269. [Google Scholar] [CrossRef]
- Louhichi, A.; Fourati, A.; Rebaï, A. IGD: A resource for intronless genes in the human genome. Gene 2011, 488, 35–40. [Google Scholar] [CrossRef]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar]
- Clancy, S. RNA splicing: Introns, exons and spliceosome. Nat. Educ. 2008, 1, 31. [Google Scholar]
- Wan, R.; Bai, R.; Zhan, X.; Shi, Y. How is precursor messenger RNA spliced by the spliceosome? Annu. Rev. Biochem. 2020, 89, 333–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhan, X.; Yan, C.; Zhang, W.; Liu, D.; Lei, J.; Shi, Y. Structures of the human spliceosomes before and after release of the ligated exon. Cell Res. 2019, 29, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Chen, W.; Moore, M.J. Spliceosomes. Curr. Biol. 2015, 25, R181–R183. [Google Scholar] [CrossRef] [PubMed]
- Norppa, A.J.; Frilander, M.J. The integrity of the U12 snRNA 3′ stem–loop is necessary for its overall stability. Nucleic Acids Res. 2021, 49, 2835–2847. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Steitz, J.A.; Dreyfuss, G.; Krainer, A.R.; Lamond, A.I.; Matera, A.G.; Padgett, R.A. Where in the cell is the minor spliceosome? Proc. Natl. Acad. Sci. USA 2008, 105, 8485–8486. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, Q.; Wang, H.; Yang, X.; Mu, H. Alternative splicing and related RNA binding proteins in human health and disease. Signal Transduct. Target. Ther. 2024, 9, 26. [Google Scholar] [CrossRef]
- Wang, S.; Sun, Z.; Lei, Z.; Zhang, H.T. RNA-binding proteins and cancer metastasis. Semin. Cancer Biol. 2022, 86, 748–768. [Google Scholar] [CrossRef]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Streitner, C.; Köster, T.; Simpson, C.G.; Shaw, P.; Danisman, S.; Brown, J.W.S.; Staiger, D. An hnRNP-like RNA-binding protein affects alternative splicing by in vivo interaction with transcripts in Arabidopsis thaliana. Nucleic Acids Res. 2012, 40, 11240–11255. [Google Scholar] [CrossRef]
- Li, D.; Yu, W.; Lai, M. Targeting serine-and arginine-rich splicing factors to rectify aberrant alternative splicing. Drug Discov. Today 2023, 28, 103691. [Google Scholar] [CrossRef]
- Busch, A.; Hertel, K.J. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip. Rev. RNA 2012, 3, 1–12. [Google Scholar] [CrossRef]
- Fisher, E.; Feng, J. RNA splicing regulators play critical roles in neurogenesis. Wiley Interdiscip. Rev. RNA 2022, 13, e1728. [Google Scholar] [CrossRef]
- Olivieri, J.E.; Dehghannasiri, R.; Wang, P.L.; Jang, S.; De Morree, A.; Tan, S.Y.; Ming, J.; Wu, A.R.; Quake, S.R.; Krasnow, M.A. RNA splicing programs define tissue compartments and cell types at single-cell resolution. Elife 2021, 10, e70692. [Google Scholar] [CrossRef]
- Badr, E.; ElHefnawi, M.; Heath, L.S. Computational Identification of Tissue-Specific Splicing Regulatory Elements in Human Genes from RNA-Seq Data. PLoS ONE 2016, 11, e0166978. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.A.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef]
- Maquat, L.E. Defects in RNA splicing and the consequence of shortened translational reading frames. Am. J. Hum. Genet. 1996, 59, 279. [Google Scholar] [PubMed]
- Nguyen, L.S.; Kim, H.-G.; Rosenfeld, J.A.; Shen, Y.; Gusella, J.F.; Lacassie, Y.; Layman, L.C.; Shaffer, L.G.; Gécz, J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 2013, 22, 1816–1825. [Google Scholar] [CrossRef]
- Tudek, A.; Schmid, M.; Jensen, T.H. Escaping nuclear decay: The significance of mRNA export for gene expression. Curr. Genet. 2019, 65, 473–476. [Google Scholar] [CrossRef]
