
Endothelial Sestrin2 Coordinates Multiple Protective Pathways to Maintain Angiogenic Function in Diabetes-Associated Endothelial Dysfunction
 ,
,  ,
,
Abstract
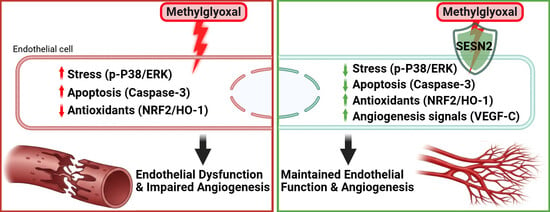
1. Introduction
2. Results
2.1. Validation of Experimental Model and SESN2 Modulation
2.2. SESN2 Expression Levels Modulate Endothelial Cell Tube Formation Capacity
2.3. SESN2 Regulates Multiple Aspects of Endothelial Cell Function Under Normal and Stress Conditions
2.4. SESN2 Promotes Angiogenesis Through Parallel Activation of NRF2/HO-1 Antioxidant Pathway and VEGF-C Expression
2.5. SESN2 Orchestrates Angiogenic Response Through Differential Regulation of AKT/mTOR Signaling
2.6. Modulation of the MAPK/ERK Signaling Pathway by SESN2
2.7. Apoptotic Response Pathway Regulation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures
4.3. Tube Formation Assay
4.4. Cell Invasion and Migration
4.5. Scratch Migration Assay
4.6. Cell Proliferation
4.7. Matrix Metalloproteinase Activity
4.8. Total RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR (qPCR)
4.9. Western Blot Analysis
4.10. VEGF ELISA
4.11. Gene Manipulation
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | Advanced Glycation End-Products |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| ANOVA | Analysis of Variance |
| BAX | BCL2-associated X protein |
| BCA | Bicinchoninic Acid |
| BCL2 | B-cell lymphoma 2 |
| BD | Becton Dickinson |
| CASP3 | Caspase-3 |
| cDNA | Complementary DNA |
| Ct | Cycle threshold |
| ΔΔCt | Delta–delta Ct method |
| Ctl | Control |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| ECL | Enhanced chemiluminescence |
| EdU | 5-Ethynyl-2′-deoxyuridine |
| EDTA | Ethylenediaminetetraacetic acid |
| EA.hy926 | Human endothelial cell line |
| eNOS (NOS3) | Endothelial nitric oxide synthase (gene NOS3) |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GPX4 | Glutathione peroxidase 4 |
| HIF-1 | Hypoxia-inducible factor-1 |
| HO-1 (HMOX1) | Heme oxygenase-1 (gene HMOX1) |
| HRP | Horseradish peroxidase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| MAPK | Mitogen-activated protein kinase |
| Matrigel | Basement membrane matrix |
| MGO | Methylglyoxal |
| MMP | Matrix metalloproteinase |
| mRNA | Messenger RNA |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| NO | Nitric oxide |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NP-40 | Nonidet P-40 |
| Oe | SESN2 overexpression |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| PI3K | Phosphoinositide 3-kinase |
| PVDF | Polyvinylidene difluoride |
| qPCR | Quantitative PCR |
| qRT-PCR | Quantitative reverse-transcription PCR |
| RFU | Relative fluorescence units |
| RIPA | Radioimmunoprecipitation assay |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| SESN2 | Sestrin2 |
| siRNA | Small interfering RNA |
| Si | SESN2 silencing |
| TBST | Tris-buffered saline with Tween-20 |
| ULK1 | Unc-51-like kinase 1 |
| VEGFA | Vascular endothelial growth factor A |
