Abstract
Regulatory T cells (Tregs) are a distinctive subset of CD4+ T cells critical in self-tolerance maintenance to prevent the development of autoimmunity. The mechanisms by which these cells provide immune regulation are numerous and, consequently, deeply involved in the pathogenesis of many autoimmune disorders. Treg-based adoptive cell transfer (ACT) therapy has generated interest as a novel, promising strategy to restore self-tolerance in autoimmunity. Polyclonal Treg-based ACT therapy was first implemented in clinical trials, presenting adequate safety profiles. Subsequent preclinical studies have shown antigen-specific Tregs to be safer and more effective than polyclonal approaches, so research has recently moved in this direction. Antigen-specificity can be conferred to Tregs by viral transduction of genes coding for engineered T cell receptors (eTCRs) or chimeric antigen receptors (CARs), with encouraging outcomes in different animal models of autoimmunity. This review focuses on the biology of Tregs, as well as on current preclinical and clinical data for Treg-based ACT in the field of autoimmunity.
1. Introduction
Immune tolerance refers to the ability of the immune system to remain unresponsive to self-antigens. When this capacity is disrupted, immune responses targeting the body’s own tissues arise, leading to what we know as autoimmune disease. Among the different cells that aid in the maintenance of this tolerance, a specialised naturally occurring subset termed CD4+ regulatory T cells (Treg) are central for the prevention of autoimmunity [1,2,3].
These autoimmune and autoinflammatory disorders pose a major burden to healthcare systems as they affect approximately 5–10% of individuals in industrialised countries [3,4,5,6]. Their heterogeneity and complexity have hampered the development of targeted therapies, as treatment must adequately suppress autoreactivity while preserving the normal functional side of the immune response [2,7,8]. Historically, first-line therapies have relied on broad immunosuppression with corticosteroids, cytotoxic drugs such as azathioprine or methotrexate, or disease-modifying antirheumatic drugs (DMARDs) like leflunomide, hydroxychloroquine, and sulfasalazine. Nonetheless, these approaches are limited by their non-specific mechanisms, chronic use, and significant side effects, which include a higher risk of infections and malignancy [5,9,10].
Biological drugs, particularly monoclonal antibodies (mAbs) directed against immune mediators such as TNF-α, have represented important advances in the treatment of some autoimmune conditions. Still, these drugs do not restore immune tolerance, and patients frequently experience treatment failure [5,11]. Consequently, there is an unmet need for strategies that actively re-establish self-tolerance [7].
In this context, adoptive cell therapy (ACT) using Treg cells has gained considerable attention as a promising therapeutic avenue in autoimmunity. In recognition of the centrality of regulatory T cells (Tregs) to immune homeostasis, the 2025 Nobel Prize in Physiology or Medicine honoured Mary E. Brunkow, Fred Ramsdell and Shimon Sakaguchi for discoveries that established FOXP3+ Tregs as key enforcers of immune tolerance. This milestone crystallised the concept that immune regulation is an active process essential to preventing autoimmunity and maintaining balance, providing the conceptual foundation for the Treg-based adoptive cell therapies discussed in this review [12,13,14,15].
2. Treg Historical Perspective
In the 1970s, Gershon and Kondo [16] identified a population distinct from helper T cells that could dampen the immune responses. These cells were initially termed “suppressor T cells”. Subsequent studies suggested heterogeneity within this population, with some subsets acting in an antigen-specific manner and others functioning more broadly [17]. However, methodological challenges and inconsistent results led to scepticism in the field, causing the “suppressor T cell” concept to fall out of favour [18,19]. At the same time, tolerance research advanced through the recognition of clonal deletion and anergy as key mechanisms [20,21,22].
Parallel experimental approaches focused on tolerance breakdown and the induction of autoimmunity. Nishizuka and Sakakura [23] demonstrated that neonatal thymectomy in mice triggered autoimmune ovarian destruction and the related production of autoantibodies. Later, Penhale et al. [24] showed that thymectomy in adult rats induced autoimmune thyroiditis, an effect that could be prevented by transferring CD4+ T cells from healthy syngeneic donors [25,26]. These findings indicated that the thymus gives rise to a subset of CD4+ T cells with suppressive functions, suggesting the coexistence of effector and regulatory CD4+ populations [27].
The next step taken was to separate these two CD4+ populations in normal naïve mice by the expression of cell surface molecules [28,29,30,31,32,33]. These attempts identified CD25 (the IL-2 receptor α-chain) as a key feature. Removal of CD25+ CD4+ cells induced severe autoimmune disease in mice, whereas reconstitution with this subset prevented disease onset [29]. This provided evidence for a distinct population of regulatory CD25+ CD4+ T cells, later designated regulatory T cells (Treg) [19].
A major breakthrough came in the early 2000s with the discovery of the transcription factor FoxP3 as the master regulator of Treg development and function [13,34,35]. The link between FOXP3 mutations and IPEX syndrome, inflammatory bowel disease, and severe allergy further underscored its essential role in immune tolerance [36,37,38].
It is now well established that Tregs are indispensable for controlling aberrant responses against self, microbial, and environmental antigens. Their dysfunction can contribute to autoimmune and allergic diseases, while their suppressive properties also facilitate tumour immune evasion, posing challenges for cancer immunotherapy [39,40].
3. Treg Biology
3.1. Treg Development, Modulation, and Types
Treg account for roughly 5–10% of circulating CD4+ T cells in peripheral blood (PB). The most widely accepted classification distinguishes between thymus-derived or natural Treg (tTreg/nTreg) and peripherally induced Treg (pTreg) [Table 1] [8].

Table 1.
Differential properties and characteristics of the different Treg subtypes.
nTreg arise in the thymus during positive selection of CD4+ single-positive thymocytes and are committed to a lineage that is functionally and phenotypically distinct from other T cell subpopulations [39,41,42]. This lineage commitment is not accomplished by simple positive selection but rather through an agonist-selection programme that depends on relatively strong self-reactive TCR signals delivered in the thymic medulla [43,44]. Within this framework, TCR signal strength operates within a window of outcomes: low/tonic signals support conventional positive selection of Tconv, very high-affinity signals trigger clonal deletion, and intermediate-to-high ‘agonist’ signals—integrated with key cytokine and co-stimulatory cues—divert cells to the Treg lineage [44]. IL-2–STAT5 signalling is indispensable for Foxp3 induction and stabilisation, with STAT5 engaging Foxp3 regulatory elements and scaling the size of the tTreg niche [45,46,47]. CD28 co-stimulation is required for tTreg development, acting in concert with TCR signals and IL-2 [48], while ICOS–ICOSL interactions fine-tune selection and support Treg differentiation/maintenance in the thymic environment [49,50]. Antigen availability is provided by AIRE- and FEZF2-dependent programmes in medullary thymic epithelial cells (mTECs) and by dendritic cells that acquire and present tissue-restricted antigens; these antigen-presentation axes coordinate deletion versus Treg diversion of strongly self-reactive clones, shaping the emergent Treg repertoire and tissue specificity [51,52,53]. Collectively, these data establish agonist selection—rather than simple positive selection—as the defining pathway for thymic Treg generation, integrating TCR signal strength with IL-2/STAT5 and CD28/ICOS co-stimulation in the context of medullary antigen presentation [43,44,45,54]. Together, these convergent signals initiate FOXP3 transcription and the early stabilisation of the Treg lineage programme, providing a mechanistic bridge from thymic selection to lineage commitment.
Building on this selection-driven signalling, the development and function of nTreg is coordinated by a transcription factor network involving FoxP3 and the establishment of a specific epigenetic landscape [14,41]. The Treg epigenome encompasses Treg-specific DNA demethylation, reinforcing expression activation and stabilisation of super-enhancers associated with Treg gene signature (FOXP3, CD25, CTLA-4, HELIOS, EOS) [2,14]. Signals downstream of IL-2/STAT5 and TCR/co-stimulation converge on chromatin to promote Treg-specific DNA demethylation, thereby consolidating the lineage programme. Within this framework, the FOXP3 CNS2 region contains a conserved CpG island (Treg-specific demethylated region, TSDR) that is hypomethylated (transcriptionally active) in nTreg but is hypermethylated and inactive in Tconv. This hypomethylated TSDR region is considered the most reliable indicator of a stable Treg lineage, as it ensures high FoxP3 levels [39].
Although FOXP3/Foxp3 expression and the Treg epigenome normally confer lineage stability, inflammatory cytokines and strong co-stimulation can bias Treg towards functional plasticity. In particular, IL-6–STAT3 signalling antagonises Treg programmes and favours Th17-like features; under sustained inflammation, some Tregs down-regulate Foxp3 and produce IL-17A or IFN-γ, a phenomenon reported in human disease and experimental models. These observations underscore that Treg identity is not immutable in hostile cytokine milieus [55,56,57].
At the molecular level, FOXP3 cis-regulatory elements, notably CNS2/TSDR, are crucial to maintain high FOXP3 expression and lineage stability [58,59]. Demethylation of the TSDR and coordinated activity of FOXP3 enhancers safeguard the Treg programme, whereas inflammatory cues that erode this landscape predispose to instability [58,59]. Experimental perturbation confirms causality: targeted TSDR demethylation stabilises FOXP3 in human T cells; conversely, loss of CNS2 function destabilises the lineage [60,61].
Beyond FOXP3, additional transcription factors (e.g., BACH2, IKZF family/Eos, C/EBP) and pathways (PI3K–AKT–mTOR) modulate the balance between suppressive and effector programmes, providing rational entry points for therapeutic interventions aimed at preserving Treg function in inflammatory settings [60,62].
In contrast, pTregs differentiate in peripheral tissues, like the intestinal mucosa or the maternal placenta, from naïve CD4+ T cells upon antigen stimulation in environments rich in TGF-β, IL-2, and retinoic acid [8,39]. Unlike nTregs, which primarily recognise self-antigens, pTregs often respond to non-self-antigens from commensal microbiota, food, or allergens, helping maintain mucosal and peripheral tolerance [2,41].
