Abstract
Chronic pain is a serious health issue, often irrationally managed by conventional analgesics. Duloxetine, a serotonin–norepinephrine reuptake inhibitor (SNRI), also effective in neuropathic and musculoskeletal pain, but the molecular mechanism of its analgesic action is still unclear. Here, we examined whether Duloxetine exerts pleiotropic effects by directly targeting phosphorylated STAT3 (pSTAT3), a key regulator of neuroinflammation and pain sensitization. Molecular docking showed that Duloxetine binds with pSTAT3 with binding energy −5.83 kcal/mol. Ruxolitinib, a JAK/STAT inhibitor used as reference, showed binding energy of −6.19 kcal/mol. Molecular dynamics (MD) simulations confirmed stable Duloxetine–pSTAT3 complexes, while MM-PBSA free energy analysis revealed more favorable binding for Duloxetine (ΔG = −15.17 kJ·mol−1) than Ruxolitinib (ΔG = −12.98 kJ·mol−1) for pSTAT3. In-vitro analyses, Western blot showed that Duloxetine significantly reduced IL-6–induced STAT3 and pSTAT3 expression in C2C12 cells in a dose-dependent manner (6.4 and 12.8 μM, *** p < 0.0001), although Ruxolitinib produced a stronger suppression. Transcriptomic analysis revealed Duloxetine-specific enrichment of mitochondrial, oxidative phosphorylation, and synaptic pathways, distinct from the immune-suppressive influence of Ruxolitinib. RNA-seq further revealed that STAT3 transcript abundance remains constant under all treatment conditions, indicating that post-transcriptional or post-translational mechanisms, such as phosphorylation-dependent activation, may be involved rather than transcriptional modulation of STAT3 in action of Ruxolitinib and Duloxetine and the formation of novel STAT3 indicating enhanced transcript diversity. The rMATS splicing analysis confirmed dose-dependent modulation, with Duloxetine promoting mild exon skipping at 6.4 μM (IncLevel 0.90 → 0.80) and recovery at 12.8 μM (0.85 → 0.86), while Ruxolitinib induced stronger exon inclusion (0.85 → 1.00,0.94), with broader transcript suppression at 6.4 μM and 12.8 μM, respectively. These findings establish Duloxetine as a dual-action therapeutic that combines neurotransmitter reuptake inhibition with pSTAT3 suppression and isoform-level transcriptomic modulation. This pleiotropic mechanism provides a rationale for its durable analgesic effects and supports repurposing in STAT3-associated disorders.
1. Introduction
Chronic pain is a prevalent, polyetiological illness that burdens patients and healthcare systems worldwide on a physical, emotional, and societal level. Conventional analgesic can work well in chronic and neuropathic pain syndromes but possess adverse effects such as gastrointestinal ulcerations and bleeds, renal insufficiency, and respiratory depression [1,2,3]. This often compels doctors towards seeking alternative treatment options, especially in case of comorbid illness in an elderly patient or late-life depression. Amongst newer options, Duloxetine, a serotonin and norepinephrine reuptake inhibitor (SNRI), has demonstrated clinical efficacy not only in mood disorders but also in pain syndromes that are chronic, such as diabetic peripheral neuropathy, fibromyalgia, and chronic musculoskeletal pain [1,2,3]. Apart from this traditional mechanism of action of SNRI, pleiotropic activity of Duloxetine implies that its analgesic activity is not necessarily attributable to monoamine modulation but could be attributable to other molecular targets.
Studies have reported the Janus kinase 2–signal transducer and activator of transcription 3 (JAK2/STAT3) pathway as a mediator of chronic pain development and maintenance [4,5]. Activation of STAT3, as pSTAT3, triggers gene expression that produces inflammation, neuronal plasticity, and glial activation that result in pain hypersensitivity and central sensitization [6,7,8,9,10]. Aberrant activation of pSTAT3 has been observed in spinal glial cells and neurons in neuropathic and inflammatory conditions; therefore, it is an attractive therapeutic target [8]. Abnormal and recurrent pSTAT3 activation has been documented in spinal microglia and astrocytes, precipitating the release of pro-inflammatory mediators. In rodent models of inflammatory and neuropathic pain, STAT3 phosphorylation blockade decreases mechanical allodynia and thermal hyperalgesia substantially, which proves its pivotal role in maintaining chronic pain states. Inhibition of such a pathway, therefore, has promising therapeutic potential for refractory and chronic pain syndromes [4,8]. To investigate Duloxetine’s potential effects on STAT3 signaling, we used an integrative approach combining In-silico molecular docking and dynamics simulations with In-vitro experimental validation [11].
To strengthen mechanistic understanding, we compared Duloxetine with Ruxolitinib, a clinically established JAK1/2 inhibitor known to suppress STAT3 phosphorylation [12]. This In-vitro analyses revealed that Duloxetine reduced pSTAT3 levels in a manner comparable to Ruxolitinib, suggesting that Duloxetine’s inhibitory effect on STAT3 may be functionally significant and therapeutically relevant. This comparison highlights Duloxetine’s potential as a dual-action agent, providing both central neurotransmitter modulation and peripheral inhibition of STAT3-mediated inflammatory signaling. The ability of Duloxetine to mimic aspects of a canonical STAT3 inhibitor underscores its value as a repurposable drug with pleiotropic therapeutic effects in chronic pain management. This study aims to elucidate the pleiotropic mechanisms of Duloxetine, with particular focus on its analgesic effects mediated through pSTAT3 inhibition, so that rationale for its prescription can be better understood, optimized, and broadened to include patients with pain, cognitive impairment, and late-life depression.
2. Results
2.1. pSTAT3 Molecular Interaction Analyses of Duloxetine and Ruxolitinib
Molecular docking analyses revealed that both Duloxetine and Ruxolitinib established stable interactions within the pSTAT3 binding pocket, but with distinct interaction profiles. Duloxetine formed a conventional hydrogen bond with GLU204, along with Pi–Pi stacking interactions with TYR176 and several Pi–alkyl contacts (LEU179, LYS180, MET200, LEU203, LEU207), indicating a balance of polar and hydrophobic stabilization. In contrast, Ruxolitinib displayed a broader hydrophobic network, characterized by carbon–hydrogen bonding with TYR176, Pi–sulfur interaction with MET200, and multiple alkyl/Pi–alkyl contacts with LEU179, LYS180, and LEU203. Binding energy of Duloxetine with PSTAT3 was −5.83 kcal/mol. Ruxolitinib demonstrated a more extensive hydrophobic stabilization pattern, consistent with its established potency as a JAK/STAT pathway inhibitor. Duloxetine, although not optimized as Ruxolitinib, showed a notable ability to occupy pSTAT3 active site, suggesting a potential modulatory effect on STAT3 signaling in addition to its known pharmacological actions. Binding energy of Ruxolitinib with pSTAT3 was −6.19 kcal/mol.
Both ligands target overlapping residues (TYR176, LEU179, LYS180, MET200, LEU203), but interaction chemistry differs. Duloxetine introduces more polar stabilization (H-bond with GLU204), while Ruxolitinib relies on stronger hydrophobic and sulfur-based contacts, supporting its tighter binding. Figure 1 represents the molecular interaction of Duloxetine and Ruxolitinib.
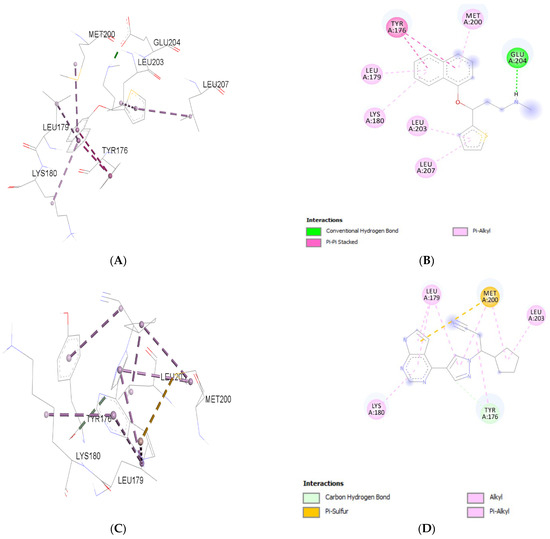
Figure 1.
Molecular interaction analysis of Duloxetine (A,B) and Ruxolitinib (C,D) with p-STAT3.