| VEGFC | Vascular endothelial growth factor C |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Zhang, B.; Li, X.; Liu, Y. Diabetes Mellitus and Cause-Specific Mortality: A Population-Based Study. Diabetes Metab. J. 2019, 43, 319–341. [Google Scholar] [CrossRef]
- Hartog, J.W.L.; Voors, A.A.; Bakker, S.J.L.; Smit, A.J.; van Veldhuisen, D.J. Advanced Glycation End-Products (AGEs) and Heart Failure: Pathophysiology and Clinical Implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef]
- Lee, J.H.; Parveen, A.; Do, M.H.; Kang, M.C.; Yumnam, S.; Kim, S.Y. Molecular Mechanisms of Methylglyoxal-Induced Aortic Endothelial Dysfunction in Human Vascular Endothelial Cells. Cell Death Dis. 2020, 11, 403. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Do, M.H.; Lee, J.H.; Wahedi, H.M.; Pak, C.; Lee, C.H.; Yeo, E.-J.; Lim, Y.; Ha, S.K.; Choi, I.; Kim, S.Y. Lespedeza Bicolor Ameliorates Endothelial Dysfunction Induced by Methylglyoxal Glucotoxicity. Phytomedicine Int. J. Phytother. Phytopharm. 2017, 36, 26–36. [Google Scholar] [CrossRef]
- Figarola, J.L.; Singhal, J.; Rahbar, S.; Awasthi, S.; Singhal, S.S. LR-90 Prevents Methylglyoxal-Induced Oxidative Stress and Apoptosis in Human Endothelial Cells. Apoptosis Int. J. Program. Cell Death 2014, 19, 776–788. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Zheng, Y.; Jiang, J.; Wang, L.; Wu, J.; Zhang, C.; Luo, M. Glucose Metabolite Methylglyoxal Induces Vascular Endothelial Cell Pyroptosis via NLRP3 Inflammasome Activation and Oxidative Stress in Vitro and in Vivo. Cell. Mol. Life Sci. CMLS 2024, 81, 401. [Google Scholar] [CrossRef]
- Zahid, M.A.; Abdelsalam, S.S.; Raïq, H.; Parray, A.; Korashy, H.M.; Zeidan, A.; Elrayess, M.A.; Agouni, A. Sestrin2 as a Protective Shield against Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 4880. [Google Scholar] [CrossRef]
- Ro, S.-H.; Semple, I.A.; Park, H.; Park, H.; Park, H.-W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 Promotes Unc-51-like Kinase 1 Mediated Phosphorylation of P62/Sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 Is a Leucine Sensor for the mTORC1 Pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef]
- Li, J.; Ren, C.; Wang, L.-X.; Yao, R.; Dong, N.; Wu, Y.; Tian, Y.; Yao, Y. Sestrin2 Protects Dendrite Cells against Ferroptosis Induced by Sepsis. Cell Death Dis. 2021, 12, 834. [Google Scholar] [CrossRef]
- Xi, X.; Chen, Q.; Ma, J.; Wang, X.; Zhang, J.; Li, Y. Sestrin2 Ameliorates Diabetic Retinopathy by Regulating Autophagy and Ferroptosis. J. Mol. Histol. 2024, 55, 169–184. [Google Scholar] [CrossRef]
- Quan, N.; Sun, W.; Wang, L.; Chen, X.; Bogan, J.S.; Zhou, X.; Cates, C.; Liu, Q.; Zheng, Y.; Li, J. Sestrin2 Prevents Age-Related Intolerance to Ischemia and Reperfusion Injury by Modulating Substrate Metabolism. FASEB J. 2017, 31, 4153–4167. [Google Scholar] [CrossRef]
- Quan, N.; Li, X.; Zhang, J.; Han, Y.; Sun, W.; Ren, D.; Tong, Q.; Li, J. Substrate Metabolism Regulated by Sestrin2-mTORC1 Alleviates Pressure Overload-Induced Cardiac Hypertrophy in Aged Heart. Redox Biol. 2020, 36, 101637. [Google Scholar] [CrossRef]