- Jacobson, A.; Peltz, S.W. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem. 1996, 65, 693–739. [Google Scholar] [CrossRef]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Means to an end: Mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdiscip. Rev. RNA 2014, 5, 183–196. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Ending the message: Poly (A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef]
- Lisitsky, I.; Klaff, P.; Schuster, G. Addition of destabilizing poly (A)-rich sequences to endonuclease cleavage sites during the degradation of chloroplast mRNA. Proc. Natl. Acad. Sci. USA 1996, 93, 13398–13403. [Google Scholar] [CrossRef]
- Gagliardi, D.; Leaver, C.J. Polyadenylation accelerates the degradation of the mitochondrial mRNA associated with cytoplasmic male sterility in sunflower. Embo J. 1999, 18, 3757–3766. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Qiu, Q.; Zhou, Q.; Ding, J.; Lu, Y.; Liu, P. Alternative polyadenylation: Methods, mechanism, function, and role in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–19. [Google Scholar] [CrossRef]
- Mitschka, S.; Mayr, C. Context-specific regulation and function of mRNA alternative polyadenylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 779–796. [Google Scholar] [CrossRef]
- Chen, W.; Jia, Q.; Song, Y.; Fu, H.; Wei, G.; Ni, T. Alternative Polyadenylation: Methods, Findings, and Impacts. Genom. Proteom. Bioinform. 2017, 15, 287–300. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomark Res. 2020, 8, 67. [Google Scholar] [CrossRef]
- Guo, S.; Lin, S. mRNA alternative polyadenylation (APA) in regulation of gene expression and diseases. Genes Dis. 2023, 10, 165–174. [Google Scholar] [CrossRef]
- Weill, L.; Belloc, E.; Bava, F.-A.; Méndez, R. Translational control by changes in poly (A) tail length: Recycling mRNAs. Nat. Struct. Mol. Biol. 2012, 19, 577–585. [Google Scholar]
- Turner, R.E.; Pattison, A.D.; Beilharz, T.H. Alternative polyadenylation in the regulation and dysregulation of gene expression. Semin. Cell Dev. Biol. 2018, 75, 61–69. [Google Scholar] [CrossRef]
- Tian, B.; Pan, Z.; Lee, J.Y. Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 2007, 17, 156–165. [Google Scholar] [CrossRef]
- Yuan, F.; Hankey, W.; Wagner, E.J.; Li, W.; Wang, Q. Alternative polyadenylation of mRNA and its role in cancer. Genes Dis. 2021, 8, 61–72. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Benne, R. RNA editing in trypanosomes. Eur. J. Biochem. 1994, 221, 9–23. [Google Scholar] [CrossRef]
- Farajollahi, S.; Maas, S. Molecular diversity through RNA editing: A balancing act. Trends Genet 2010, 26, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhu, L.; Gao, Y.; Wang, Y.; Li, P. RNA editing enzymes: Structure, biological functions and applications. Cell Biosci. 2024, 14, 34. [Google Scholar] [CrossRef]
- Simpson, L.; Emeson, R.B. RNA editing. Annu. Rev. Neurosci. 1996, 19, 27–52. [Google Scholar] [CrossRef]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef]
- Slotkin, W.; Nishikura, K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013, 5, 105. [Google Scholar] [CrossRef]
- Gallo, A.; Vukic, D.; Michalík, D.; O’Connell, M.A.; Keegan, L.P. ADAR RNA editing in human disease; more to it than meets the I. Hum. Genet. 2017, 136, 1265–1278. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef]
- Raghava Kurup, R.; Oakes, E.K.; Manning, A.C.; Mukherjee, P.; Vadlamani, P.; Hundley, H.A. RNA binding by ADAR3 inhibits adenosine-to-inosine editing and promotes expression of immune response protein MAVS. J. Biol. Chem. 2022, 298, 102267. [Google Scholar] [CrossRef]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Prados-Carvajal, R.; Fernández-Ávila, M.J.; Silva, S.; Silvestris, D.A.; Endara-Coll, M.; Rodríguez-Real, G.; Domingo-Prim, J.; Mejías-Navarro, F.; Romero-Franco, A.; et al. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun. 2021, 12, 5512. [Google Scholar] [CrossRef]