Additional subsets with regulatory properties, also induced in the periphery, are Th3 and Tr1 cells. Th3 cells, first defined by Weiner, emerge in the gut following oral antigen exposure and produce TGF-β and IL-4 [61], while Tr1 cells, as termed by Cottrez and Groux, arise under IL-10-rich conditions and are defined by high IL-10 secretion with minimal IL-2 and IL-4 production [63,64]. In contrast to nTregs, Th3 and Tr1 subsets contribute to tolerance toward food, microbial, and even some-self antigens [65,66,67,68].
Besides these naturally occurring Treg subtypes, a third group of regulatory cells that are artificially induced in vitro under non-inflammatory conditions could be established; these are termed “induced Tregs” (iTregs) [69]. These in vitro-induced regulatory cells are likely similar to their pTreg counterparts, as their induction from FoxP3- T cells is achieved through incubation conditions that resemble those that promote pTreg generation [70,71,72]. It is noteworthy that these iTreg may exhibit greater instability in inflammatory environments, raising questions about their therapeutic applicability [73].
While CD4+ Treg cells have been extensively studied, their CD8+ counterparts remain far less understood [74], although their potential contribution to the therapy field of autoimmunity makes them an ideal candidate to explore. Evidence indicates that CD8+ Treg are a heterogeneous group with diverse phenotypes, origins, and mechanisms of action. Among them, FoxP3+ CD8+ CD28low CD8+, CD103+ CD8+, and CD8αα+ CD4− T cells have been described [75,76,77,78,79,80,81].
FoxP3+ CD8+ Treg cells are rare under steady-state conditions in both mice and humans [82], but they can expand under pathological circumstances such as transplantation, autoimmunity, or cancer [74,83,84,85,86,87]. Their developmental origin is not fully resolved, though recent studies suggest they may be generated peripherally rather than exclusively in the thymus [88]. As with CD4+ Treg, their induction relies on TCR signalling along with co-stimulatory and cytokine-mediated signals, particularly IL-2 and TGF-β.
Functionally, CD8+ Treg suppress immune responses through both contact-dependent and cytokine-mediated mechanisms. They can inhibit effector T cell activity via high IL-2 consumption, production of IL-10, TGF-β, and IFN-γ, or by competing for γ-chain cytokines [89]. Additionally, some CD8+ Treg subsets display cytotoxic properties, resembling conventional CD8+ effector T cells, although the relevance of this function in vivo remains uncertain [74].
3.2. CD4+ Treg-Mediated Suppression
Treg employ a broad repertoire of suppressive strategies [Figure 1], including direct cell–cell interactions, secretion of immunomodulatory cytokines, and modulation of the microenvironment [41].
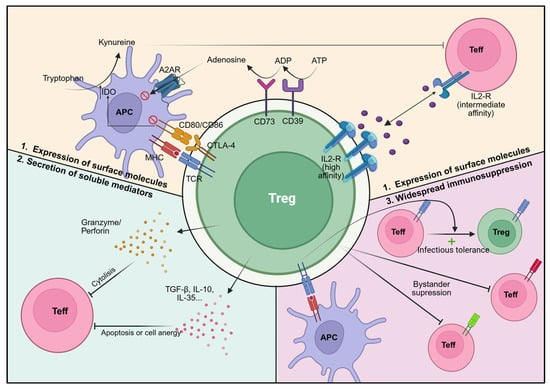
Figure 1.
Treg-mediated suppression mechanisms. Foxp3+/FOXP3+ Treg (green) suppress Teff (red) and other immune cells through multiple, non-mutually exclusive pathways: (1) Expression of surface molecules: CTLA-4, which binds to CD80/CD86 on antigen presenting cells (purple), leading to IDO expression and tryptophan deprivation, which is essential for Teff function; CD39 and CD73, which mediate adenosine release via conversion of ATP and ADP, respectively; CD25, which allows expression of high-affinity IL-2R and IL-2 deprivation from Teff cells; (2) Secretion of soluble factors: immunosuppressive factors such as anti-inflammatory cytokines (TGF-β, IL-10, and IL-35) or cytolytic factors such as granzyme and perforin release; (3) Regulating other T cells: Treg activation via antigen-specific recognition induces suppressive effects on Teff of different specificities (bystander suppression), as well as propagation of their immune regulatory properties to neighbouring cells (infectious tolerance). Created with BioRender.com (accessed on 1 July 2025).
A defining feature of Treg cells, especially upon activation, is their inability to produce IL-2 due to FoxP3-driven transcriptional repression. Instead, they depend on exogenous IL-2 for survival, which they efficiently capture via constitutive expression of the high-affinity IL-2 receptor (CD25). By consuming IL-2, Treg deprive conventional T cells (Tconv) of this cytokine, limiting their proliferation and differentiation into effectors [41].
Another major mechanism involves cytotoxic T-lymphocyte antigen 4 (CTLA-4), expressed constitutively by Treg. CTLA-4 binds with high affinity to CD80/CD86 on antigen-presenting cells (APCs), preventing CD28-mediated co-stimulation of effector T cells. Additionally, CTLA-4 removes CD80/CD86 from the APC surface through transendocytosis and induces indoleamine-2,3-dioxygenase (IDO) in APCs, which depletes tryptophan and further inhibits effector T cell activity [41].
Treg also secrete anti-inflammatory cytokines such as IL-10, TGF-β, and IL-35, which directly suppress effector T cells. In addition, they can mediate cytotoxicity via granzyme/perforin-dependent mechanisms [9,41]. Expression of the ectoenzymes CD39 and CD73 enables the breakdown of extracellular ATP into adenosine, which binds A2A receptors on effector cells, exerting suppressive effects [90].
Beyond direct inhibition, Treg can act indirectly by fostering the development of additional regulatory populations, a phenomenon referred to as “infectious tolerance”. Once activated by specific antigens, Treg can exert bystander suppression over effector T cells of different specificities, extending their regulatory influence beyond the initiating antigen [91,92].
Interestingly, Tregs have also been implicated in tissue repair and regeneration. They can produce growth factors such as amphiregulin, keratinocyte growth factor, and neuregulin-1, contributing to regeneration in muscle, lung, intestine, and other tissues [93]. Furthermore, Treg cells modulate neutrophil survival and cytokine production, protect against inflammation-induced tissue damage, and influence processes such as angiogenesis and metabolic homeostasis [72,93,94,95].
4. Treg in Human Autoimmune Disease
4.1. IPEX Syndrome and IPEX-like Monogenic Diseases
Mutations of Treg signature genes (FOXP3, IL2R, and CTLA4) impair Treg development and function, resulting in severe autoimmunity [2,96].
FOXP3 mutations produce intense Treg-specific dysfunction, resulting in IPEX syndrome, which is a rare early-onset condition characterised by multiorgan autoimmunity. IPEX syndrome is often fatal in the first years of life and typically includes the triad: neonatal-onset type 1 diabetes (T1D), eczematous dermatitis, and enteropathy [96]. To date, only long-term immunosuppression or bone marrow transplantation have been effective therapies, carrying significant side effects. However, novel approaches, including targeted gene editing of FOXP3, are being investigated as potential treatment options [97].
Similarly, CD25 deficiency and CTLA-4 haploinsufficiency can lead to severe autoimmune syndromes (IPEX-like syndromes), which reconfirm the important role of CTLA-4 and CD25 expression in Treg function [2].
4.2. Treg in Multifactorial and Polygenic Autoimmune Diseases
Common polygenic autoimmune diseases, such as T1D or multiple sclerosis (MS), afflict 5–10% of the population worldwide and are caused by chronic immune responses against the host’s cells, subsequently culminating in tissue damage. These autoimmune disorders develop from interactions between environmental triggers and genetic polymorphisms that induce a loss of self-tolerance [9].
Since the discovery of Treg indispensable role in self-tolerance, efforts have been made to detect Treg abnormalities in autoimmunity [8,39]. In the last decade, an increasing number of papers reported either functional or numerical anomalies regarding Treg population in multiple autoimmune diseases, including T1D, MS, autoimmune hepatitis (AIH), rheumatoid arthritis (RA), vitiligo, and IBD, among others [98,99,100,101,102].
The observed Treg defective function in autoimmune diseases involves their inability to inhibit Teff activation and proliferation, as well as Teff secretion of inflammatory cytokines [98,100]. The decreased capacity of Treg in regulating autoreactive cells depends on many factors, but their downregulated FoxP3 protein expression (Treg instability) seems to be uniformly present [39,103].
5. Treg-Based Therapies in Autoimmunity
Treg’s crucial role in maintaining self-tolerance, and therefore, controlling autoimmune responses, reveals the clinical potential of Treg-based therapies. They can find broad applications in restoring self-tolerance in autoimmune disorders and inducing tolerance to allogeneic tissues upon solid organ and hematopoietic stem cell transplantation. Treg-based therapies are being investigated with promising outcomes in the transplantation field [90], but this review will only discuss their application in autoimmunity.
Novel therapeutic approaches include either ACT of ex vivo expanded Treg or immunomodulatory interventions to enhance Treg expansion in vivo. Both applications can potentially promote Treg-mediated immune regulation in a non-specific (polyclonal) or antigen-specific way [Figure 2] [9,104].
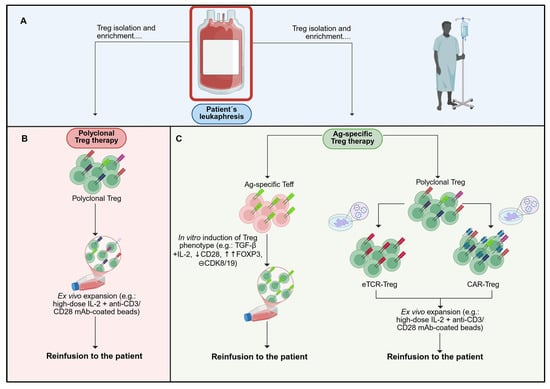
Figure 2.