2.2. STAT3 Molecular Interaction Analyses with Duloxetine
Molecular interaction analyses revealed distinct binding profiles of Duloxetine and Ruxolitinib with STAT3. Duloxetine primarily engaged residues LYS591, ARG609, SER636, PRO639, VAL637, and GLU638. Its binding was stabilized by one conventional hydrogen bond with GLU638, in addition to Pi–cation (LYS591, ARG609), Pi–alkyl (PRO 639), and amide–Pi stacking (VAL 637) interactions. These findings suggest that Duloxetine stabilizes STAT3 through a combination of polar and aromatic interactions. Binding energy of Duloxetine with STAT3 was −5.24 kcal/mol.
In contrast, Ruxolitinib demonstrated stronger polar interactions by forming conventional hydrogen bonds with ILE634, ARG 609, and LYS 591 and carbon hydrogen bond with SER636 and GLU594. It also showed Pi–cation interaction with LYS591 and hydrophobic (alkyl/Pi–alkyl) interactions with surrounding residues PRO639 (Figure 2). Binding energy of Ruxolitinib with STAT3 was −6.06 kcal/mol. This indicates that Ruxolitinib engages STAT3 through a hydrogen-bond–dominant binding pattern, which may contribute to stronger affinity compared to Duloxetine. Table 1 provides details about molecular interaction.
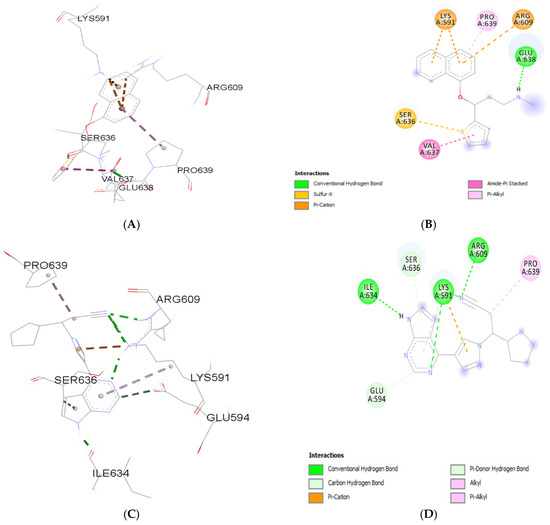
Figure 2.
Molecular interaction analysis of Duloxetine (A,B) and Ruxolitinib (C,D) with STAT3.

Table 1.
Molecular interaction analyses.
2.3. MD Simulation Analyses with Duloxetine and Ruxolitinib
MD simulations were conducted to assess the dynamic stability of STAT3 and pSTAT3 complexes with Duloxetine and Ruxolitinib. Root mean square deviation (RMSD) profiles demonstrated that both STAT3 and pSTAT3 complexes achieved equilibrium during simulation, with fluctuations remaining within a narrow range. Notably, STAT3–Ruxolitinib and pSTAT3–Ruxolitinib exhibited lower RMSD deviations compared to Duloxetine complexes, indicating a more stable conformation of protein–ligand complexes. Solvent Accessible Surface Area (SASA) values remained consistent across the trajectory for both STAT3 and pSTAT3. However, Ruxolitinib-bound forms displayed slightly reduced solvent exposure, suggesting a more compact structural arrangement relative to Duloxetine-bound complexes. Root mean square fluctuation (RMSF) analyses revealed that the binding of both ligands reduced local flexibility of STAT3 and pSTAT3. Furthermore, Ruxolitinib provided stronger stabilization by minimizing residue fluctuations more effectively than Duloxetine.
Radius of Gyration (Rg) values for all complexes were stable within the range of ~3.35–3.45 nm. Both STAT3–Ruxolitinib and pSTAT3–Ruxolitinib consistently maintained marginally lower Rg values, supporting more compact structures. Finally, hydrogen bond analyses highlighted a marked difference: Ruxolitinib formed a greater number of persistent hydrogen bonds with both STAT3 and pSTAT3 throughout 10 ns trajectory, whereas Duloxetine interactions were fewer and more transient, Figure 3.
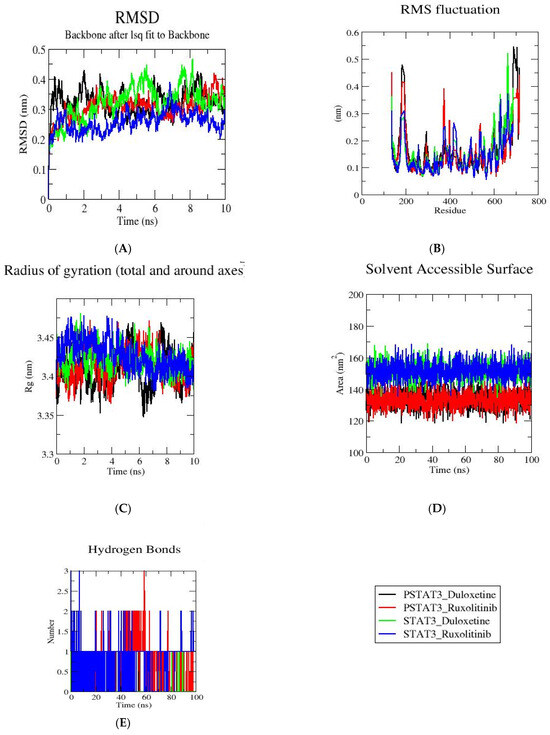
Figure 3.
STAT3 complexes with Duloxetine and Ruxolitinib along a 10 ns trajectory were analyzed using molecular dynamics simulations. (A) RMSD profiles demonstrate general stability of Duloxetine and Ruxolitinib. (B) RMSF plots demonstrate residue fluctuations in presence of Duloxetine and Ruxolitinib. (C) Radius of gyration for Duloxetine and Ruxolitinib. (D) SASA values Duloxetine and Ruxolitinib. (E) Hydrogen bond analysis of Duloxetine and Ruxolitinib.
Overall, MD results confirm that Ruxolitinib stabilizes STAT3 and pSTAT3 more effectively than Duloxetine, as reflected by lower RMSD, reduced solvent exposure, enhanced rigidity, and stronger hydrogen-bonding interactions. Table 2 provides details of the MD simulation.
Table 2.
MD simulation analyses.
2.4. Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) Analyses for pSTAT3 and STAT3 with Duloxetine and Ruxolitinib
MM-PBSA calculations were carried out to compute the binding free energies of Duloxetine and Ruxolitinib with both phosphorylated STAT3 (pSTAT3) and non-phosphorylated STAT3 (STAT3). Results are summarized in Table 3.
Table 3.
MM-PBSA analyses for pSTAT3 and STAT3 with Duloxetine and Ruxolitinib.
For pSTAT3 complexes, Duloxetine showed a slightly more favorable total binding free energy (ΔG = −15.17 kJ·mol−1) than Ruxolitinib (ΔG = −12.98 kJ·mol−1). In both cases, van der Waals interactions were the major favorable contributors (−20.29 and −26.27 kJ·mol−1, respectively). Ruxolitinib had a stronger electrostatic component (ΔEEL = −14.61 kJ·mol−1 vs. −19.16 kJ·mol−1 for Duloxetine), but this advantage was offset by a larger unfavorable solvation energy (ΔGSOLV = +27.89 kJ·mol−1).
In contrast, for STAT3 complexes, Duloxetine formed an unfavorable binding free energy (ΔG = +9.78 kJ·mol−1), whereas Ruxolitinib maintained weak but favorable binding (ΔG = −6.39 kJ·mol−1). However, Ruxolitinib and Duloxetine both showed strong van der Waals and electrostatic interactions (ΔEvdW = −23.54 to −19.84 kJ·mol−1; ΔEEL = −22.18 to −40.98 kJ·mol−1). These contributions were outweighed by highly unfavorable solvation energies (ΔGSOLV = +55.51 and +54.44 kJ·mol−1 for Duloxetine and Ruxolitinib, respectively). This suggests that solvation penalties significantly reduce the binding affinity of both ligands to unphosphorylated STAT3.
Overall, these findings suggest that both Duloxetine and Ruxolitinib bind more favorably to phosphorylated STAT3 compared with non-phosphorylated STAT3, with Duloxetine exhibiting the most stable interaction among the tested complexes.
2.5. C2C12 Cell Culture and MTT Assay
C2C12 murine myoblasts grew normally under standard culture conditions, displaying typical spindle-shaped morphology and reaching ~70–80% confluency before experimental treatment. MTT assay demonstrated a concentration-dependent reduction in cell viability following exposure to Duloxetine and Ruxolitinib. At lower concentrations (0.2–3.2 µM), both drugs caused minimal cytotoxicity. At highest tested concentration (12.8 µM), Duloxetine-treated cells maintained ~80–90% viability, whereas Ruxolitinib-treated cells showed reduced viability of ~70–80% relative to untreated controls. Figures are provided in Supplementary File.