- Booth, B.J.; Nourreddine, S.; Katrekar, D.; Savva, Y.; Bose, D.; Long, T.J.; Huss, D.J.; Mali, P. RNA editing: Expanding the potential of RNA therapeutics. Mol. Ther. 2023, 31, 1533–1549. [Google Scholar] [CrossRef]
- Brennicke, A.; Marchfelder, A.; Binder, S. RNA editing. FEMS Microbiol. Rev. 1999, 23, 297–316. [Google Scholar] [CrossRef]
- Deogharia, M.; Gurha, P. The “guiding” principles of noncoding RNA function. Wiley Interdiscip. Rev. RNA 2022, 13, e1704. [Google Scholar] [CrossRef]
- Soemedi, R.; Cygan, K.J.; Rhine, C.L.; Glidden, D.T.; Taggart, A.J.; Lin, C.-L.; Fredericks, A.M.; Fairbrother, W.G. The effects of structure on pre-mRNA processing and stability. Methods 2017, 125, 36–44. [Google Scholar] [CrossRef]
- Haraksingh, R.R.; Snyder, M.P. Impacts of variation in the human genome on gene regulation. J. Mol. Biol. 2013, 425, 3970–3977. [Google Scholar] [CrossRef]
- Marian, A.J. Clinical Interpretation and Management of Genetic Variants. JACC Basic Transl. Sci. 2020, 5, 1029–1042. [Google Scholar] [CrossRef]
- Jones, B.L.; Swallow, D.M. The impact of cis-acting polymorphisms on the human phenotype. Hugo J. 2011, 5, 13–23. [Google Scholar] [CrossRef]
- Deka, B.; Chandra, P.; Singh, K.K. Functional roles of human Up-frameshift suppressor 3 (UPF3) proteins: From nonsense-mediated mRNA decay to neurodevelopmental disorders. Biochimie 2021, 180, 10–22. [Google Scholar] [CrossRef]
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Gabel, A.M.; Belleville, A.E.; Thomas, J.D.; Pineda, J.M.B.; Bradley, R.K. APC mutations dysregulate alternative polyadenylation in cancer. Genome Biol. 2024, 25, 255. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Saika, W.; Inoue, D. Minor introns impact on hematopoietic malignancies. Exp. Hematol. 2024, 132, 104173. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Gonzalez, C.; Legnini, I.; Holec, S.; Arrigoni, L.; Ozbulut, H.C.; Mateos, F.; Koppstein, D.; Rybak-Wolf, A.; Bönisch, U.; Rajewsky, N.; et al. Sites of transcription initiation drive mRNA isoform selection. Cell 2023, 186, 2438–2455.e2422. [Google Scholar] [CrossRef] [PubMed]
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Plass, C.; Huang, T.H.-M. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008, 24, 167–177. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Hughes, J.R.; Garrick, D.; Lynch, M.D.; Sharpe, J.A.; Sloane-Stanley, J.A.; McGowan, S.J.; De Gobbi, M.; Hosseini, M.; Vernimmen, D. Intragenic enhancers act as alternative promoters. Mol. Cell 2012, 45, 447–458. [Google Scholar] [CrossRef]
- Yuan, J.; Tong, Y.; Wang, L.; Yang, X.; Liu, X.; Shu, M.; Li, Z.; Jin, W.; Guan, C.; Wang, Y. A compendium of genetic variations associated with promoter usage across 49 human tissues. Nat. Commun. 2024, 15, 8758. [Google Scholar] [CrossRef]
- Pan, K.; Guo, G.; Xue, X. mRNA modifications: Clinical clarification and significance. Epigenomics 2021, 13, 1901–1903. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Wu, C. Alternative Splicing and Isoforms: From Mechanisms to Diseases. Genes 2022, 13, 401. [Google Scholar] [CrossRef]