Schematic representation of polyclonal and antigen-specific Treg manufacturing workflows. Current Treg-based ACT products can be categorised by antigen specificity. (A) Isolation of Treg is typically based on surface markers (CD4+ CD25high CD127low), optionally enriching the naïve subset (CD45RA+), using magnetic selection or FACS. (B) Polyclonal Treg products retain a broad TCR repertoire, reflecting that of the starting Treg population. Following isolation, ex vivo expansion is performed to achieve clinically relevant doses while preserving the regulatory phenotype and function. (C) Antigen-specific products are generated via two principal routes: (i) selection of antigen-enriched T cell precursors (e.g., Teff/Tconv bearing a defined TCR) followed by induction of a Treg phenotype under appropriate conditions (e.g., FOXP3-directed conversion); or (ii) genetic redirection of isolated Treg to express synthetic antigen receptors, namely engineered TCRs (eTCR; MHC-restricted, peptide–MHC recognition) or chimeric antigen receptors (CAR; MHC-independent), conferring specificity to a defined target antigen. Colour/key: panel A (isolation), panel B (polyclonal/blue), panel C (antigen-specific/orange). Created with BioRender.com (accessed on 1 July 2025).
In vivo immunomodulatory interventions were first attempted in the treatment of AIDs. They enhance Treg-mediated function in a polyclonal or antigen-specific manner according to the mechanism of action targeted. On one side, the polyclonal approach includes interventions that promote Treg-specific IL-2 signalling [105] (e.g., low-dose IL-2, mutant IL-2 and IL-2/anti-IL-2 mAb complexes), interventions that preferentially inhibit pathogenic T cells over Treg (e.g., anti-CD3 mAbs and mTOR inhibitors) or activation of Treg-co-stimulatory receptors (e.g., tumour necrosis factor receptor 2, TNFR2), or even gut microbiome transplantation [72,106,107]. On the other side, the antigen-specific approach comprises tolerogenic dendritic cell-based therapies (e.g., antigen-specific DCs engineered to overexpress 1α-hydroxylase in combination with non-toxic zinc concentration [108] or antigen-specific tolerogenic vaccines (e.g., protein/peptide- and DNA/RNA-based vaccines) [7,9,104,109,110,111]. This review only focuses on Treg-based ACT.
6. Treg-Based ACT in Autoimmunity
The therapeutic potential of Treg-based ACT in autoimmunity has been demonstrated in multiple preclinical models of autoimmune disorders, which have promoted advances in their clinical development, with polyclonal Treg therapy most widely used in clinical trials [92]. Nowadays, around 14 clinical trials (active and completed) have tested the safety and efficacy of Treg-based ACT in different autoimmune diseases [Table 2]. There are currently four main products developed for Treg-based ACT: polyclonal Treg, endogenous antigen-specific Treg, antigen-specific induced Treg (iTreg), and engineered Treg (eTreg) [Figure 2] [92,104,112].
Table 2.
Completed and ongoing clinical trials of Treg-based adoptive cell therapy in autoimmunity as published on clinicaltrials.gov, clinicaltrialsregister.eu, and isrctn.com (accessed between 1 May and 20 September 2025).
6.1. Polyclonal Treg Therapy
Treg can be isolated from either peripheral blood (PB) or umbilical cord blood (UCB). Those from PB have a low precursor frequency, making them difficult to extract and expand effectively [119]. In addition, peripheral Treg cells are unstable in PB and can be transformed into Teff cells. In contrast, Treg cells from UCB of allogeneic donors have a higher percentage of naïve T cells, maintaining a highly activated state with immunosuppressive function [119]. They also have a unique homing capacity, ideal in ACT for autoimmune disorders [119]. A third promising source besides PB and UCB could be neonatal thymus, as they are often discarded in paediatric cardiac surgery and contain more undifferentiated Treg cells than UCB and PB, which, indeed, have a stronger suppressive capacity [120]. Nevertheless, this source needs to be further investigated [3,8].
Despite the potential of UCB and allogeneic sources, current Treg-based ACT predominantly utilises expanded autologous Treg from PB because of its accessibility [116]. Two primary methods: magnetic-activated cell sorting (MACS) and fluorescence-activated cell sorting (FACS). In the first one, from the isolated mononuclear cell population via the standard gradient centrifugation technique, further purification is needed to sort out desired cells with immunosuppressive capacities. This is achieved through the employment of a magnetic field that distinguishes and separates cells attached to magnetic beads. In this sense, there is a first step of CD8+ and CD19+ cell depletion (double negative selection) followed by enrichment of the CD25+ fraction (positive selection). This MACS-based method is in accordance with the specifications of GMP production, as it works in a closed system, but its accuracy is suboptimal, reaching only 80% of FoxP3+ cells in the final product [5,8,90,92,121].
Instead, another advanced method to isolate Treg is a flow cytometry-based purification (FACS), where Treg differentiation is based on fluorescent labelling. Therefore, it is possible to select this population according to CD4+ CD25high CD127low expression. This ensures a superior separation purity (>99%), but the technique is limited by the slower processing speed and the inability to guarantee aseptic operation, making it difficult to meet the requirements of GMP production [8,114,119,122]. Nevertheless, novel closed and GMP-compliant cell sorting systems, such as the MACSQuant® Tyto® (Miltenyi Biotec, Bergisch Gladbach, Germany) or the CGX10 Cell Isolation System (Sony Biotechnology), have recently been developed to enable sterile, clinical-grade FACS-based Treg isolation. Although the resulting cells are relatively pure after isolation, the existence of contaminating Teff cells may outgrow Tregs over time [123,124]. Thus, an additional selection of the naïve (CD45RA+) population can enhance purity by eliminating activated T cells that transiently upregulate CD25 [124]
Secondly, post-Treg-isolation, the cell count is usually insufficient; thus, expansion in cell cultures is required to achieve optimal concentration for therapeutic applications, making it the primary challenge of Treg therapies. Polyclonal Treg isolated are usually expanded ex vivo using high-dose IL-2 and anti-CD3/CD28 mAb-coated beads [125,126]. It is important to bear in mind that a high purity of the initial Treg cell population ensures to increase in Treg yield, and some researchers have studied their suppressive effect. The incorporation of rapamycin into the cell culture is especially relevant when the Treg population has been purified by MACS, as it helps to compensate for the considerably reduced initial purity [112]. Nonetheless, it has been shown [127] that rapamycin is not needed when Treg cells are purified using FACS methods with selection of CD4+ CD25hihg CD127low Treg cells. It is noteworthy that the process of expanding Tregs not only expands the number of Tregs but also has been shown to correct impairments of Treg function [114,128,129]. Finally, polyclonal Treg are reinfused intravenously as an autologous Treg-based ACT [8,90,92,130].
The first data on the use of polyclonal Tregs was focused on graft-versus-host disease (GvHD) in 2009 [122] and Crohn’s disease in 2012 [131]. Now, polyclonal Treg therapy has been or is currently being tested in patients with T1D, AIH, IBD, systemic lupus erythematosus (SLE), systemic sclerosis (SS), pemphigus, and Guillain–Barré syndrome [Table 2]. Moreover, completed phase I clinical trials in T1D, MS, IBD, and SLE have revealed that autologous polyclonal Treg therapy is well tolerated and safe [114,115,116,117,118,119].
Particularly in T1D, where most studies have been carried out, recent phase l clinical trials have assessed and proved the safety and feasibility of polyclonal Treg therapy to reverse recent T1D onset [114,119,120,132]. Specifically, Bluestone et al. found [114] that these expanded polyclonal Tregs maintained their Treg cell state and polyclonality, and they could be detected in circulation after a year post-transfusion. A similar follow-up phase I study, followed by IL-2 administration, was confounded by the non-Treg effects of IL-2 [120]. Marek-Trzonkowska et al. [132] also demonstrated safety with possible efficacy in a phase I new-onset T1D study with a limited number of children. Nonetheless, the small cohort sizes and differences in the age and disease stage of the treated cohorts may have contributed to the variability in results between these studies [119].
The safety and efficacy of polyclonal Treg therapy were also tested in the first and recent randomised placebo-controlled phase II clinical trial (Sanford Project T-Rex Study, NCT02691247) [133]. This was a large, well-powered study to evaluate a range of Treg doses in a blinded manner, including a placebo group. It was found that better C-peptide preservation was achieved when the cells presented a lower in vitro fold expansion. On the other hand, higher doses of cells with a higher in vitro fold expansion did not correlate with better outcomes. Hypothetically, with lower in vitro fold expansion, Tregs are more functional in vivo or may have an enhanced ability to expand further in vivo. It is difficult to speculate whether the mixed results respond to variances in patients and study design (e.g., different dosages) or if the therapy is inefficient in its current setting. Thus, these findings highlight the need to better understand Treg expansion biology and consider new avenues for the improvement of current ACT Treg therapies [119].
6.2. Polyclonal vs. Antigen-Specific Treg Therapies
As previously mentioned, various clinical trials have been performed employing polyclonal Treg therapy, providing relevant lessons on production procedures; cell dosages; cell circulation in patients; and Tregs’ stability, safety, tolerability, and clinical efficacy (e.g., prolonged C-peptide levels in T1D trials) [1,114,118]. Nevertheless, the current difficulties in building improved nTreg ACT include antigen specificity, functional stability, and survivability [105].
Various preclinical models of organ transplantation and autoimmune diseases show that antigen-specific Tregs enriched by in vitro antigen stimulation are superior to polyclonal Tregs [105,134,135,136,137]. Antigen-specific Treg predominantly localise at the site of antigen presentation, reducing the risk of generalised/off-target immunosuppression and making them both more effective and safer than polyclonal Treg for ACT [104,138]. In addition, the increased trafficking of antigen-specific Treg to inflamed target tissues probably permits the infusion of lower cell numbers [92,104]. For all these reasons, in the last years, research has moved towards the development of antigen-specific approaches to treat autoimmune disease, but no clinical trials using such ACT Tregs are ongoing in the autoimmunity field.
The purification and expansion of endogenous disease-relevant autoantigen-specific Tregs remain technically challenging owing to their very low frequency in PB, as most of them are localised in the target tissue, limiting their accessibility. The endogenous antigen-specific Treg approach cannot be adapted for the treatment of autoimmune diseases [92,104]. Antigen-specific Treg has been assessed to prevent graft rejection in transplantation using APC from the graft donor to specifically stimulate alloreactive-specific Treg from the recipient in vitro, producing a differentiation into effector-type nTregs (CD45RAlo CD25hi FoxP3hi), which are Bcl-2lo and prone to die by apoptosis upon TCR re-stimulation. This could explain why the transfer of a large number of antigen-specific nTregs generally does not incur general immunosuppression [105,139].