These findings indicate that both Duloxetine and Ruxolitinib modestly reduce C2C12 cell viability, with Ruxolitinib exerting a comparatively stronger inhibitory effect at higher concentrations. MTT assay results are presented in Supplementary File.
2.6. Transcriptomic Clustering of Duloxetine and Ruxolitinib
Analysis of STAT3 (XLOC_008057; chr11:100777631–100861443) expression across treatment groups showed that Ruxolitinib at 12.8 μM (log2FC = 0.38) and 6.4 μM (log2FC = 0.71) did not alter STAT3 expression compared with control. Likewise, Duloxetine at 6.4 μM (log2FC = −0.20) and 12.8 μM (log2FC = −0.04) had no significant effect. Findings show that STAT3 transcript abundance remains constant under all treatment conditions, indicating that post-transcriptional or post-translational mechanisms, such as phosphorylation-dependent activation, may be involved rather than transcriptional modulation of STAT3 in action of Ruxolitinib and Duloxetine. These findings confirm Duloxetine phosphorylation inhibitory potential reported by other studies [13]. A volcano plot of different treatment conditions is provided in Supplementary File.
Hierarchical clustering and heatmap analysis of differentially expressed transcripts demonstrated distinct gene expression profiles following IL6 treatment combined with Duloxetine or Ruxolitinib at two different concentrations (12.8 μM and 6.4 μM). Findings demonstrate two major gene clusters. First and larger cluster contained transcripts that were upregulated (depicted in orange red), and this induction was consistent across both drugs, indicating a broadly shared transcriptional activation program. Upregulated genes included stress-response and signaling regulators such as Ndrg2, Enpp2, Madd, Ucp2, Robo1, and Rbbdl2. The second cluster contained a smaller group of transcripts strongly downregulated after treatment, particularly in Ruxolitinib-exposed cells, as shown in blue shading. Representative suppressed genes included Tle2, Adgrl1, Plekhg2, Col6a1, Shd, and Scr.
Interestingly, samples treated with varying dosages of Ruxolitinib and Duloxetine grouped together, indicating that the transcriptional effects of both drugs are dose consistent. Inter-drug comparison showed that groups treated with Ruxolitinib formed a separate branch from groups treated with Duloxetine, indicating compound-specific variations in gene regulation. However, combined data show that whereas both medications significantly reprogram transcription when used in conjunction with IL6, Duloxetine mainly increases gene activation, while Ruxolitinib induces a higher suppressive response. Ruxolitinib’s stronger suppressive action may account for its effectiveness in treating resistant graft-vs-host disease (GVHD), polycythemia vera, and myelofibrosis, Figure 4.
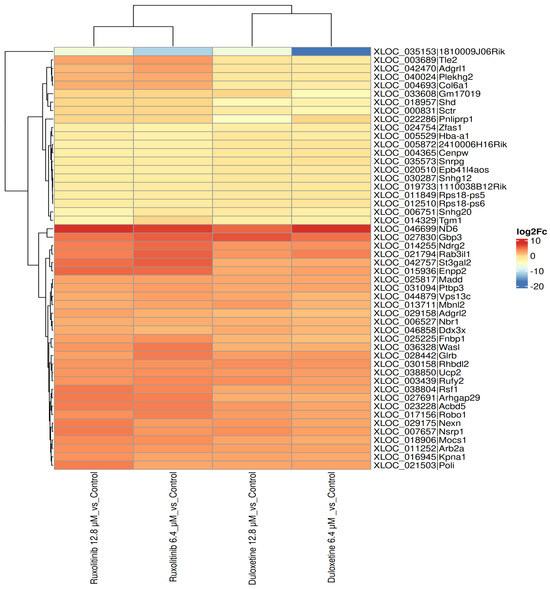
Figure 4.
Heatmap representation of differential gene expression profiles (log2 fold change) in IL-6 treated samples exposed to Duloxetine or Ruxolitinib at 12.8 μM and 6.4 μM compared with untreated controls. Each column represents a treatment condition, and each row corresponds to a transcript. The color index ranges from –20 to +10. Hierarchical clustering identified two major gene clusters: Downregulated group (blue) predominantly suppressed under Ruxolitinib treatment and an upregulated group (orange–red) commonly induced across both drugs. Duloxetine- and Ruxolitinib-treated samples clustered separately, indicating drug-specific transcriptional signatures, although both concentrations within each drug formed tight subclusters, reflecting dose consistency. Collectively, these findings suggest Duloxetine and Ruxolitinib followed by IL6 pharmacological intervention produces distinct but overlapping gene expression programs, with Ruxolitinib exerting a comparatively stronger downregulatory influence.
2.7. Heatmap Analysis of STAT3 Isoforms Across Duloxetine and Ruxolitinib Treatments
Heatmap analysis of STAT3 isoforms revealed distinct, dose-dependent expression patterns across treatment groups. Canonical isoforms showed relatively stable expression under both Duloxetine and Ruxolitinib treatments, indicating that baseline STAT3 transcription remains largely unchanged. In contrast, several novel isoforms, annotated with class code “j”, exhibited pronounced differential regulation, emphasizing impact of alternative splicing in response to pharmacological intervention.
For Duloxetine, lower dose (6.4 µM) produced modest changes in isoform expression, with select novel isoforms showing slight up or downregulation. At higher dose (12.8 µM), several novel isoforms were found to be upregulated, suggesting that increased drug exposure promotes transcript diversity and selectively enhances specific splice variants. Similarly, Ruxolitinib demonstrated dose-dependent modulation of STAT3 isoforms: Ruxolitinib at 6.4 µM dose elicited minor changes in novel isoforms, while at 12.8 µM dose, it induced stronger differential regulation, with certain novel transcripts significantly upregulated, indicating that JAK-STAT pathway inhibition can influence STAT3 splicing in a dose-specific manner.
Overall, these results show that the expression of the STAT3 isoform is modulated in a dose-dependent pattern by both Duloxetine and Ruxolitinib, with novel transcripts contributing substantially to observed variability (Table 4). These patterns suggest that pharmacological interventions can differentially influence STAT3 transcript diversity, potentially impacting downstream signaling and functional cellular responses, Figure 5.
Table 4.
Transcript assembly and genomic coordinates of STAT3 isoforms.
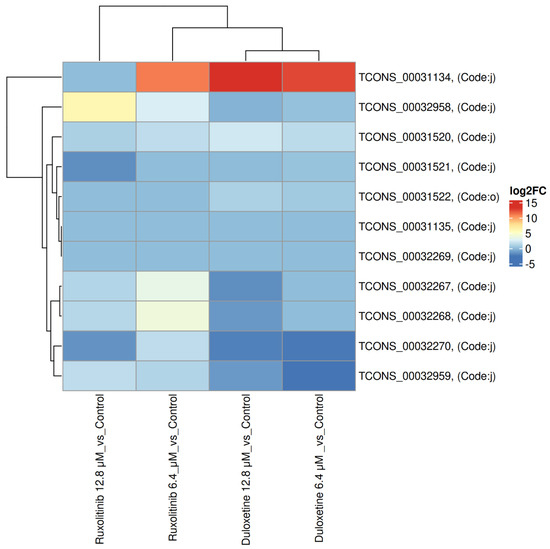
Figure 5.
Heatmap of STAT3 isoform expression across Duloxetine and Ruxolitinib treatments. Heatmap depicting expression patterns of STAT3 isoforms under treatment with Duloxetine (6.4 and 12.8 µM) and Ruxolitinib (6.4 and 12.8 µM). The color index ranges from –5 to +15. Canonical isoforms remained relatively stable across treatments, whereas novel isoforms (class code “j”) showed pronounced differential regulation. Duloxetine at 12.8 µM induced marked upregulation of specific novel isoforms, while 6.4 µM dose produced modest changes. Ruxolitinib exhibited a similar dose-dependent effect, with stronger upregulation of certain novel isoforms at 12.8 µM. These data indicate that both pharmacological interventions modulate STAT3 isoform expression in a dose- and treatment-specific manner, highlighting potential impact of transcript diversity on downstream signaling.