- Ding, S.; Ma, N.; Liu, H.; Tang, M.; Mei, J. Sesn2 Attenuates the Damage of Endothelial Progenitor Cells Induced by Angiotensin II through Regulating the Keap1/Nrf2 Signal Pathway. Aging 2020, 12, 25505–25527. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, Y.; Li, Y. Sestrin2 Overexpression Attenuates Focal Cerebral Ischemic Injury in Rat by Increasing Nrf2/HO-1 Pathway-Mediated Angiogenesis. Neuroscience 2019, 410, 140–149. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in Life, Disease and Medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Wan, G.; Xu, Z.; Xiang, X.; Zhang, M.; Jiang, T.; Chen, J.; Li, S.; Wang, C.; Yan, C.; Yang, X.; et al. Elucidation of Endothelial Progenitor Cell Dysfunction in Diabetes by RNA Sequencing and Constructing lncRNA–miRNA–mRNA Competing Endogenous RNA Network. J. Mol. Med. 2022, 100, 1569–1585. [Google Scholar] [CrossRef]
- Sawada, N.; Arany, Z. Metabolic Regulation of Angiogenesis in Diabetes and Aging. Physiology 2017, 32, 290–307. [Google Scholar] [CrossRef]
- Jarisarapurin, W.; Kunchana, K.; Chularojmontri, L.; Wattanapitayakul, S.K. Unripe Carica Papaya Protects Methylglyoxal-Invoked Endothelial Cell Inflammation and Apoptosis via the Suppression of Oxidative Stress and Akt/MAPK/NF-κB Signals. Antioxidants 2021, 10, 1158. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Yang, Q.; Xu, C.; Zheng, Y.; Wang, L.; Wu, J.; Zeng, M.; Luo, M. Metformin Prevents Methylglyoxal-Induced Apoptosis by Suppressing Oxidative Stress in Vitro and in Vivo. Cell Death Dis. 2022, 13, 29. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO System in Tumor Growth, Angiogenesis and Metabolism—Targeting HO-1 as an Anti-Tumor Therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.-H. The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging. Oxid. Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Dirat, B.; Laurent, K.; Puissant, A.; Auberger, P.; Budanov, A.; Tanti, J.-F.; Bost, F. Sestrin2 Integrates Akt and mTOR Signaling to Protect Cells against Energetic Stress-Induced Death. Cell Death Differ. 2013, 20, 611–619. [Google Scholar] [CrossRef]
- Fatima, M.T.; Hasan, M.; Abdelsalam, S.S.; Sivaraman, S.K.; El-Gamal, H.; Zahid, M.A.; Elrayess, M.A.; Korashy, H.M.; Zeidan, A.; Parray, A.S.; et al. Sestrin2 Suppression Aggravates Oxidative Stress and Apoptosis in Endothelial Cells Subjected to Pharmacologically Induced Endoplasmic Reticulum Stress. Eur. J. Pharmacol. 2021, 907, 174247. [Google Scholar] [CrossRef]
- Uruski, P.; Mikuła-Pietrasik, J.; Drzewiecki, M.; Budkiewicz, S.; Gładki, M.; Kurmanalina, G.; Tykarski, A.; Książek, K. Diverse Functional Responses to High Glucose by Primary and Permanent Hybrid Endothelial Cells in Vitro. J. Mol. Cell. Cardiol. 2021, 156, 1–6. [Google Scholar] [CrossRef]
- Wang, D.; Chen, Z.; Wai Kan Yeung, A.; Atanasov, A.G. Differences between Common Endothelial Cell Models (Primary Human Aortic Endothelial Cells and EA.Hy926 Cells) Revealed through Transcriptomics, Bioinformatics, and Functional Analysis. Curr. Res. Biotechnol. 2021, 3, 135–145. [Google Scholar] [CrossRef]