Thus, alternative strategies are needed to produce antigen-specific Tregs for a particular antigen in the autoimmunity field, such as the generation of antigen-specific Treg in vitro by transformation of antigen-specific Teff into cells with suppressive capacity (antigen-specific iTreg cells) or redirecting polyclonal Treg by genetic insertion of synthetic antigen receptors (engineered Treg cells, eTreg) [104].
6.3. Antigen-Specific Induced Treg Therapy
FoxP3+ Treg can be induced from Teff in vitro, provided they are stimulated under conditions of high IL-2 and TGF-β levels, generating in vitro iTreg [39]. This could have the advantage over nTregs of in vitro preparation of a large number of antigen-specific iTregs from CD4+ Tconvs, serving as a potential source to produce antigen-specific iTregs for ACT [104,105]. By definition, Teff-derived (or Tconv-derived) iTregs retain the native T cell receptor (TCR) of the precursor cell; therefore, their antigen specificity is dictated by that original TCR. As a consequence, polyclonal Teff-to-iTreg conversions yield a repertoire-wide mixture of specificities mirroring the diversity of the starting TCR pool, whereas induction from antigen-enriched precursors produces oligoclonal or monoclonal iTregs with focused specificity.
However, in contrast to nTreg, it has become clear that iTregs are phenotypically and functionally more unstable, especially under inflammatory conditions. This is clinically relevant as iTreg may regain their Teff features in vivo and enhance autoimmune response. This instability of TGF-β-induced iTreg could be mainly explained because of their incomplete Treg-type epigenomic changes at FOXP3 [39,105,140]. Nevertheless, it has been shown that deprivation of the CD28 co-stimulatory signal at an early stage of iTreg generation can establish Treg-specific DNA hypomethylation at Treg signature genes, allowing production of stable antigen-specific iTreg [105,141].
Other strategies to reprogram Teff into Treg include inhibition of cyclin-dependent kinases (CDK) 8/19 signalling pathways in combination with TGF-β [141,142], the use of ascorbate (vitamin C) that could facilitate de novo hypomethylation of Foxp3 CNS2 in iTregs through Tet activation [143,144], the use of epigenetic regulators (e.g., inhibition of HDAC6 [145,146,147]), and transgenic overexpression of the transcription factors FOXP3, HELIOS and BACH2 via lentiviral vectors or lately via more advanced genetic tools (e.g., CRISPR-Cas9 genome editing systems) [97,142,148]. Indeed, enforced expression of FOXP3 on conventional CD4+ T cells establishes an effectively stable Foxp3+ Treg phenotype and associated suppressive functions, even under inflammatory conditions [5,97,148,149]. It has been seen that the combination of several of these methods can be applied. For example, the combined use of an anti-CD3, a CDK8/19 inhibitor, and TGFβ resulted in a 10-fold increase in CD25high FoxP3+ Tregs in cultured CD4+ T lymphocytes compared to unstimulated lymphocytes due to the transdifferentiation of antigen-specific effector T lymphocytes into Tregs [150].
All these approaches could confer immunosuppressive functions to Teff, preserving their Treg-like properties in vivo when transferred into different autoimmunity preclinical models, but their clinical utility in common autoimmune diseases must be determined [105,142,148]. Indeed, a registered clinical trial (NCT05241444), which is still recruiting patients, aims to test in 30 participants with IPEX syndrome the infusion of autologous CD4+ T cells, which have undergone lentiviral-mediated gene transfer of the healthy human FOXP3 gene [151].
6.4. Engineered Treg Therapy
Another approach to produce antigen-specific Treg is to redirect polyclonal Treg by genetically introducing, through lentiviral/retroviral vectors, synthetic receptors that recognise a target antigen of interest. These receptors can take the form of engineered TCRs (eTCRs) or chimeric antigen receptors (CARs) [Figure 3]. New strategies to generate these engineered Treg consist of FOXP3 genetic transduction in combination with the introduction of eTCRs or CARs to convert Teff into immunosuppressive antigen-specific Treg-like cells [152,153]. In most preclinical and translational studies, however, CAR-/TCR-modified Tregs are derived from natural Tregs (CD4+ CD25+ CD127low FOXP3+) that are isolated and then genetically engineered, whereas Teff-to-Treg conversion via FOXP3 plus a CAR/eTCR remains an alternative, more experimental route [154].
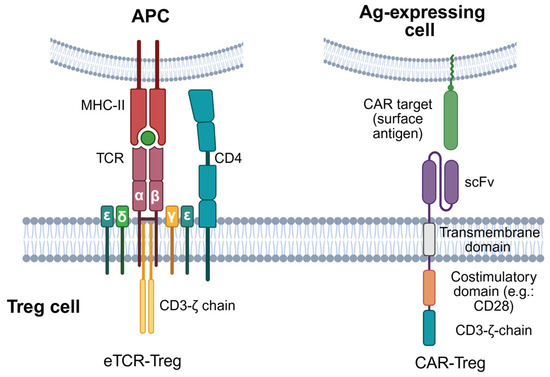
Figure 3.
Synthetic antigen-specific receptors in engineered Treg therapy. Antigen-specific Treg can be engineered by transducing genes encoding synthetic TCRs targeting antigenic peptides in the context of MHC class I or class II (pMHC-I or -II) molecules, or CARs enabling direct antigen recognition in a pMHC-independent manner. Engineered TCRs, same as endogenous TCRs, are composed of TCRα and TCRβ chain heterodimers binding to specific pMHC-I/II complexes and associated with a CD3 complex composed of three signalling subunits: one CD3ζ homodimer, one CD3εδ heterodimer, and one CD3γε heterodimer. CARs are hybrid antigen receptors made up of an extracellular scFv capable of binding a target antigen; a transmembrane domain; and an intracellular part, including activating motifs from CD3ζ and co-stimulatory receptors (e.g., CD28 or 4-1BB/CD137), with CD28 being the most current used co-stimulatory domain in CAR-Treg. Created with BioRender.com (accessed on 1 July 2025).
On one hand, eTCRs are produced through the integration of genes that code for TCR α and β chains specific for an antigen of interest. This heterodimeric nature of TCRs makes them complex to engineer, as the introduced subunits may recombine with the endogenous TCRα or TCRβ chains. This difficulty raises the risk of generating TCRs with unknown specificities and off-target effects [112]. Numerous strategies have been envisaged to overcome this problem. One is the insertion of extra cysteine residues in the constant region of eTCRα and eTCRβ chains to form a disulfide bond between the two to promote preferential pairing of the transduced chains [155,156]. Another strategy is the replacement of the human constant region in TCR chains with a mouse constant region, as interspecies pairing of TCR chains has never been observed [157,158]. Nonetheless, the current most promising approach is to apply genome editing tools to engineer the endogenous TCR locus. This could be achieved through knocking out one or both endogenous TCR chains or, more elegantly, by the replacement of the endogenous TCR with the new TCR by knocking it into the TCRα constant region locus (TRAC) [159].
Given their effectiveness and safety in cancer, CARs are considered an encouraging strategy for the clinical administration of antigen-specific Treg in autoimmune disorders [160,161]. CARs are formed by the artificial fusion of different protein motifs, and basically involve 3 major components: (1) an extracellular domain, commonly derived from the single chain variable fragment (scFv) of a mAb that specifically recognises the desired antigen in an MHC-independent manner; (2) a transmembrane domain; and (3) an intracellular domain, which grants the signal transduction for cell activation. Diverse CAR generations have been described, differing in the number/type of intracellular domains. However, the second-generation CAR has been the most used in CAR-Treg designs, especially the one that integrates the co-stimulatory molecule CD28 as an intracellular domain [162], which seems to be essential for potent CAR-Treg functions. The optimal design of CAR-Treg remains under investigation; however, tailored signalling architectures may be required to accommodate differences in the affinity of, and the propensity for self-aggregation amongst, scFv [112]. Separately, it should be noted that CD19-directed CAR-T cells used to treat autoimmune disease are typically conventional T cells rather than Tregs, and multiple early clinical reports (including case series and prospective cohorts) have shown high rates of remission in refractory conditions such as SLE, supporting the concept of B-cell depletion/immune reset without the need for a Treg phenotype [163,164,165].
The pioneers of CAR-Treg research were Megan Levings et al. They developed a CAR specific to HLA-A2 to prevent rejection of solid grafts. The experiments showed that Treg cells with the A2-CAR construct retained their phenotype and ability to suppress the immune response before, during, and after stimulation, thereby proving the concept of using CAR-Tregs in immune-mediated responses [72,166]. After this solid proof, CAR-Tregs have been used in the treatment of various disease models [Table 3]. Excitingly, the first-in-human study of a genetically engineered Treg product (NCT04817774) is currently ongoing as part of the STEADFAST phase I/II trial to test CAR-Tregs specific for HLA-A2 in renal transplantation [167]. Encouragingly, intermediate results from this study showed that CAR-Tregs were well tolerated in the 6 patients dosed across 4 dose levels [168].
Table 3.
CAR- and eTCR-based Treg therapies in preclinical models of autoimmune diseases.
Likewise, TCRs have been used to generate TCR-Tregs for the treatment of various AID models [Table 3] such as T1DM, MS, acquired factor VII deficiency… [91,177,178,179] and have shown their efficacy. Interestingly, a recent study demonstrated the possibility of developing Sm-specific TCRs and the production of Sm-specific Tregs for the treatment of SLE patients positive for anti-Sm and HLA-DR15. In this work, Sm-Tregs effectively suppressed inflammatory responses and inhibited disease progression in a humanised mouse model of lupus nephritis [180].