2.8. Replicate Multivariate Analysis of Transcript Splicing (rMATSv4.30) Junction Analysis of STAT3 Isoforms Across Duloxetine and Ruxolitinib Treatments
Splicing analyses further demonstrated distinct treatment-dependent effects. In the case of Duloxetine, splicing modulation was subtle across the chr11:100780712–100784112 region. Control samples exhibited an IncLevel of 0.90, which decreased slightly to 0.80 under Duloxetine 6.4 µM but returned close to baseline (0.86) under Duloxetine 12.8 µM, suggesting mild and reversible effects on exon usage. In event spanning chr11:100780712–100780754 and downstream exons at chr11:100780304–100780366/416, control samples showed substantial exon inclusion (IncLevel 0.85), whereas Ruxolitinib at 6.4 µM resulted in complete inclusion (IncLevel 1.00), suggesting enhanced splicing fidelity. For event linking chr11:100780712–100780754 to chr11:100783900–100784112, control samples again showed strong inclusion (IncLevel 0.90), but Ruxolitinib at 12.8 µM reduced inclusion to 0.94, indicating a dose-dependent suppression of exon incorporation.
Overall, rMATS junction analyses validate heatmap findings by demonstrating that both Duloxetine and Ruxolitinib reshape STAT3 isoform diversity in a dose-dependent manner. While Duloxetine induces modest exon skipping at low concentrations and enhanced inclusion of novel variants at higher doses, Ruxolitinib exerts a stronger and more uniform effect, driving increased inclusion across multiple splice junctions. These findings emphasize role of novel “j” isoforms as key contributors to STAT3 variability and suggest that drug-induced alternative splicing may act as an additional regulatory mechanism with significant consequences for STAT3-mediated signaling, Figure 6.
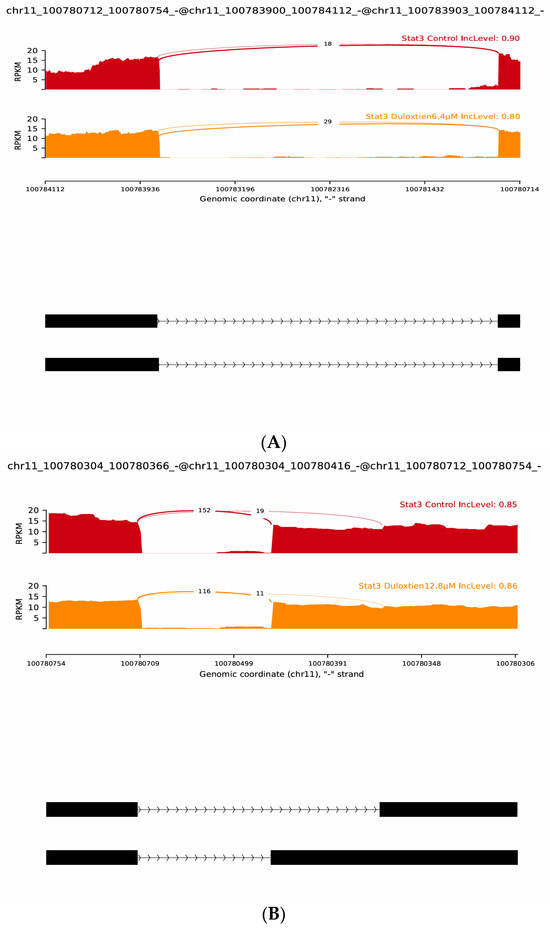
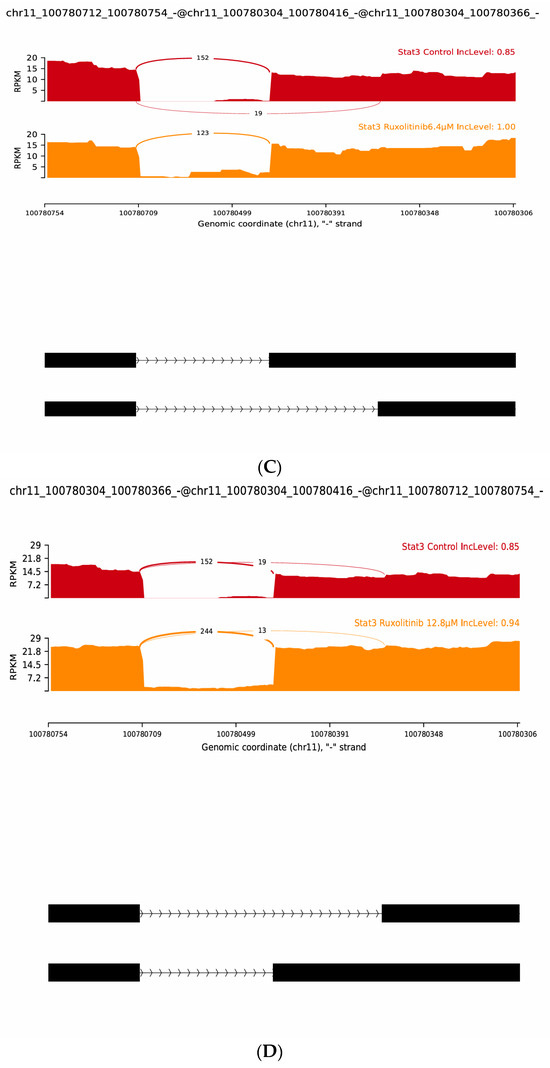
Figure 6.
Representative rMATS sashimi plots of STAT3 alternative splicing events under Duloxetine and Ruxolitinib treatments. (A) Control versus Duloxetine 6.4 µM showing reduced exon inclusion at chr11:100780712–100784112 (IncLevel 0.90 → 0.80). (B) Control versus Duloxetine 12.8 µM showing slight inclusion increase at chr11:100780304–100780754 (IncLevel 0.85 → 0.86). (C) Control versus Ruxolitinib 6.4 µM showing enhanced exon inclusion at chr11:100780712–100780366 (IncLevel 0.85 → 1.00). (D) Control versus Ruxolitinib 12.8 µM showing inclusion gain at chr11:100780304–100780754 (IncLevel 0.85 → 0.94). Together, these analyses highlight dose-dependent and drug-specific regulation of STAT3 splicing, with novel isoforms contributing significantly to transcriptomic diversity.
2.9. GO and Pathway Enrichment Profiles of Duloxetine and Ruxolitinib
To explore the molecular impact of Duloxetine and Ruxolitinib at two concentrations (6.4 µM and 12.8 µM), we performed Gene Ontology (GO) enrichment and KEGG pathway analyses in comparison with untreated controls. Duloxetine treatment at 6.4 µM was associated with significant enrichment of pathways related to mitochondrial activity, oxidative phosphorylation, and protein quality control. Notably, GO categories highlighted alterations in cellular respiration and electron transport chain activity, while KEGG pathways emphasized modulation of neuroactive ligand–receptor interactions and metabolic signaling. At the higher concentration of 12.8 µM, the impact of Duloxetine became more extensive, with enrichment of processes involved in apoptotic signaling, unfolded protein response, and transcriptional regulation. Importantly, KEGG analysis at this dose revealed activation of MAPK and calcium signaling pathways as well as synaptic regulatory mechanisms, suggesting a dose-dependent shift toward stress-responsive and signaling networks.
In contrast, Ruxolitinib demonstrated a distinct enrichment pattern consistent with its known pharmacological actions. At 6.4 µM, it prominently affected cytokine-mediated signaling, immune regulation, and JAK–STAT–associated processes. KEGG analysis further underscored significant involvement of cytokine–cytokine receptor interactions and JAK–STAT signaling. Increasing dose to 12.8 µM amplified these effects and extended enrichment profile to include inflammatory responses, apoptotic regulation, and cell cycle control. Correspondingly, KEGG pathways at this concentration highlighted PI3K–Akt signaling, p53 pathway, and additional immune-related signaling cascades, indicating broader immunomodulatory and stress-regulatory effects at higher drug exposure.
Taken together, comparative analyses showed that Duloxetine predominantly modulated neuronal, mitochondrial, and metabolic pathways, whereas Ruxolitinib targeted immune and cytokine-driven networks. Both drugs exhibited clear dose-dependent effects, with higher concentrations leading to engagement of apoptosis, transcriptional regulation, and cell cycle control. Nevertheless, overlap between Duloxetine- and Ruxolitinib-associated pathways was minimal, underscoring distinct mechanistic profiles: Duloxetine exerted its influence primarily through neuro-metabolic and stress-related signaling, while Ruxolitinib acted chiefly through immunomodulatory and cytokine-regulated pathways, Figure 7.