- Osman, A.; El-Gamal, H.; Pasha, M.; Zeidan, A.; Korashy, H.M.; Abdelsalam, S.S.; Hasan, M.; Benameur, T.; Agouni, A. Endoplasmic Reticulum (ER) Stress-Generated Extracellular Vesicles (Microparticles) Self-Perpetuate ER Stress and Mediate Endothelial Cell Dysfunction Independently of Cell Survival. Front. Cardiovasc. Med. 2020, 7, 584791. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A Comparative Morphometric Analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Pasha, M.; Sivaraman, S.K.; Frantz, R.; Agouni, A.; Munusamy, S. Metformin Induces Different Responses in Clear Cell Renal Cell Carcinoma Caki Cell Lines. Biomolecules 2019, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, S.S.; Zahid, M.A.; Ghanem, S.K.; Khan, A.; Parray, A.; Agouni, A. Sestrin2 Suppression Promotes Endothelial–Mesenchymal Transition and Exacerbates Methylglyoxal-Induced Endothelial Dysfunction. Int. J. Mol. Sci. 2024, 25, 13463. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| MMP2 | TACAGGATCATTGGCTACACACC | GGTCACATCGCTCCAGACT |
| MMP9 | TGTACCGCTATGGTTACACTCG | GGCAGGGACAGTTGCTTCT |
| NOS3 | TGATGGCGAAGCGAGTGAAG | ACTCATCCATACACAGGACCC |
| VEGFA | AGGGCAGAATCATCACGAAGT | AGGGTCTCGATTGGATGGCA |
| VEGFC | GAGGAGCAGTTACGGTCTGTG | TCCTTTCCTTAGCTGACACTTGT |
| KDR | GGCCCAATAATCAGAGTGGCA | CCAGTGTCATTTCCGATCACTTT |
| BAX | CCCGAGAGGTCTTTTTCCGAG | CCAGCCCATGATGGTTCTGAT |
| BCL2 | GGTGGGGTCATGTGTGTGG | CGGTTCAGGTACTCAGTCATCC |
| CASP3 | CATGGAAGCGAATCAATGGACT | CTGTACCAGACCGAGATGTCA |
| HMOX1 | AAGACTGCGTTCCTGCTCAAC | AAAGCCCTACAGCAACTGTCG |
| NQO1 | GAAGAGCACTGATCGTACTGGC | GGATACTGAAAGTTCGCAGGG |
| SESN2 | CCTCTGGGCGAGTAGACAAC | GGAGCCTACCAGGTAAGAACA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahid, M.A.; Parray, A.; Rathore, H.A.; Khan, A.; Agouni, A. Endothelial Sestrin2 Coordinates Multiple Protective Pathways to Maintain Angiogenic Function in Diabetes-Associated Endothelial Dysfunction. Int. J. Mol. Sci. 2025, 26, 11396. https://doi.org/10.3390/ijms262311396
Zahid MA, Parray A, Rathore HA, Khan A, Agouni A. Endothelial Sestrin2 Coordinates Multiple Protective Pathways to Maintain Angiogenic Function in Diabetes-Associated Endothelial Dysfunction. International Journal of Molecular Sciences. 2025; 26(23):11396. https://doi.org/10.3390/ijms262311396
Chicago/Turabian StyleZahid, Muhammad Ammar, Aijaz Parray, Hassaan Anwer Rathore, Abbas Khan, and Abdelali Agouni. 2025. "Endothelial Sestrin2 Coordinates Multiple Protective Pathways to Maintain Angiogenic Function in Diabetes-Associated Endothelial Dysfunction" International Journal of Molecular Sciences 26, no. 23: 11396. https://doi.org/10.3390/ijms262311396
APA StyleZahid, M. A., Parray, A., Rathore, H. A., Khan, A., & Agouni, A. (2025). Endothelial Sestrin2 Coordinates Multiple Protective Pathways to Maintain Angiogenic Function in Diabetes-Associated Endothelial Dysfunction. International Journal of Molecular Sciences, 26(23), 11396. https://doi.org/10.3390/ijms262311396