Although preclinical evidence has supported the development of eTCR- and CAR-Treg therapies [Table 3], these engineered receptors hold different functional features that could represent advantages/disadvantages depending on the context. While CARs present a greater affinity for their specific antigen than eTCRs, CARs require a higher density of antigen to trigger their activation. Hence, eTCR may be more useful for clinical settings where the disease-relevant antigen is lowly expressed, whereas CARs are probably more useful in autoimmune disorders associated with highly expressed antigens on target tissue and low expression on normal tissues [92,161]. Furthermore, antigen recognition by eTCR-Treg is MHC-restricted and thereby only recognises pMHC complexes and requires matching of the patient’s MHC genotype. MHC restriction and the necessity to identify/isolate antigen-specific and disease-relevant TCRs may limit the translation of eTCR-Treg into the clinic and may be the reason why CARs are more widely studied [104]. Instead, CAR-Treg can recognise soluble/surface antigens in an MHC-independent manner, enabling the specific detection of antigens of diverse nature (whole proteins, glycolipids, gangliosides…) and expanding their application to a larger number of patients (no matching of MHC genotype required) [9,161].
7. Challenges and Future Perspectives on the Field of Treg Therapies
7.1. Challenges in TCR-Treg-Based Therapies
A key challenge in developing eTCR-Treg therapies is the identification of suitable target antigens that are expressed on specific HLA alleles [72,176,181]. In this sense, there are three major determinants. First, it is necessary to explore whether TCRs from Tregs or Teff cells are more promising sources for their isolation [182]. Second, it may be necessary to identify those TCRs that are restricted by widely expressed HLA molecules. Third, sequence data from both TCRα and TCRβ will be needed to determine TCR specificity [72,112].
Another limitation in choosing the proper TCR is selecting the affinity with which it will bind to the pMHC complex. In this regard, both low- and high-affinity TCRs have been compared in studies, which have shown that they can be positively [177] and negatively [152] correlated with immunosuppressive function. Nonetheless, this association is dependent on a more complex pattern. For instance, Sprouse et al. showed [183] that TCRs with high- and low-affinity TCRs migrated to the pancreas, though executing differential functions. The ones with high affinity were demonstrated to activate classical pathways (CTLA-4, IL-10, etc.), and the ones with low affinity expressed proteins associated with tissue repair.
7.2. Challenges in CAR-Treg-Based Therapies
One restraint of CAR-Treg is their ability to accomplish suppressor functions by interacting with tissue-specific autoantigens [184]. Therefore, if these antigens are expressed not only in the site of an autoimmune reaction but also in other healthy tissues, the immune response may be less effective and lead to systemic hyperactivation of CAR-Treg that, in turn, can cause a nonspecific immunosuppressive reaction [185]. On this basis, several modifications of the classical CAR-Treg product have been proposed and studied to overcome these limitations.
An optimisation of the CAR-Treg product to overcome this problem could be the manufacturing of universal CAR-Tregs (UniCAR-Tregs) [72]. These are designed based on universal tumour-targeted CAR-T cells (UniCAR-T), which are two-component systems. The first component is a universal CAR-T that, instead of interacting with human surface antigens, interacts with a concrete peptide motif. This motif is then contained in the second component, a soluble adapter called the targeting module (TM) [186]. TM are bispecific molecules that link UniCAR-T/Treg cells to target cells. Thus, UniCAR-T/Treg could be turned on and off by dosing the TM [187], ensuring the maintenance of the CAR-T/Treg as inert until they encounter the appropriate target module. As far as we are aware, two groups have tested these constructs in Tregs, proving their high versatility and easy reprogrammability [145,188].
In addition to UniCARs, there are numerous other CAR constructions that are currently being investigated, each of which offers its unique benefits (Figure 4). Since Grada et al. [189] first developed the idea of dual targeting cancer cells using bispecific TanCARs, the application of this concept to Treg to Tregs has gained attention. Another potential strategy could be the replacement of the scFv of the CAR with the antigen itself to regulate B-cells that are antigen-specific, which is known as B-cell-targeting Ab receptors (BARs). Furthermore, to target combinations of antigens that are widely expressed throughout the body, the incorporation of SynNotch receptors may prove valuable. These receptors incorporate a two-step positive feedback circuit that comprises a priming signalling receptor devoid of activating-signalling capacity, which, upon antigen recognition, releases a transcription factor, thus initiating the expression of a CAR targeting another antigen [145].
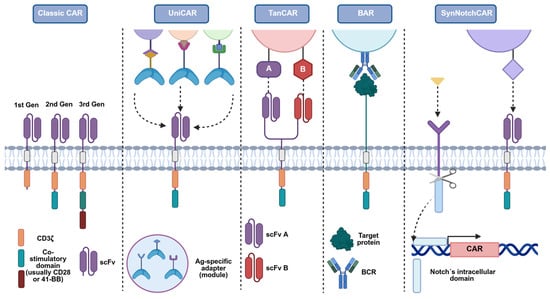
Figure 4.
Design variants of the classical CAR to address antigen-specificity and control constraints in CAR-Treg therapy. Schematic comparison of modular receptor architectures built on the classical CAR backbone (scFv–hinge–transmembrane–co-stimulatory domain–CD3ζ). Each format offers distinct advantages and trade-offs for specificity, controllability, and safety in Treg applications. Classic CAR: single scFv confers fixed, MHC-independent recognition of a defined surface antigen. UniCAR: a two-component system in which cells express a “universal” CAR that recognises a short peptide epitope; adapter molecules (bearing that epitope and an antigen-binding moiety) retarget the cells to different antigens and enable on/off, dose-tunable control. TanCAR: a bispecific receptor with two extracellular scFv, allowing recognition of two antigens (A and B) and the possibility of combinatorial (OR/AND-like) targeting to improve precision. BAR/CAAR: the scFv is replaced by the autoantigen itself to engage autoreactive B-cell receptors (BCRs) on pathogenic B-cell clones, enabling selective recognition and potential deletion of disease-relevant B cells. SynNotch-CAR: a two-step circuit in which a ligand (yellow)-specific SynNotch receptor (purple) signalling-deficient for activation induces de novo CAR expression upon sensing a first cue, thereby restricting CAR activity spatially/temporally to sites where both cues are present. Figure adapted from Ref. [145]. Created with BioRender.com (accessed on 1 July 2025).
The idea of addressing these diseases in an antigen-specific way is attractive, but there remains the paramount obstacle of the search for the concrete antigen, where in many clinical scenarios, the pathogenic antigen is unknown, or multiple antigens are involved, with the potential for epitope spreading [190,191]. The exploration of new targets will greatly benefit these cellular therapies.
One way to address the above limitation is targeting a T cell activation-associated antigen, resulting in an engineered Treg product capable of suppressing a wide range of autoimmune disorders [192]. As demonstrated by the group of Rui X. [191], a prime candidate target may be OX40L (CD252), a TNF superfamily protein expressed on activated DCs, B cells, and endothelial cells [193], which constitutes a critical co-stimulatory signal for T cell activation through binding to its cognate receptor OX40 (CD134) during antigen presentation and helps in the development of T follicular helper (Tfh) cells and autoreactive B cells [194,195,196]. Even though this group developed the cell product for the GvHD caused by stem cell transplantation, the fact that OX40L is overexpressed in RA [197,198], systemic sclerosis [199,200], and SLE [194,201,202], makes it and apparently ideal target to consider in future investigations.
This notion of targeting inflammatory mediators for the production of CAR-Tregs was further developed in other studies [154,203], and the concept of a CAR was converted into what is known as artificial immune receptors (AIRs). AIR-Treg development may help to solve the problem of choosing antigens for Treg-based therapies, as these inflammation biosensors are targeted at inflammatory mediators, including TNF-α, TNF-like ligand 1A… instead of antigens [203]. The structure of an AIR may resemble that of a CAR as they are composed intracellularly of a CD3ζ chain and a CD28 co-stimulatory signalling domain, and extracellularly by the receptor to the inflammatory ligand [72].
7.3. Challenges in Engineering Strategies of Artificial Receptors and Treg Functionality
A critical specific limitation of eTCR and CAR approaches in Treg-based ACT is the current use of viral vectors to insert the receptor transgene. Apart from manufacturing complexities, viral vectors imply random transgene integration, which carries the risk of oncogenic changes. Instead, the arrival of non-viral genome editing technologies, such as CRISPR-Cas9, has enabled alternative strategies to potentially improve the safety, efficacy, and manufacturing of genetically modified antigen-specific T cells [92]. In addition, novel episomal vector systems have been developed in which the incorporation of a scaffold matrix attachment region (S/MAR) into the vector DNA ensures persistent mitotic stability, resistance to epigenetic silencing, and efficient expression of the gene of interest [204,205]. Recently, human CAR-T cells were generated by nano-nS/MARt vectors, and they mediated efficient tumour killing [154,206]. Nonetheless, application in Tregs and clinical studies has yet to be carried out.
In addition to antigen specificity via eTCR/CAR, Treg cell migration into disease-specific inflamed sites could be reinforced by transgenic introduction of chemokine receptors, which might restrict their activation to target tissue, thereby limiting the risk of off-target activity. For instance, patients suffering from MS may benefit from Treg expressing CXCR3, as studies have suggested that T cells require its expression to brain homing [207].
Insights into Treg plasticity under inflammatory conditions argue for product and regimen designs that actively promote FOXP3 stability and resist Th1/Th17 deviation. First, source selection matters: CD45RA+ (naïve) nTregs display superior epigenetic stability and a lower propensity to acquire effector cytokines compared with memory-phenotype Tregs, which translates into more reliable expansion and function after infusion [208,209]. Second, ex vivo culture conditions can be tuned to preserve lineage commitment: IL-2–STAT5 support together with mTOR inhibition (rapamycin) maintains FOXP3 and suppressive function during expansion, while excessive PI3K–AKT–mTOR signalling correlates with loss of the Treg programme [209,210]. Approaches that favour FOXP3-CNS2/TSDR demethylation—including vitamin C/TET-facilitation in iTreg settings—further consolidate lineage stability [60,211]. Third, co-interventions in vivo can buffer hostile cytokine milieus: low-dose IL-2 preferentially amplifies Tregs via STAT5 and has shown acceptable safety across multiple autoimmune indications, whereas IL-6/STAT3 pathway blockade mitigates Treg-to-Th17 drift in inflamed tissues [212,213,214]. Taken together, stability-aware sourcing, manufacturing, and adjunct therapy can be systematically combined to increase the odds of durable Treg engraftment, function, and clinical benefit in autoimmune settings.