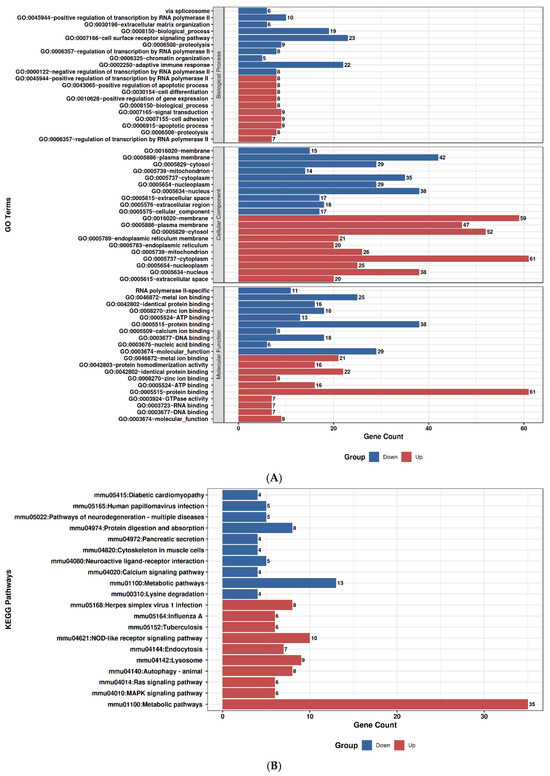
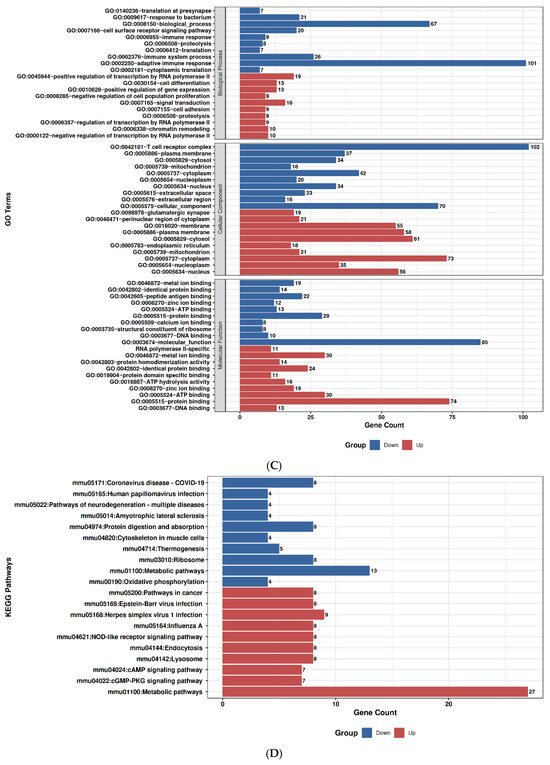
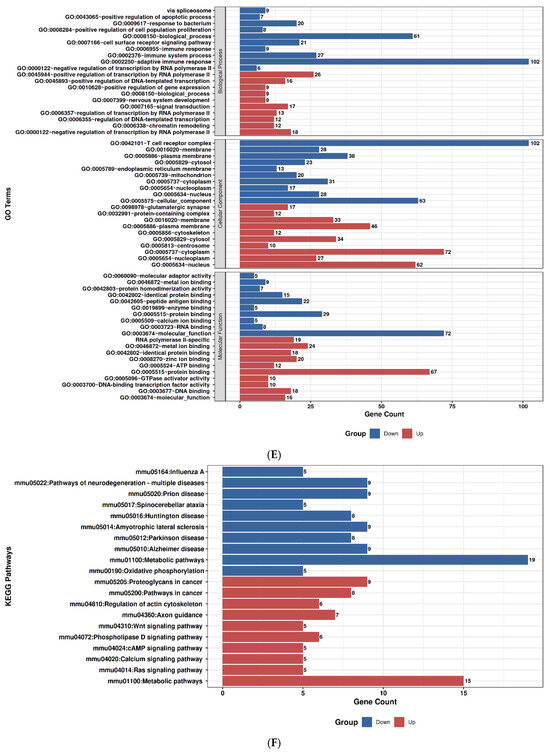
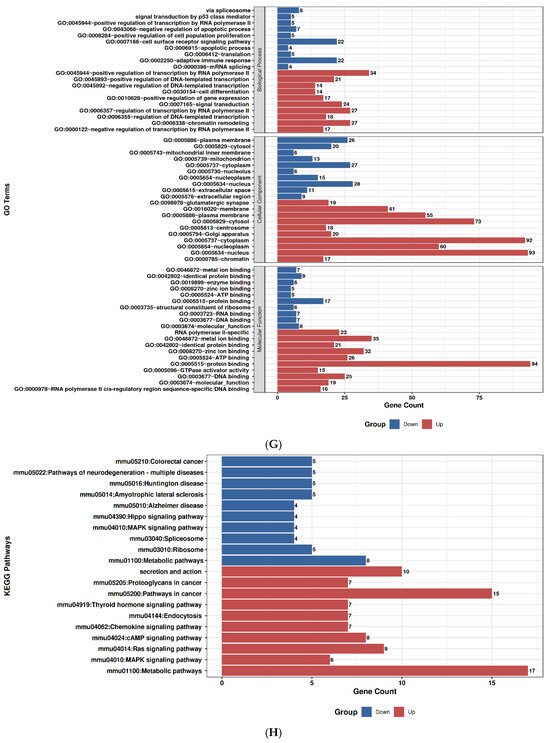
Figure 7.
Gene Ontology (GO) and KEGG pathway enrichment analysis of Duloxetine and Ruxolitinib at two concentrations compared with control. (A) Duloxetine 6.4 µM–GO enrichment. (B) Duloxetine 6.4 µM–KEGG enrichment. (C) Duloxetine 12.8 µM–GO enrichment. (D) Duloxetine 12.8 µM–KEGG enrichment. (E) Ruxolitinib 6.4 µM–GO enrichment. (F) Ruxolitinib 6.4 µM–KEGG enrichment. (G) Ruxolitinib 12.8 µM–GO enrichment. (H) Ruxolitinib 12.8 µM–KEGG enrichment.
2.10. Duloxetine Suppresses Il-6–Induced STAT3 Phosphorylation in C2C12 Cells
In both pSTAT3 and STAT3 expression analyses, treatment with Duloxetine and Ruxolitinib led to a marked reduction compared to control groups. DMSO did not cause any significant change when compared with control, confirming that the solvent had no effect on protein expression. pSTAT3 and STAT3 levels were significantly reduced by Duloxetine administration at 12.8 µM (p < 0.0001), and while a substantial suppression was also observed at 6.4 µM (p < 0.0001), the effect was a bit smaller, suggesting a dose-dependent inhibition. Similarly, Ruxolitinib treatment at both 12.8 µM and 6.4 µM caused a profound reduction in pSTAT3 and STAT3 expression (p < 0.0001). Notably, Ruxolitinib exhibited a stronger inhibitory effect in comparison with Duloxetine at corresponding concentrations, with most prominent reduction observed at 12.8 µM. Overall, these results demonstrate that both Duloxetine and Ruxolitinib effectively inhibit total STAT3 and its phosphorylated form, with Ruxolitinib exerting a more potent suppressive effect compared to Duloxetine, Figure 8. Raw files of Western blot are provided in the Supplementary File.
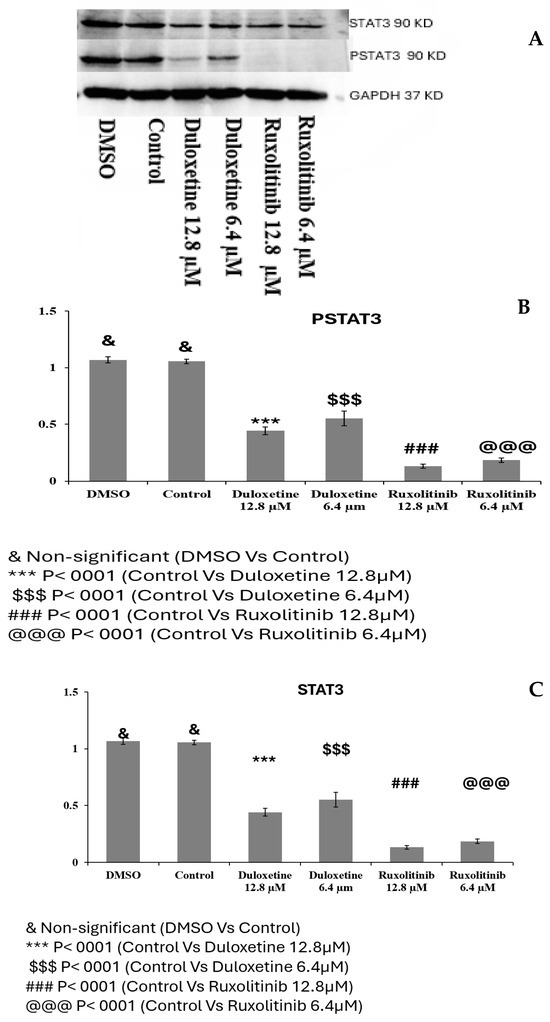
Figure 8.