- Peng, Q.; Zhou, Y.; Oyang, L.; Wu, N.; Tang, Y.; Su, M.; Luo, X.; Wang, Y.; Sheng, X.; Ma, J.; et al. Impacts and mechanisms of alternative mRNA splicing in cancer metabolism, immune response, and therapeutics. Mol. Ther. 2022, 30, 1018–1035. [Google Scholar] [CrossRef]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Cappannini, A.; Ray, A.; Purta, E.; Mukherjee, S.; Boccaletto, P.; Moafinejad, S.N.; Lechner, A.; Barchet, C.; Klaholz, B.P.; Stefaniak, F.; et al. MODOMICS: A database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 2023, 52, D239–D244. [Google Scholar] [CrossRef] [PubMed]
- Levi, O.; Arava, Y.S. RNA modifications as a common denominator between tRNA and mRNA. Curr. Genet 2021, 67, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fan, Z. New insights into the interaction between m6A modification and lncRNA in cancer drug resistance. Cell Prolif. 2024, 57, e13578. [Google Scholar] [CrossRef]
- Liu, W.-W.; Zheng, S.-Q.; Li, T.; Fei, Y.-F.; Wang, C.; Zhang, S.; Wang, F.; Jiang, G.-M.; Wang, H. RNA modifications in cellular metabolism: Implications for metabolism-targeted therapy and immunotherapy. Signal Transduct. Target. Ther. 2024, 9, 70. [Google Scholar] [CrossRef]
- Gao, B.; Guo, J.; She, C.; Shu, A.; Yang, M.; Tan, Z.; Yang, X.; Guo, S.; Feng, G.; He, L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet 2001, 28, 386–388. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, J. RNA 5-methylcytosine modification and its emerging role as an epitranscriptomic mark. RNA Biol. 2021, 18, 117–127. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Li, J.; Pei, Y.; Zhou, R.; Tang, Z.; Yang, Y. Regulation of RNA N6-methyladenosine modification and its emerging roles in skeletal muscle development. Int. J. Biol. Sci. 2021, 17, 1682. [Google Scholar] [CrossRef]
- Cai, T.; Atteh, L.L.; Zhang, X.; Huang, C.; Bai, M.; Ma, H.; Zhang, C.; Fu, W.; Gao, L.; Lin, Y. The N6-methyladenosine modification and its role in mRNA metabolism and gastrointestinal tract disease. Front. Surg. 2022, 9, 819335. [Google Scholar] [CrossRef]
- Qi, Z.; Zhang, C.; Jian, H.; Hou, M.; Lou, Y.; Kang, Y.; Wang, W.; Lv, Y.; Shang, S.; Wang, C.; et al. N1-Methyladenosine modification of mRNA regulates neuronal gene expression and oxygen glucose deprivation/reoxygenation induction. Cell Death Discov. 2023, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yi, X.; Hou, M.; Li, Q.; Li, X.; Lu, L.; Qi, E.; Wu, M.; Qi, L.; Jian, H.; et al. The landscape of m(1)A modification and its posttranscriptional regulatory functions in primary neurons. Elife 2023, 12, e85324. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Xu, K.; Lee, J.H. Biological roles of the RNA m6A modification and its implications in cancer. Exp. Mol. Med. 2022, 54, 1822–1832. [Google Scholar] [CrossRef]
- Cui, L.; Ma, R.; Cai, J.; Guo, C.; Chen, Z.; Yao, L.; Wang, Y.; Fan, R.; Wang, X.; Shi, Y. RNA modifications: Importance in immune cell biology and related diseases. Signal Transduct. Target. Ther. 2022, 7, 334. [Google Scholar] [CrossRef]
- Delaunay, S.; Helm, M.; Frye, M. RNA modifications in physiology and disease: Towards clinical applications. Nat. Rev. Genet. 2024, 25, 104–122. [Google Scholar] [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef]
- Shi, H.; Chai, P.; Jia, R.; Fan, X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol. Cancer 2020, 19, 78. [Google Scholar] [CrossRef]
- Parker, M.T.; Soanes, B.K.; Kusakina, J.; Larrieu, A.; Knop, K.; Joy, N.; Breidenbach, F.; Sherwood, A.V.; Barton, G.J.; Fica, S.M. m6A modification of U6 snRNA modulates usage of two major classes of pre-mRNA 5’splice site. Elife 2022, 11, e78808. [Google Scholar] [CrossRef]
- Bilal, A.; Alarfaj, F.K.; Khan, R.A.; Suleman, M.T.; Long, H. m5c-iEnsem: 5-methylcytosine sites identification through ensemble models. Bioinformatics 2024, 41, btae722. [Google Scholar] [CrossRef]