Regarding engineered Treg therapies’ safety, CAR-Treg seem to be likely safer than conventional CAR-T therapies, as it has been proven that they do not elicit tumour lysis syndrome, haemotoxicity, or cytokine release syndrome [145]. Nonetheless, there are some risks. One potential off-target effect could result from their immunosuppressive effects, such as an increased risk of infection or cancer. However, all phase I/II clinical trials using Treg ACT have demonstrated the safety of this therapy [215]. Another likely concern would be the contamination of Treg products with Tconv cells, exacerbating the underlying autoimmune or alloimmune disease [145]. In addition, some in vitro studies reported that CAR-stimulated Treg might exhibit cytotoxic activity, which needs to be deeply studied to avoid unexpected side effects [216]. To resolve these theoretical issues, one approach used for conventional CAR-T therapy could be translated into the CAR-Treg field. This is the implementation of an “OFF” switch to eliminate CAR-Treg without disturbing the immune system. That could be achieved by the utilisation of a kinase inhibitor, such as dasatinib, or an enzyme inhibitor, to reversibly lead to CAR degradation [145,217]. Moreover, for eTCR/CAR-Treg therapies, genetic approaches may also be used to induce Treg expression of suicide cassettes, which can be activated if an adverse event occurs (e.g., cell instability, off-target toxicity, immune suppression), allowing rapid elimination of the engineered Treg [92,218].
8. Manufacturability and Scalability of Treg Therapies
The production of autologous Treg-based cell therapies continues to face significant challenges that limit their application beyond rare disorders such as acute lymphoblastic leukaemia and multiple myeloma. Expanding these therapies to more prevalent autoimmune diseases, including systemic lupus erythematosus and multiple sclerosis, introduces considerable industrial, logistical, and financial hurdles [219].
While the cell therapy industry has made progress in establishing standardised practices for commercial CAR-T production, the generation of Tregs remains in its early stages. This is largely due to their low abundance in circulation and the lack of a unique surface marker to facilitate their selective isolation, making Tregs among the most technically demanding cell types to manufacture [220,221].
Across autologous therapies, three major challenges persist: scalability, dose determination, and cost. Scaling up Treg production is particularly difficult, as current processes are labour-intensive and rely on open operations with highly specialised equipment. Developing automated, integrated, and closed manufacturing systems will be essential to improve efficiency and patient throughput. Additionally, accurate dose generation is critical, since Tregs proliferate more slowly than CAR-T cells in vivo, necessitating sufficient cell yields for therapeutic effect. Finally, the high cost of cell therapies—driven by single-use reagents, specialised equipment, skilled labour, and extensive analytical testing—remains a key barrier, and addressing these factors will be important for broader clinical accessibility [220].
What is more, patients with autoimmune conditions often require long-term treatment, which can adversely affect T cell numbers and function, further complicating the collection and production of high-quality autologous Treg products. In this context, allogeneic, “off-the-shelf” cell therapies derived from universal donors may offer a viable solution to overcome the inherent limitations of autologous approaches [222].
9. Conclusions
Treg cells play a crucial role in the maintenance of self-tolerance. The mechanisms by which they provide immune regulation are multiple and complex, and their dysfunction has been demonstrated to contribute to the pathogenesis/development of multiple autoimmune diseases, but they also take part in other processes, such as graft rejection or cancer immune evasion mechanisms.
Treg-based ACT has generated enthusiasm for the treatment of autoimmune disorders, where current treatments often alleviate symptoms and promote episodes of remission but do not cure the disease. Instead, Treg-based ACT is believed to provide a potential cure for autoimmunity by durably establishing tolerogenic cell populations to eliminate autoreactivity.
Polyclonal Treg therapy was the first-implemented Treg-based ACT in human clinical trials, showing correct safety profiles and limited efficacy. However, several preclinical studies have revealed that antigen-specific Tregs, including engineered Tregs (CARs or eTCRs), are safer and more effective. Thus, research has moved towards the development of antigen-specific approaches in recent years, with encouraging outcomes in multiple preclinical studies.
Nevertheless, to develop safe and effective antigen-specific Treg therapies, further studies of human Treg biology in physiological and pathological conditions are required, applying new approaches to characterise different Treg subsets, as well as better strategies to identify disease-relevant autoantigens and Treg anomalies. All of this, together with the current efforts to develop useful tools to engineer Tregs, will be essential to enhance and optimise Treg-based ACT.
Author Contributions
Conceptualisation, E.G., G.M.-S. and F.L.; writing—original draft preparation, A.L. and G.M.-S.; writing—review and editing, E.G., G.M.-S., and F.L.; supervision, F.L.; funding acquisition, F.L. All authors have read and agreed to the published version of the manuscript.
Funding
F.L. is supported by the Spanish Ministerio de Economía y Competitividad (MINECO; PID2019-106658RB-I00), co-financed by European Development Regional Fund (ERDF) “A way to achieve Europe”, and Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR; 2017/SGR/1582) from Generalitat de Catalunya.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors are indebted to Marcos Isamat for his expert manuscript review and editing.
Conflicts of Interest
F.L. is the founder and ad honorem scientific advisor of Sepsia Therapeutics SL and inventor of patent EP2138259. The authors declare no conflict of interest.
Abbreviations
AOM-DSS, azoxymethane-dextran sodium sulphate; CEA, carcinoembryonic antigen; CIA, collagen-induced arthritis; Co-stim: co-stimulation domain; CNS, central nervous system; CV, citrullinated vimentin; EAE, experimental autoimmune encephalomyelitis; GAD65, glutamic acid decarboxylase; GD3, ganglioside D3; HLA, human leukocyte antigen; IGRP, islet-specific glucose-6-phosphatase-related protein; MBP, myelin basic protein; mBSA, methylated bovine serum albumin; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; NF-M, neurofilament-medium; NOD, non-obese diabetic; OVA, ovalbumin; PLP, proteolipid protein; PPI, pre-proinsulin; TNBS, 2,4,6-trinitrobenzene sulphonic acid; TNP, 2,4,6-trinitrophenol.
References
- Pisetsky, D.S. Pathogenesis of Autoimmune Disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.T.; Frazee, C.S.; Herman, P.J.; Chen, S.; Chen, A.; Kuo, Y.; Ellebrecht, C.T. Current Advancements in Cellular Immunotherapy for Autoimmune Disease. Semin. Immunopathol. 2025, 47, 7. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.W. The Increasing Prevalence of Autoimmunity and Autoimmune Diseases: An Urgent Call to Action for Improved Understanding, Diagnosis, Treatment, and Prevention. Curr. Opin. Immunol. 2023, 80, 102266. [Google Scholar] [CrossRef]
- Bulliard, Y.; Freeborn, R.; Uyeda, M.J.; Humes, D.; Bjordahl, R.; de Vries, D.; Roncarolo, M.G. From Promise to Practice: CAR T and Treg Cell Therapies in Autoimmunity and Other Immune-Mediated Diseases. Front. Immunol. 2024, 15, 1509956. [Google Scholar] [CrossRef]
- Conrad, N.; Misra, S.; Verbakel, J.; Verbeke, G.; Molenberghs, G.; Taylor, P.; Mason, J.; Sattar, N.; Mcmurray, J.; Mcinnes, I.; et al. Op0007 incidence, prevalence and co-occurrence of autoimmune disorders, trends over time and by age, sex and socioeconomic status. a population-based study in 22 million individuals. Ann. Rheum. Dis. 2023, 82, 5–6. [Google Scholar] [CrossRef]
- Moorman, C.D.; Sohn, S.J.; Phee, H. Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T Cell Therapy, and IL-2 Therapy. Front. Immunol. 2021, 12, 657768. [Google Scholar] [CrossRef]
- Yu, L.; Fu, Y.; Miao, R.; Cao, J.; Zhang, F.; Liu, L.; Mei, L.; Ou, M. Regulatory T Cells and Their Derived Cell Pharmaceuticals as Emerging Therapeutics Against Autoimmune Diseases. Adv. Funct. Mater. 2024, 34, 2405133. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Glück, T.; Kiefmann, B.; Grohmann, M.; Falk, W.; Straub, R.H.; Schölmerich, J. Immune Status and Risk for Infection in Patients Receiving Chronic Immunosuppressive Therapy. J. Reumatol. 2005, 32, 1473–1480. [Google Scholar]