Effect of Duloxetine and Ruxolitinib on pSTAT3 and STAT3 expression in C2C12 cells. C2C12 cells were treated with Duloxetine (12.8 µM and 6.4 µM) and Ruxolitinib (12.8 µM and 6.4 µM), (A) Western blot showing expression of pSTAT3 and STAT3. (B) Relative expression of pSTAT3 (C) Relative expression of STAT3. DMSO-treated cells showed no significant change compared with untreated control, confirming that solvent had no effect. Duloxetine treatment significantly reduced pSTAT3 and STAT3 expression in a dose-dependent manner, with greatest suppression observed at 12.8 µM (*** p < 0.0001 vs. control; $$$ p < 0.0001 vs. control). Ruxolitinib caused a more pronounced inhibition than Duloxetine at both concentrations, with lowest levels of pSTAT3 and STAT3 observed at 12.8 µM (### p < 0.0001; @@@ p < 0.0001 vs. control). Data are presented as mean ± SEM of independent experiments.
3. Discussion
This study provides integrative In-silico and In-vitro evidence that Duloxetine, an SNRI widely used to treat depressive disorder and for chronic pain, exerts a pleiotropic mechanism of action by directly targeting phosphorylated STAT3 (pSTAT3) signaling in addition to its canonical role in neurotransmitter reuptake inhibition. By combining molecular docking, molecular dynamics (MD) simulations, MM-PBSA binding energy estimations, cellular assays, and transcriptomic profiling, we establish Duloxetine as a dual-function therapeutic agent with potential advantages over selective JAK/STAT inhibitors such as Ruxolitinib [13]. However, earlier Duloxetine efficacy to prevent activation of p38 phosphorylation has been reported [13]. Our docking studies demonstrated that Duloxetine binds stably within pSTAT3 pocket through a combination of hydrogen bond (notably GLU204) and hydrophobic interactions, although its binding profile is different from Ruxolitinib, which relied more extensively on hydrophobic stabilization. Additionally, common interacting residues for STAT3 was LYS 591, which stabilized with Pi-cation LYS 591.
MD simulations further confirmed that both drugs achieve equilibrium with pSTAT3, STAT3, but Ruxolitinib produced a more compact and stable protein–ligand complex with lower RMSD and higher hydrogen bond persistence. Nonetheless, MM-PBSA free energy analyses revealed that Duloxetine exhibits more favorable binding to pSTAT3 (ΔG = −15.17 kJ·mol−1) than Ruxolitinib (ΔG = −12.98 kJ·mol−1). This suggests that Duloxetine selectively stabilizes interactions with activated STAT3, providing a potential mechanism for targeted inhibition of pathological STAT3 signaling.
In-vitro, Duloxetine efficiently and dose-dependently prevented phosphorylation of STAT3 in C2C12 cells caused by IL-6. Duloxetine significantly reduced pSTAT3 levels in Western blot at both 6.4 µM and 12.8 µM, although it has been observed that Ruxolitinib had strong inhibitory impact at same dose. Furthermore, MTT viability assays revealed that Duloxetine maintained higher cell survival (~80–90% at 12.8 µM) compared with Ruxolitinib (~70–80%), indicating that Duloxetine achieves STAT3 suppression with less cytotoxicity. This balance between efficacy and cellular safety highlights a key translational benefit of Duloxetine over more potent but less tolerable JAK/STAT inhibitors.
In addition, RNA sequencing uncovered drug specific gene expression programs. Duloxetine appears to modulate mitochondrial function, oxidative phosphorylation, metabolic pathways, and synaptic signaling, suggesting a potential link between its effects and neuroenergetic and stress-response processes. Specifically, long treatment with Duloxetine has been shown to influence mitochondrial dynamics and energy metabolism in neural tissues [13,14,15,16]. Additionally, the antidepressant Fluoxetine’s impact on synaptic signaling pathways and mitochondrial features has been observed in a previous study [17]. While direct evidence of Duloxetine’s interaction with STAT3 is limited, its modulation of related pathways, such as the IL-6 signaling pathway, suggests a potential involvement in the JAK/STAT3 pathway [2,13]. Furthermore, Duloxetine’s effects on oxidative stress and apoptosis [2,13,16,18], as well as its influence on neuroplasticity and synaptic signaling [2,13,18], support the hypothesis that Duloxetine may enhance neuroenergetic and stress-response processes through mechanisms that could involve STAT3 signaling. Ruxolitinib predominantly suppressed immune- and cytokine-related signaling, consistent with its pharmacological role as a JAK1/2 inhibitor [12,19]. Heatmap and clustering analyses confirmed these distinct signatures: Duloxetine promoted gene activation and stress-response regulators (e.g., Ndrg2, Ucp2), while Ruxolitinib induced stronger downregulation of structural and regulatory genes (e.g., Tle2, Col6a1).
A novel finding of this study is the isoform-level regulation of STAT3. Duloxetine promoted transcript diversity, particularly at higher concentrations, by enhancing the expression of several novel splice variants, notably the “j” class, consistent with reports that antidepressants can modulate alternative splicing [20,21]. In contrast, Ruxolitinib induced more pronounced exon inclusion events, as revealed by rMATS junction analysis, promoting splicing fidelity while limiting transcript diversity. These observations suggest that both Duloxetine and Ruxolitinib reshape STAT3 signaling not only through inhibition of phosphorylation but also via modulation of alternative splicing. Notably, Duloxetine appears to favor isoform diversification, potentially broadening its regulatory influence on downstream signaling pathways, whereas Ruxolitinib may reinforce specific splicing patterns, leading to more constrained functional outcomes.
Collectively, these findings offer a theory in which Duloxetine couples its classical role as an SNRI with direct suppression of pSTAT3 activation and regulation of STAT3 transcript diversity. Duloxetine exerts its effects through interconnected pathways involving neurotransmission, mitochondrial metabolism, and stress-response regulation, which may underline its sustained clinical efficacy in chronic pain syndromes with both neuropathic and inflammatory components. Furthermore, its favorable balance of efficacy and cell viability suggests a wider therapeutic window compared to selective JAK inhibitors. Future studies should extend validation to neuronal and glial cell types, as well as in vivo models of chronic pain. Moreover, while RNA-seq revealed robust isoform-level regulation, functional characterization of these novel STAT3 splice variants is important to determine its specific roles in signaling and pain modulation.
4. Materials and Methods
4.1. Structural Preparation for Molecular Docking
To investigate the potential interaction of Duloxetine with molecular targets implicated in chronic pain, crystal structures of PSTAT3 and STAT3 were retrieved from the Protein Data Bank (PDB). All water molecules were removed from crystallographic structures to prevent interference with ligand binding. Hydrogen atoms, Kollman united charges, and solvation parameters were added for the molecular docking analyses. Docking was conducted using AutoDock 4.2, employing Lamarckian Genetic Algorithm to explore binding conformations. For each protein–ligand system, 50 conformational poses were generated, and pose with lowest binding energy was selected for molecular dynamics simulation [11,22].
Ruxolitinib, a known JAK/STAT pathway inhibitor, was docked in parallel as a reference to validate docking results of Duloxetine, a comparative benchmark for Duloxetine’s binding affinities and interaction profiles.