- Luo, J.; Xu, T.; Sun, K. N6-Methyladenosine RNA Modification in Inflammation: Roles, Mechanisms, and Applications. Front. Cell Dev. Biol. 2021, 9, 670711. [Google Scholar] [CrossRef]
- Ochiai, Y.; Clifton, B.E.; Le Coz, M.; Terenzio, M.; Laurino, P. SUPREM: An engineered non-site-specific m6A RNA methyltransferase with highly improved efficiency. Nucleic Acids Res. 2024, 52, 12158–12172. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Single-base m(6)A profiling. Nat. Cell Biol. 2022, 24, 403. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.S.; Akanksha, S.; Ravindran, A.; Oyelami, F.; Ip, C.K.; Gupta, P.; Villanueva, J.; King, H.E.; Grootveld, A.; Blackburn, J. The conserved landscape of RNA modifications and transcript diversity across mammalian evolution. bioRxiv 2024, 11. [Google Scholar] [CrossRef]
- Dedon, P.C.; Begley, T.J. A System of RNA Modifications and Biased Codon Use Controls Cellular Stress Response at the Level of Translation. Chem. Res. Toxicol. 2014, 27, 330–337. [Google Scholar] [CrossRef]
- Gilbert, W.V.; Nachtergaele, S. mRNA Regulation by RNA Modifications. Annu. Rev. Biochem. 2023, 92, 175–198. [Google Scholar] [CrossRef]
- Licht, K.; Jantsch, M.F. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J. Cell Biol. 2016, 213, 15–22. [Google Scholar] [CrossRef]
- He, F.; Guo, Q.; Jiang, G.-x.; Zhou, Y. Comprehensive analysis of m6A circRNAs identified in colorectal cancer by MeRIP sequencing. Front. Oncol. 2022, 12, 927810. [Google Scholar] [CrossRef]
- Sağlam, B.; Akgül, B. An Overview of Current Detection Methods for RNA Methylation. Int. J. Mol. Sci. 2024, 25, 3098. [Google Scholar] [CrossRef]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef]
- Nombela, P.; Miguel-López, B.; Blanco, S. The role of m6A, m5C and Ψ RNA modifications in cancer: Novel therapeutic opportunities. Mol. Cancer 2021, 20, 18. [Google Scholar] [CrossRef]
- Marasco, L.E.; Kornblihtt, A.R. The physiology of alternative splicing. Nat. Rev. Mol. Cell Biol. 2023, 24, 242–254. [Google Scholar] [CrossRef]
- Modrek, B.; Resch, A.; Grasso, C.; Lee, C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001, 29, 2850–2859. [Google Scholar] [CrossRef] [PubMed]
- Dieudonné, F.-X.; O’Connor, P.B.; Gubler-Jaquier, P.; Yasrebi, H.; Conne, B.; Nikolaev, S.; Antonarakis, S.; Baranov, P.V.; Curran, J. The effect of heterogeneous Transcription Start Sites (TSS) on the translatome: Implications for the mammalian cellular phenotype. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C.; Bailey, T.L.; Kasukawa, T.; Mignone, F.; Kummerfeld, S.K.; Madera, M.; Sunkara, S.; Furuno, M.; Bult, C.J.; Quackenbush, J. Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biol. 2006, 3, 40–48. [Google Scholar] [CrossRef]
- Li, S.; Mason, C.E. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genom. Hum. Genet. 2014, 15, 127–150. [Google Scholar] [CrossRef]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene regulation by non-coding RNAs. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 16–32. [Google Scholar] [CrossRef]
- Dhamija, S.; Menon, M.B. Non-coding transcript variants of protein-coding genes—What are they good for? RNA Biol. 2018, 15, 1025–1031. [Google Scholar] [CrossRef]
- Mockenhaupt, S.; Makeyev, E.V. Non-coding functions of alternative pre-mRNA splicing in development. Proc. Semin. Cell Dev. Biol. 2015, 47, 32–39. [Google Scholar]
- George, T.P.; Subramanian, S.; Supriya, M. A brief review of noncoding RNA. Egypt. J. Med. Hum. Genet. 2024, 25, 98. [Google Scholar] [CrossRef]
- Chen, L.-L.; Kim, V.N. Small and long non-coding RNAs: Past, present, and future. Cell 2024, 187, 6451–6485. [Google Scholar] [CrossRef]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar]