- Singh, S.; George, J.; Boland, B.S.; Vande, C.N.; Sandborn, W.J. Primary Non-Response to Tumor Necrosis Factor Antagonists Is Associated with Inferior Response to Second-Line Biologics in Patients with Inflammatory Bowel Diseases: A Systematic Review and Meta-Analysis. J. Crohn's Colitis 2018, 12, 635–643. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Pillars Article: Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Pillars Article: Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.-A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a New Forkhead/Winged-Helix Protein, Scurfin, Results in the Fatal Lymphoproliferative Disorder of the Scurfy Mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef]
- Gershon, R.K.; Kondo, K. Cell Interactions in the Induction of Tolerance: The Role of Thymic Lymphocytes. Immunology 1970, 18, 723–737. [Google Scholar]
- Green, D.R.; Flood, P.M.; Gershon, R.K. Immunoregulatory T-Cell Pathways. Annu. Rev. Immunol. 1983, 1, 439–461. [Google Scholar] [CrossRef]
- Kronenberg, M.; Steinmetz, M.; Kobori, J.; Kraig, E.; Kapp, J.A.; Pierce, C.W.; Sorensen, C.M.; Suzuki, G.; Tada, T.; Hood, L. RNA Transcripts for I-J Polypeptides Are Apparently Not Encoded between the I-A and I-E Subregions of the Murine Major Histocompatibility Complex. Proc. Natl. Acad. Sci. USA 1983, 80, 5704–5708. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Wing, K.; Miyara, M. Regulatory T Cells—A Brief History and Perspective. Eur. J. Immunol. 2007, 37, S116–S123. [Google Scholar] [CrossRef]
- Kappler, J.W.; Roehm, N.; Marrack, P. T Cell Tolerance by Clonal Elimination in the Thymus. Cell 1987, 49, 273–280. [Google Scholar] [CrossRef]
- Kisielow, P.; Blüthmann, H.; Staerz, U.D.; Steinmetz, M.; von Boehmer, H. Tolerance in T-Cell-Receptor Transgenic Mice Involves Deletion of Nonmature CD4+8+ Thymocytes. Nature 1988, 333, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Goodnow, C.C.; Cyster, J.G.; Hartley, S.B.; Bell, S.E.; Cooke, M.P.; Healy, J.I.; Akkaraju, S.; Rathmell, J.C.; Pogue, S.L.; Shokat, K.P. Self-Tolerance Checkpoints in B Lymphocyte Development. Adv. Immunol. 1995, 59, 279–368. [Google Scholar] [CrossRef]
- Nishizuka, Y.; Sakakura, T. Thymus and Reproduction: Sex-Linked Dysgenesia of the Gonad after Neonatal Thymectomy in Mice. Science 1969, 166, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Penhale, W.J.; Farmer, A.; McKenna, R.P.; Irvine, W.J. Spontaneous Thyroiditis in Thymectomized and Irradiated Wistar Rats. Clin. Exp. Immunol. 1973, 15, 225–236. [Google Scholar] [PubMed]
- Sakaguchi, S.; Takahashi, T.; Nishizuka, Y. Study on Cellular Events in Post-Thymectomy Autoimmune Oophoritis in Mice. II. Requirement of Lyt-1 Cells in Normal Female Mice for the Prevention of Oophoritis. J. Exp. Med. 1982, 156, 1577–1586. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Takahashi, T.; Nishizuka, Y. Study on Cellular Events in Postthymectomy Autoimmune Oophoritis in Mice. I. Requirement of Lyt-1 Effector Cells for Oocytes Damage after Adoptive Transfer. J. Exp. Med. 1982, 156, 1565–1576. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N. Organ-Specific Autoimmune Disease Induced in Mice by Elimination of T Cell Subsets. V. Neonatal Administration of Cyclosporin A Causes Autoimmune Disease. J. Immunol. 1989, 142, 471–480. [Google Scholar] [CrossRef]
- Asano, M.; Toda, M.; Sakaguchi, N.; Sakaguchi, S. Autoimmune Disease as a Consequence of Developmental Abnormality of a T Cell Subpopulation. J. Exp. Med. 1996, 184, 387–396. [Google Scholar] [CrossRef]
- Sakaguchi, S. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of selftolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- McKeever, U.; Mordes, J.P.; Greiner, D.L.; Appel, M.C.; Rozing, J.; Handler, E.S.; Rossini, A.A. Adoptive Transfer of Autoimmune Diabetes and Thyroiditis to Athymic Rats. Proc. Natl Acad. Sci. USA 1990, 87, 7618–7622. [Google Scholar] [CrossRef]
- Powrie, F.; Mason, D. OX-22high CD4+ T Cells Induce Wasting Disease with Multiple Organ Pathology: Prevention by the OX-22low Subset. J. Exp. Med. 1990, 172, 1701–1708. [Google Scholar] [CrossRef]
- Smith, H. Tolerance mechanism in experimental ovarian and gastric autoimmune disease. J. Immunol. 1992, 149, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, S.; Izumi, Y.; Yoshioka, T.; Yagi, H.; Tsujimura, T.; Tarutani, O.; Kohno, Y.; Murakami, S.; Hamaoka, T.; Fujiwara, H. Autoimmune Thyroiditis Induced in Mice Depleted of Particular T Cell Subsets. I: Requirement of Lyt-1dull L3T4bright Normal T Cells for the Induction of Thyroiditis. J. Immunol. 1988, 141, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Liston, A. (Ed.) Regulatory T Cells in Health and Disease, 1st ed.; Academic Press: Cambridge, MA, USA, 2015; Volume 129, ISBN 978-0-12-803415-6. [Google Scholar]
- Khattri, R.; Cox, T.; Yasayko, S.-A.; Ramsdell, F. An Essential Role for Scurfin in CD4+CD25+ T Regulatory Cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.A.; Blaeser, F.; Ho, N.; Lederman, H.M.; Voulgaropoulos, C.; Helms, C.; Bowcock, A.M. JM2, Encoding a Fork Head–Related Protein, Is Mutated in X-Linked Autoimmunity–Allergic Disregulation Syndrome. J. Clin. Investig. 2000, 106, R75–R81. [Google Scholar] [CrossRef]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.-L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome Is the Human Equivalent of Mouse Scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) Is Caused by Mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T Cells in Autoimmune Disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef]
- Shan, F.; Somasundaram, A.; Bruno, T.C.; Workman, C.J.; Vignali, D.A.A. Therapeutic Targeting of Regulatory T Cells in Cancer. Trends Cancer 2022, 8, 944–961. [Google Scholar] [CrossRef]
- Grover, P.; Goel, P.N.; Greene, M.I. Regulatory T Cells: Regulation of Identity and Function. Front. Immunol. 2021, 12, 750542. [Google Scholar] [CrossRef]
- Gootjes, C.; Zwaginga, J.J.; Roep, B.O.; Nikolic, T. Defining Human Regulatory T Cells beyond FOXP3: The Need to Combine Phenotype with Function. Cells 2024, 13, 941. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.L; Sjaastad, L.E.; Farrar, M.A. Regulatory T Cell Development in the Thymus. J. Immunol. 2019, 203, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W.J.; Hsieh, C.-S. A Two-Step Process for Thymic Regulatory T Cell Development. Immunity 2008, 28, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 Receptor Beta-Dependent STAT5 Activation Is Required for the Development of Foxp3+ Regulatory T Cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef]
- Dikiy, S.; Li, J.; Bai, L.; Jiang, M.; Janke, L.; Zong, X.; Hao, X.; Hoyos, B.; Wang, Z.-M.; Xu, B.; et al. A distal Foxp3 enhancer enables interleukin-2 dependent thymic Treg cell lineage commitment for robust immune tolerance. Immunity 2021, 54, 931–946.e11. [Google Scholar] [CrossRef]
- Mahmud, S.A.; Manlove, L.S.; Farrar, M.A. Interleukin-2 and STAT5 in regulatory T cell development and function. JAK-STAT 2013, 2, e23154. [Google Scholar] [CrossRef]
- Salomon, B.; Lenschow, D.J.; Rhee, L.; Ashourian, N.; Singh, B.; Sharpe, A.; Bluestone, J.A. B7/CD28 Costimulation Is Essential for the Homeostasis of the CD4+CD25+ Immunoregulatory T Cells That Control Autoimmune Diabetes. Immunity 2000, 12, 431–440. [Google Scholar] [CrossRef]
- Hayes, E.T.; Mittelsteadt, K.L.; Campbell, D.J. ICOS signaling limits regulatory T cell accumulation and function in visceral adipose tissue. J. Exp. Med. 2020, 218. [Google Scholar] [CrossRef]
- Nazzal, D.; Gradolatto, A.; Truffault, F.; Bismuth, J.; Berrih-Aknin, S. Human Thymus Medullary Epithelial Cells Promote Regulatory T-Cell Generation by Stimulating Interleukin-2 Production via ICOS Ligand. Cell. Death Dis. 2014, 5, e1420. [Google Scholar] [CrossRef]
- Malchow, S.; Leventhal, D.S.; Nishi, S.; Fischer, B.I.; Shen, L.; Paner, G.P.; Amit, A.S.; Kang, C.; Geddes, J.E.; Allison, J.P.; et al. Aire-Dependent Thymic Development of Tumor-Associated Regulatory T Cells. Science 2013, 339, 1219–1224. [Google Scholar] [CrossRef]
- Perry, J.S.A.; Lio, C.-W.J.; Kau, A.L.; Nutsch, K.; Yang, Z.; Gordon, J.I.; Murphy, K.M.; Hsieh, C.-S. Distinct Contributions of Aire and Antigen-Presenting-Cell Subsets to the Generation of Self-Tolerance in the Thymus. Immunity 2014, 41, 414–426. [Google Scholar] [CrossRef]
- Takaba, H.; Morishita, Y.; Tomofuji, Y.; Danks, L.; Nitta, T.; Komatsu, N.; Kodama, T.; Takayanagi, H. Fezf2 Orchestrates a Thymic Program of Self-Antigen Expression for Immune Tolerance. Cell 2015, 163, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.L.; Mahmud, S.A.; Sjaastad, L.E.; Williams, J.B.; Spanier, J.A.; Simeonov, D.R.; Ruscher, R.; Huang, W.; Proekt, I.; Miller, C.N.; et al. Thymic Regulatory T Cells Arise via Two Distinct Developmental Programs. Nat. Immunol. 2019, 20, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Hafler, D.A. The Plasticity of Human Treg and Th17 Cells and Its Role in Autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Castillo, E.; García-Rasilla, V.Y.; García-Patiño, M.G.; Licona-Limón, P. Stability and Plasticity of Regulatory T Cells in Health and Disease. J. Leukoc. Biol. 2024, 116, 33–53. [Google Scholar] [CrossRef]