4.2. Molecular Dynamics (Md) Simulations
To refine docking results and evaluate dynamic stability of protein–ligand complexes, molecular dynamics (MD) simulations were performed using GROMACS 2020.6 with the CHARMM36 force field. Top-ranked docking poses of Duloxetine and control ligand Ruxolitinib bound to pSTAT3 and STAT3 were used for MD simulation. Protein–ligand complexes were solvated with simple point charge (SPC) water molecules, and counter ions (Na+ or Cl−) were added to neutralize systems. Energy minimization was carried out to eliminate unfavorable van der Waals interactions between atoms. Each complex was equilibrated for 10 ns in two steps: (i) under constant number of particles, volume, and temperature (NVT ensemble), and (ii) under constant number of particles, pressure, and temperature (NPT ensemble) at 310 K, mimicking physiological temperature. The LINear Constraint Solve (LINCS) algorithm was applied to constrain covalent bond lengths. Subsequently, 10 ns MD production run was carried out for each system to investigate stability and conformational dynamics of Duloxetine and Ruxolitinib with pSTAT3 and STAT3. Trajectory analysis was performed using GROMACS utilities (gmx rms, gmx rmsf, gmx gyrate, gmx sasa, gmx hbond) to evaluate root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonding interactions [11,22,23].
4.3. Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) Analysis
The MM-PBSA computations were carried out to enhance molecular docking results and acquire more accurate estimations of binding free energies. The GROMACS 2020.6 package with the CHARMM36 force field was used to perform molecular dynamics (MD) simulations in explicit solvent on docked protein–ligand complexes.
Resulting trajectories were analyzed with g_mmpbsa tool to estimate binding free energy (ΔG_bind) according to the following equation:
where ΔG bind is the binding free energy of receptor (pSTAT3, STAT3) with Duloxetine and Ruxolitinib. G protein represents (pSTAT3, STAT3), whereas G ligand represents binding energy of Duloxetine and Ruxolitinib [11,23].
ΔG bind = G complex − (G Protein + G Ligand)
4.4. Invitro Functional Testing
4.4.1. C2C12 Cell Culture
C2C12 mouse myoblast cells (American Type Culture Collection, ATCC, Manassas, VA, USA; Cat. No. CRL-1772™) were grown in standard conditions in DMEM culture medium containing 10% FBS and 1% penicillin-streptomycin. The cells were maintained at 37 °C in a humidified incubator under 5% CO2 for appropriate densities prior to experimental treatment [24].
4.4.2. MTT Assay
Cell viability was assessed using MTT assay kit (Abcam, Waltham, MA, USA, cat. no. ab197010). Duloxetine and Ruxolitinib were treated at various concentrations ranging from 0.2 to 12.8 µM for C2C12 cells. In addition to this, inflammatory cytokine activation was also carried out by utilizing IL-6 at a final concentration of 100 ng/mL each. After treatment period, MTT reagent was added to culture medium and incubated to allow for formation of formazan crystals that were subsequently dissolved and assayed spectrophotometrically at 570 nm. Relative cell viability was measured in comparison to untreated controls [18,24].
4.4.3. Cell Line Treatment, RNA Isolation and Western Blot Analysis
C2C12 cells were cultured under standard conditions and subjected to pharmacological treatments. Cells were pretreated with Duloxetine and Ruxolitinib at the concentration of 6.4 µM and 12.6 µM, followed by stimulation with interleukin-6 (IL-6, 100 ng/mL) for the designated time periods of 40 min. After treatment, total RNA was isolated using RNA isolation kit (Qiagen, Germantown, MD, USA, Cat. No. 217004) according to the manufacturer’s protocol, and RNA integrity was confirmed by spectrophotometric analysis.
For protein expression studies, Western blot was performed on C2C12 cells following treatment with Duloxetine, Ruxolitinib, and IL-6. Total protein was extracted using RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was determined by Pierce TM BCA pierce assay kit (ThermoFisher Scientific, Waltham, MA, USA, Catalog number 23225). The equal amounts of protein were resolved by 10% SDS-PAGE before and transferred to PVDF membranes. Membranes were blocked with 5% BSA and probed with primary antibodies against target proteins of interest, followed by incubation with HRP-conjugated secondary antibodies. Protein bands were visualized using enhanced chemiluminescence and quantified densitometrically, as per previously optimized methods [17,24,25]. Statistical analyses were performed using SPSS software version 22.0 (IBM Corp., Armonk, NY, USA). The Student’s t-test was used, and differences were considered statistically significant at p < 0.05.
4.4.4. RNA Sequencing
To evaluate transcriptional alterations, RNA sequencing (RNA-seq) was conducted. C2C12 cells were treated with Duloxetine and Ruxolitinib with 6.4 µM and 12.8 µM for 24 h, and IL6 (100 ng/mL) was given to stimulate PSTAT3. High-quality RNA samples were used for library preparation and sequenced on an Illumina platform. Differential gene expression analyses were carried out to determine transcriptomic changes induced by treatments. Pathway enrichment analyses were performed to identify significantly altered biological processes and molecular signaling networks [17].
Raw RNA-sequencing reads obtained from treated and control C2C12 cells were subjected to a comprehensive bioinformatics pipeline. Initial quality of reads was assessed using FastQC v0.11.9, and low-quality bases and adapter sequences were removed using Trimmomatic. v0.39. High-quality reads were aligned to mouse reference genome (mm39) using HISAT2 v2.2.1, and aligned reads were sorted and indexed with SAMtools v1.17. Gene expression quantification was performed using feature counts with annotation from NCBI RefSeq mouse genome. Differential gene expression analysis between experimental groups was carried out using DESeq2 in R. Genes with an −log10 (p-value) < 0.05 and a log2 fold change ≥ |1| were considered significantly differentially expressed [26].
To classify assembled transcripts relative to reference annotation, gffcompare was used. This program compares predicted transcripts with reference genome and assigns class codes that describe the relationship to known isoforms [27]. Through this approach, novel isoforms were detected primarily as transcripts with code “j”, which represent new splice variants of known genes. Similarly, novel exons were identified in transcripts with class codes such as “i” or “o”, indicating the presence of expressed exonic regions not annotated in reference genome. These codes include the following:
| Code | Meaning |
| = | Exact match with reference transcript |
| C | Contained in reference transcript |
| J | Potential novel isoform (shares at least one splice junction with reference) |
| I | Fully within a reference intron |
| O | Generic exonic overlap with reference (partial match) |
| U | Unknown/intergenic—likely a novel gene |
| X | Exonic overlap on opposite strand |
| E | Single exon transcript overlapping a reference exon |
| P | Possible polymerase run-on transcript |
| S | Opposite strand, intronic |
| . | No class code assigned |
4.4.5. Replicate Multivariate Analysis of Transcript Splicing (rMATS) Junction Analyses
The rMATSv4.3.0 was used to conduct alternative splicing analysis. Each biological sample’s aligned BAM files and GTF annotation (Ensembl GRCm38/mm10 GTF) were used for analyses to find differences in exon and splice-junction alterations [28,29].
5. Conclusions
This study establishes Duloxetine pleiotropic mechanism as pain relief via inhibiting pSTAT3 signaling while maintaining favorable cell viability and inducing transcriptomic reprogramming. By combining monoamine reuptake inhibition with direct modulation of STAT3 phosphorylation and splicing, Duloxetine offers a mechanistic rationale for its durable analgesic effects in chronic pain. These insights open avenues for repurposing Duloxetine in pain conditions with inflammatory and neuroenergetic components, suggesting potential benefits in combination strategies with selective STAT3 modulators. However, lack of In-vivo and clinical confirmation is a key limitation of this study. Since In- silico findings cannot demonstrate relationships, they only support In-vitro evidence of Duloxetine’s inhibitory effect on pSTAT3.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms262110432/s1.