- Röther, S.; Meister, G. Small RNAs derived from longer non-coding RNAs. Biochimie 2011, 93, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ortín, J.E.; Alepuz, P.; Chávez, S.; Choder, M. Eukaryotic mRNA decay: Methodologies, pathways, and links to other stages of gene expression. J. Mol. Biol. 2013, 425, 3750–3775. [Google Scholar] [CrossRef]
- Ross, J. mRNA stability in mammalian cells. Microbiol. Rev. 1995, 59, 423–450. [Google Scholar] [CrossRef]
- Wagner, E.; Lykke-Andersen, J. mRNA surveillance: The perfect persist. J. Cell Sci. 2002, 115, 3033–3038. [Google Scholar] [CrossRef]
- Meyer, S.; Temme, C.; Wahle, E. Messenger RNA turnover in eukaryotes: Pathways and enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 197–216. [Google Scholar] [CrossRef]
- Nagarajan, V.K.; Jones, C.I.; Newbury, S.F.; Green, P.J. XRN 5′→ 3′ exoribonucleases: Structure, mechanisms and functions. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 590–603. [Google Scholar] [CrossRef]
- Mukherjee, D.; Gao, M.; O’Connor, J.P.; Raijmakers, R.; Pruijn, G.; Lutz, C.S.; Wilusz, J. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J. 2002, 21, 165–174. [Google Scholar] [CrossRef]
- Woodward, L.A.; Mabin, J.W.; Gangras, P.; Singh, G. The exon junction complex: A lifelong guardian of mRNA fate. Wiley Interdiscip. Rev. RNA 2017, 8, e1411. [Google Scholar] [CrossRef]
- Gupta, P.; Li, Y.-R. Upf proteins: Highly conserved factors involved in nonsense mRNA mediated decay. Mol. Biol. Rep. 2018, 45, 39–55. [Google Scholar] [CrossRef]
- Atkinson, G.C.; Baldauf, S.L.; Hauryliuk, V. Evolution of nonstop, no-go and nonsense-mediated mRNA decay and their termination factor-derived components. BMC Evol. Biol. 2008, 8, 1–18. [Google Scholar] [CrossRef]
- Alagar Boopathy, L.R.; Beadle, E.; Garcia-Bueno Rico, A.; Vera, M. Proteostasis regulation through ribosome quality control and no-go-decay. Wiley Interdiscip. Rev. RNA 2023, 14, e1809. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, S.; Belmonte, J.C.I.; Zhang, W.; Qu, J.; Liu, G.-H. Emerging role of RNA m6A modification in aging regulation. Curr. Med. 2022, 1, 8. [Google Scholar] [CrossRef]
- Wang, K.; Wu, D.; Zhang, H.; Das, A.; Basu, M.; Malin, J.; Cao, K.; Hannenhalli, S. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Sci. Rep. 2018, 8, 10929. [Google Scholar] [CrossRef]
- Stegeman, R.; Weake, V.M. Transcriptional Signatures of Aging. J. Mol. Biol. 2017, 429, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Debès, C.; Papadakis, A.; Grönke, S.; Karalay, Ö.; Tain, L.S.; Mizi, A.; Nakamura, S.; Hahn, O.; Weigelt, C.; Josipovic, N.; et al. Ageing-associated changes in transcriptional elongation influence longevity. Nature 2023, 616, 814–821. [Google Scholar] [CrossRef]
- López-Domínguez, J.A.; Rodríguez-López, S.; Ahumada-Castro, U.; Desprez, P.Y.; Konovalenko, M.; Laberge, R.M.; Cárdenas, C.; Villalba, J.M.; Campisi, J. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging 2021, 13, 13380–13392. [Google Scholar] [CrossRef]
- Michalova, E.; Vojtesek, B.; Hrstka, R. Impaired pre-mRNA processing and altered architecture of 3’untranslated regions contribute to the development of human disorders. Int. J. Mol. Sci. 2013, 14, 15681–15694. [Google Scholar] [CrossRef]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2010, 220, 152–163. [Google Scholar] [CrossRef]