Author Contributions
Conceptualization, S.A.H.A.; methodology, S.A.H.A.; software, S.A.H.A.; validation, S.A.H.A., J.Y.W. and G.A.; formal analysis, S.A.H.A.; investigation, S.A.H.A., J.Y.W., X.Z. and G.A.; resources, S.A.H.A., J.Y.W. and G.A.; data curation, S.A.H.A., J.Y.W. and G.A.; writing—S.A.H.A.; J.Y.W., G.A.; original draft preparation, S.A.H.A.; review and editing, S.A.H.A., J.Y.W., X.Z. and G.A.; visualization, S.A.H.A.; supervision, J.Y.W., X.Z. and G.A.; project administration, J.Y.W. and G.A.; funding acquisition, J.Y.W. and G.A. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to acknowledge the partial support provided by the Lyon Research Aging Program, the Reynolds Institute on Aging, and the University of Arkansas for Medical Sciences.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article and Supplementary Material. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Huang, Y.; Chen, H.; Chen, S.-R.; Pan, H.-L. Duloxetine and Amitriptyline Reduce Neuropathic Pain by Inhibiting Primary Sensory Input to Spinal Dorsal Horn Neurons via α1- and α2-Adrenergic Receptors. ACS Chem. Neurosci. 2023, 14, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, M.K.; Helmy, S.A.; Badran, D.I.; Zaitone, S.A. Neuroprotective Effect of Duloxetine in a Mouse Model of Diabetic Neuropathy: Role of Glia Suppressing Mechanisms. Life Sci. 2018, 205, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Obata, H. Strategies to Treat Chronic Pain and Strengthen Impaired Descending Noradrenergic Inhibitory System. Int. J. Mol. Sci. 2019, 20, 822. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, E.; Rivat, C.; Pommier, B.; Mauborgne, A.; Pohl, M. JAK/STAT3 Pathway Is Activated in Spinal Cord Microglia after Peripheral Nerve Injury and Contributes to Neuropathic Pain Development in Rat. J. Neurochem. 2008, 107, 50–60. [Google Scholar] [CrossRef]
- Liu, X.; Tian, Y.; Lu, N.; Gin, T.; Cheng, C.H.K.; Chan, M.T.V. Stat3 Inhibition Attenuates Mechanical Allodynia through Transcriptional Regulation of Chemokine Expression in Spinal Astrocytes. PLoS ONE 2013, 8, e75804. [Google Scholar] [CrossRef]
- Park, J.; Lee, C.; Kim, Y.T. Effects of Natural Product-Derived Compounds on Inflammatory Pain via Regulation of Microglial Activation. Pharmaceuticals 2023, 16, 941. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Wu, M.; Ru, Y.; Meng, Y.; Zhang, X.; Wang, F.; Xia, Z.; Yang, L.; Zhai, Y.; Li, G.; et al. A Novel Compound DBZ Alleviates Chronic Inflammatory Pain and Anxiety-like Behaviors by Targeting the JAK2-STAT3 Signaling Pathway. J. Biol. Chem. 2025, 301, 110223. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, C.S.; Seo, K.J.; Kim, I.Y.; Han, S.; Youn, I.; Yune, T.Y. IL-6/JAK2/STAT3 Axis Mediates Neuropathic Pain by Regulating Astrocyte and Microglia Activation after Spinal Cord Injury. Exp. Neurol. 2023, 370, 114576. [Google Scholar] [CrossRef]
- Tang, Q.; Shen, Q.; Wu, L.; Feng, X.; Liu, H.; Wu, B.; Huang, X.; Wang, G.; Li, Z.; Liu, Z. STAT3 Signal That Mediates the Neural Plasticity Is Involved in Willed-Movement Training in Focal Ischemic Rats. J. Zhejiang Univ. Sci. B 2016, 17, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Berta, T.; Nedergaard, M. Glia and Pain: Is Chronic Pain a Gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef]
- Abdi, S.A.H.; Ali, A.; Sayed, S.F.; Nagarajan, S.; Abutahir; Alam, P.; Ali, A. Sunscreen Ingredient Octocrylene’s Potency to Disrupt Vitamin D Synthesis. Int. J. Mol. Sci. 2022, 23, 10154. [Google Scholar] [CrossRef]
- Song, H.; Cui, Y.; Zhang, L.; Cao, G.; Li, L.; Li, G.; Jia, X. Ruxolitinib Attenuates Intimal Hyperplasia via Inhibiting JAK2/STAT3 Signaling Pathway Activation Induced by PDGF-BB in Vascular Smooth Muscle Cells. Microvasc. Res. 2020, 132, 104060. [Google Scholar] [CrossRef]
- Meng, J.; Zhang, Q.; Yang, C.; Xiao, L.; Xue, Z.; Zhu, J. Duloxetine, a Balanced Serotonin-Norepinephrine Reuptake Inhibitor, Improves Painful Chemotherapy-Induced Peripheral Neuropathy by Inhibiting Activation of p38 MAPK and NF-κB. Front. Pharmacol. 2019, 10, 365. [Google Scholar] [CrossRef]
- Edtmayer, S.; Witalisz-Siepracka, A.; Zdársky, B.; Heindl, K.; Weiss, S.; Eder, T.; Dutta, S.; Graichen, U.; Klee, S.; Sharif, O.; et al. A Novel Function of STAT3β in Suppressing Interferon Response Improves Outcome in Acute Myeloid Leukemia. Cell Death Dis. 2024, 15, 369. [Google Scholar] [CrossRef]
- Jetsonen, E.; Didio, G.; Suleymanova, I.; Teino, I.; Castrén, E.; Umemori, J. Chronic Treatment with Fluoxetine Regulates Mitochondrial Features and Plasticity-Associated Transcriptomic Pathways in Parvalbumin-Positive Interneurons of Prefrontal Cortex. Neuropsychopharmacology 2025, 50, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.C.; Paladini, M.S.; Brüning, C.A.; Spero, V.; Cattaneo, M.G.; Racagni, G.; Papp, M.; Riva, M.A.; Molteni, R. Involvement of the IL-6 Signaling Pathway in the Anti-Anhedonic Effect of the Antidepressant Agomelatine in the Chronic Mild Stress Model of Depression. Int. J. Mol. Sci. 2022, 23, 12453. [Google Scholar] [CrossRef]
- Abdi, S.A.H.; Afjal, M.A.; Najmi, A.K.; Raisuddin, S. S-Allyl Cysteine Ameliorates Cyclophosphamide-Induced Downregulation of Urothelial Uroplakin IIIa with a Concomitant Effect on Expression and Release of CCL11and TNF-α in Mice. Pharmacol. Rep. 2018, 70, 769–776. [Google Scholar] [CrossRef]
- Jang, S.-K. Duloxetine Enhances the Sensitivity of Non-Small Cell Lung Cancer Cells to EGFR Inhibitors by REDD1-Induced MTORC1/S6K1 Suppression. Am. J. Cancer Res. 2024, 14, 1087–1100. [Google Scholar] [CrossRef]
- Febvre-James, M.; Lecureur, V.; Augagneur, Y.; Mayati, A.; Fardel, O. Repression of Interferon β-Regulated Cytokines by the JAK1/2 Inhibitor Ruxolitinib in Inflammatory Human Macrophages. Int. Immunopharmacol. 2018, 54, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Adeosun, S.O.; Zhao, X.; Hill, R.; Zheng, B.; Reddy, R.; Su, X.; Meyer, J.; Mosley, T.; Wang, J.M. ERβ Agonist Alters RNA Splicing Factor Expression and Has a Longer Window of Antidepressant Effectiveness than Estradiol after Long-Term Ovariectomy. J. Psychiatry Neurosci. 2019, 44, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.R.; Oh, D.H.; Kim, S.H.; Jung, K.H.; Das, N.D.; Chai, Y.G. Fluoxetine Increases the Expression of NCAM140 and PCREB in Rat C6 Glioma Cells. Psychiatry Investig. 2012, 9, 180. [Google Scholar] [CrossRef]
- Mazumder, R.; Morampudi, K.S.; Motwani, M.; Vasudevan, S.; Goldman, R. Proteome-Wide Analysis of Single-Nucleotide Variations in the N-Glycosylation Sequon of Human Genes. PLoS ONE 2012, 7, e36212. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S.A.H.; Ali, A.; Sayed, S.F.; Ali, A.; Abadi, S.S.H.; Tahir, A.; Afjal, M.A.; Rashid, H.; Aly, O.M.; Nagarajan, S. Potential of Paracetamol for Reproductive Disruption: Molecular Interaction, Dynamics, and MM-PBSA Based In-Silico Assessment. Toxicol. Mech. Methods 2023, 33, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S.A.H.; Najmi, A.K.; Raisuddin, S. Cyclophosphamide-induced Down-Regulation of Uroplakin II in the Mouse Urinary Bladder Epithelium Is Prevented by S-Allyl Cysteine. Basic Clin. Pharmacol. Toxicol. 2016, 119, 598–603. [Google Scholar] [CrossRef]
- Swiderski, K.; Thakur, S.S.; Naim, T.; Trieu, J.; Chee, A.; Stapleton, D.I.; Koopman, R.; Lynch, G.S. Muscle-Specific Deletion of SOCS3 Increases the Early Inflammatory Response but Does Not Affect Regeneration after Myotoxic Injury. Skelet. Muscle 2016, 6, 36. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. RMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S.A.H.; Azhar, G.; Zhang, X.; Sharma, S.; Hafeez, M.; Wei, J.Y. Alternative Splicing of Serum Response Factor Reveals Isoform-Specific Remodeling in Cardiac Diseases. Genes 2025, 16, 947. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).