- Smalheiser, N.R. Proteins in unexpected locations. Mol. Biol. Cell 1996, 7, 1003–1014. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Bungeroth, M.; Appenzeller, S.; Regulin, A.; Völker, W.; Lorenzen, I.; Grötzinger, J.; Pendziwiat, M.; Kuhlenbäumer, G. Differential aggregation properties of alpha-synuclein isoforms. Neurobiol. Aging 2014, 35, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.R.; Swanson, M.S. Mechanisms of RNA-mediated disease. J. Biol. Chem. 2009, 284, 7419–7423. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.; Timothy, K.W.; Golden, A. Update on the Molecular Genetics of Timothy Syndrome. Front. Pediatr. 2021, 9, 668546. [Google Scholar] [CrossRef]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef]
- Maziuk, B.; Ballance, H.I.; Wolozin, B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Front. Mol. Neurosci. 2017, 10, 89. [Google Scholar] [CrossRef]
- Campagne, S. U1 snRNP Biogenesis Defects in Neurodegenerative Diseases. ChemBioChem 2024, 25, e202300864. [Google Scholar] [CrossRef]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflügers Arch. -Eur. J. Physiol. 2018, 470, 995–1016. [Google Scholar] [CrossRef]
- Escribano, O.; Beneit, N.; Rubio-Longás, C.; López-Pastor, A.; Gómez-Hernández, A. The role of insulin receptor isoforms in diabetes and its metabolic and vascular complications. J. Diabetes Res. 2017, 2017, 1403206. [Google Scholar] [CrossRef]
- Yang, H.D.; Nam, S.W. Pathogenic diversity of RNA variants and RNA variation-associated factors in cancer development. Exp. Mol. Med. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Hong, S.E.; Kneissl, J.; Cho, A.; Kim, M.J.; Park, S.; Lee, J.; Woo, S.; Kim, S.; Kim, J.-S.; Kim, S.Y. Transcriptome-based variant calling and aberrant mRNA discovery enhance diagnostic efficiency for neuromuscular diseases. J. Med. Genet. 2022, 59, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Ji, J. N6-methyladenosine (m6A) RNA modification in cancer stem cells. Stem Cells 2020, 38, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Malka-Tunitsky, N.; Sas-Chen, A. Role of RNA modifications in cancer metastasis. Curr. Opin. Genet. Dev. 2024, 87, 102232. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, M.C.; van Roon-Mom, W.; Aartsma-Rus, A.; N = 1 Collaborative. Possibilities and limitations of antisense oligonucleotide therapies for the treatment of monogenic disorders. Commun. Med. 2024, 4, 6. [Google Scholar] [CrossRef]
- Barrie, E.S.; Smith, R.M.; Sanford, J.C.; Sadee, W. mRNA transcript diversity creates new opportunities for pharmacological intervention. Mol. Pharmacol. 2012, 81, 620–630. [Google Scholar] [CrossRef]
- Pozo, F.; Rodriguez, J.M.; Vázquez, J.; Tress, M.L. Clinical variant interpretation and biologically relevant reference transcripts. NPJ Genom. Med. 2022, 7, 59. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, Y.; Vo, K.; Shila, S.; Paul, A.; Dahiya, V.; Fields, P.E.; Rumi, M.A.K. mRNA Transcript Variants Expressed in Mammalian Cells. Int. J. Mol. Sci. 2025, 26, 1052. https://doi.org/10.3390/ijms26031052
Sharma Y, Vo K, Shila S, Paul A, Dahiya V, Fields PE, Rumi MAK. mRNA Transcript Variants Expressed in Mammalian Cells. International Journal of Molecular Sciences. 2025; 26(3):1052. https://doi.org/10.3390/ijms26031052
Chicago/Turabian StyleSharma, Yashica, Kevin Vo, Sharmin Shila, Anohita Paul, Vinesh Dahiya, Patrick E. Fields, and M. A. Karim Rumi. 2025. "mRNA Transcript Variants Expressed in Mammalian Cells" International Journal of Molecular Sciences 26, no. 3: 1052. https://doi.org/10.3390/ijms26031052
APA StyleSharma, Y., Vo, K., Shila, S., Paul, A., Dahiya, V., Fields, P. E., & Rumi, M. A. K. (2025). mRNA Transcript Variants Expressed in Mammalian Cells. International Journal of Molecular Sciences, 26(3), 1052. https://doi.org/10.3390/ijms26031052