- Cheng, H.; Nan, F.; Ji, N.; Ma, X.; Zhang, J.; Liang, H.; Zhang, W.; Nakatsukasa, H.; Zhang, H.; Jin, W.; et al. Regulatory T Cell Therapy Promotes TGF-β and IL-6-Dependent pro-Inflammatory Th17 Cell Generation by Reducing IL-2. Nat. Commun. 2025, 16, 7644. [Google Scholar] [CrossRef]
- Kressler, C.; Gasparoni, G.; Nordström, K.; Hamo, D.; Salhab, A.; Dimitropoulos, C.; Tierling, S.; Reinke, P.; Volk, H.-D.; Walter, J.; et al. Targeted De-Methylation of the FOXP3-TSDR Is Sufficient to Induce Physiological FOXP3 Expression but Not a Functional Treg Phenotype. Front. Immunol. 2021, 11, 609891. [Google Scholar] [CrossRef]
- Zong, X.; Hao, X.; Xu, B.; Crawford, J.C.; Wright, S.; Li, J.; Zhang, Y.; Bai, L.; He, M.; Jiang, M.; et al. Foxp3 Enhancers Synergize to Maximize Regulatory T Cell Suppressive Capacity. J. Exp. Med. 2021, 218, e20202415. [Google Scholar] [CrossRef]
- Chen, Y.; Kuchroo, V.K.; Inobe, J.; Hafler, D.A.; Weiner, H.L. Regulatory T Cell Clones Induced by Oral Tolerance: Suppression of Autoimmune Encephalomyelitis. Science 1994, 265, 1237–1240. [Google Scholar] [CrossRef]
- Lee, S.; Park, K.; Kim, J.; Min, H.; Seong, R.H. Foxp3 Expression in Induced Regulatory T Cells Is Stabilized by C/EBP in Inflammatory Environments. EMBO Rep 2018, 19, e45995. [Google Scholar] [CrossRef]
- Ramón-Vázquez, A.; Flood, P.; Cashman, T.L.; Patil, P.; Ghosh, S. T Lymphocyte Plasticity in Chronic Inflammatory Diseases: The Emerging Role of the Ikaros Family as a Key Th17-Treg Switch. Autoimmun. Rev. 2025, 24, 103735. [Google Scholar] [CrossRef] [PubMed]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+T-Cell Subset Inhibits Antigen-Specific T-Cell Responses and Prevents Colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Cottrez, F.; Hurst, S.D.; Coffman, R.L.; Groux, H. T Regulatory Cells 1 Inhibit a Th2-Specific Response In Vivo. J. Immunol. 2000, 165, 4848–4853. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Weaver, C.T.; Lazenby, A.; Elson, C.O. Bacterial-Reactive T Regulatory Cells Inhibit Pathogenic Immune Responses to the Enteric Flora. J. Immunol. 2002, 169, 6112–6119. [Google Scholar] [CrossRef]
- Wildbaum, G.; Netzer, N.; Karin, N. Tr1 Cell–Dependent Active Tolerance Blunts the Pathogenic Effects of Determinant Spreading. J. Clin. Investig. 2002, 110, 701–710. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, C.; Lee, W.-H.; Yun, P.; Snow, P.; Liu, C.-P. Induction of Autoantigen-Specific Th2 and Tr1 Regulatory T Cells and Modulation of Autoimmune Diabetes. J. Immunol. 2003, 171, 733–744. [Google Scholar] [CrossRef]
- Satoguina, J.; Mempel, M.; Larbi, J.; Badusche, M.; Löliger, C.; Adjei, O.; Gachelin, G.; Fleischer, B.; Hoerauf, A. Antigen-Specific T Regulatory-1 Cells Are Associated with Immunosuppression in a Chronic Helminth Infection (Onchocerciasis). Microbes Infect. 2002, 4, 1291–1300. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Vignali, D.A.A.; Rudensky, A.Y.; Niec, R.E.; Waldmann, H. The Plasticity and Stability of Regulatory T Cells. Nat. Rev. Immunol. 2013, 13, 461–467. [Google Scholar] [CrossRef]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-Mediated Induction of a Potent Regulatory T Cell Population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef]
- Fisher, M.S.; Sennikov, S.V. T-Regulatory Cells for the Treatment of Autoimmune Diseases. Front. Immunol. 2025, 16. [Google Scholar] [CrossRef]
- Shevach, E.M.; Thornton, A.M. TTregs, PTregs, and ITregs: Similarities and Differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Larson, J.H.; Blazar, B.R.; Abdi, R.; Bromberg, J.S. Foxp3+CD8+ Regulatory T Cells: Bona Fide Tregs with Cytotoxic Function. Trends Immunol. 2025, 46, 324–337. [Google Scholar] [CrossRef]
- Shimokawa, C.; Kato, T.; Takeuchi, T.; Ohshima, N.; Furuki, T.; Ohtsu, Y.; Suzue, K.; Imai, T.; Obi, S.; Olia, A.; et al. CD8+ Regulatory T Cells Are Critical in Prevention of Autoimmune-Mediated Diabetes. Nat. Commun. 2020, 11, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Liao, W.; Liu, Y.; Yang, M.; Ma, C.; Wu, H.; Zhao, M.; Zhang, X.; Qiu, Y.; Lu, Q.; et al. TGF-β and Eomes Control the Homeostasis of CD8+ Regulatory T Cells. J. Exp. Med. 2021, 218, e20200030. [Google Scholar] [CrossRef]
- Li, H.; Zhou, L.; Jia, Y.; Wang, R.; Zhu, J.; Zhang, X.; Xu, W.; Liu, S.; He, Q.; Li, X. Plasmacytoid Dendritic Cells Mediate the Tolerogenic Effect of CD8+regulatory T Cells in a Rat Tolerant Liver Transplantation Model. Transpl. Immunol. 2022, 70, 101508. [Google Scholar] [CrossRef] [PubMed]
- Xystrakis, E. Identification of a novel natural regulatory CD8 T-cell subset and analysis of its mechanism of regulation. Blood 2004, 104, 3294–3301. [Google Scholar] [CrossRef]
- Fan, J.-N.; Ho, H.; Chiang, B.-L. Characterization of Novel CD8+ Regulatory T Cells and Their Modulatory Effects in Murine Model of Inflammatory Bowel Disease. Cell. Mol. Life Sci. 2024, 81, 327. [Google Scholar] [CrossRef]
- Sheng, H.; Marrero, I.; Maricic, I.; Fanchiang, S.S.; Zhang, S.; Sant’Angelo, D.B.; Kumar, V. Distinct PLZF+CD8αα+ Unconventional T Cells Enriched in Liver Use a Cytotoxic Mechanism to Limit Autoimmunity. J. Immunol. 2019, 203, 2150–2162. [Google Scholar] [CrossRef]
- Plaumann, J.; Engelhardt, M.; Awwad, M.H.S.; Echchannaoui, H.; Amman, E.; Raab, M.S.; Hillengass, J.; Halama, N.; Neuber, B.; Müller-Tidow, C.; et al. IL-10 Inducible CD8+ Regulatory T-Cells Are Enriched in Patients with Multiple Myeloma and Impact the Generation of Antigen-Specific T-Cells. Cancer Immunol. Immunother. 2018, 67, 1695–1707. [Google Scholar] [CrossRef]
- Churlaud, G.; Pitoiset, F.; Jebbawi, F.; Lorenzon, R.; Bellier, B.; Rosenzwajg, M.; Klatzmann, D. Human and Mouse CD8+CD25+FOXP3+ Regulatory T Cells at Steady State and during Interleukin-2 Therapy. Front. Immunol. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Robb, R.J.; Lineburg, K.E.; Kuns, R.D.; Wilson, Y.A.; Raffelt, N.C.; Olver, S.D.; Varelias, A.; Alexander, K.A.; Teal, B.E.; Sparwasser, T.; et al. Identification and Expansion of Highly Suppressive CD8+FoxP3+ Regulatory T Cells after Experimental Allogeneic Bone Marrow Transplantation. Blood 2012, 119, 5898–5908. [Google Scholar] [CrossRef] [PubMed]
- Beres, A.J.; Haribhai, D.; Chadwick, A.C.; Gonyo, P.J.; Williams, C.B.; Drobyski, W.R. CD8+ Foxp3+ Regulatory T Cells Are Induced during Graft-versus-Host Disease and Mitigate Disease Severity. J. Immunol. 2012, 189, 464–474. [Google Scholar] [CrossRef]
- Pellegrino, M.; Crinò, A.; Rosado, M.M.; Fierabracci, A. Identification and Functional Characterization of CD8+ T Regulatory Cells in Type 1 Diabetes Patients. PLoS ONE 2019, 14, e0210839. [Google Scholar] [CrossRef]
- Lozano, T.; Conde, E.; Martín-Otal, C.; Navarro, F.; Lasarte-Cia, A.; Nasrallah, R.; Alignani, D.; Gorraiz, M.; Sarobe, P.; Romero, J.P.; et al. TCR-Induced FOXP3 Expression by CD8+ T Cells Impairs Their Anti-Tumor Activity. Cancer Lett. 2022, 528, 45–58. [Google Scholar] [CrossRef]
- Lin, L.; Dai, F.; Wei, J.; Chen, Z. Influences of CD8+ Tregs on Peripheral Blood Mononuclear Cells from Allergic Rhinitis Patients. Laryngoscope 2021, 131, 316–323. [Google Scholar] [CrossRef]
- Badr, M.E.; Zhang, Z.; Tai, X.; Singer, A. CD8 T Cell Tolerance Results from Eviction of Immature Autoreactive Cells from the Thymus. Science 2023, 382, 534–541. [Google Scholar] [CrossRef]
- Vignali, D.A. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- Malviya, M.; Saoudi, A.; Bauer, J.; Fillatreau, S.; Liblau, R. Treatment of Experimental Autoimmune Encephalomyelitis with Engineered Bi-Specific Foxp3+ Regulatory CD4+ T Cells. J. Autoimmun. 2020, 108, 102401. [Google Scholar] [CrossRef]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg Cell-Based Therapies: Challenges and Perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef]
- Taams, L.S.; van Amelsfort, J.M.R.; Tiemessen, M.M.; Jacobs, K.M.G.; de Jong, E.C.; Akbar, A.N.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Modulation of Monocyte/Macrophage Function by Human CD4+CD25+ Regulatory T Cells. Hum. Immunol. 2005, 66, 222–230. [Google Scholar] [CrossRef]
- Lewkowicz, N.; Klink, M.; Mycko, M.P.; Lewkowicz, P. Neutrophil—CD4+ CD25+ T Regulatory Cell Interactions: A Possible New Mechanism of Infectious Tolerance. Immunobiology 2013, 218, 455–464. [Google Scholar] [CrossRef]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M. From IPEX Syndrome to FOXP3 Mutation: A Lesson on Immune Dysregulation. Ann. N. Y. Acad. Sci. 2018, 1417, 5–22. [Google Scholar] [CrossRef]
- Goodwin, M.; Lee, E.; Lakshmanan, U.; Shipp, S.; Froessl, L.; Barzaghi, F.; Passerini, L.; Narula, M.; Sheikali, A.; Lee, C.M.; et al. CRISPR-Based Gene Editing Enables FOXP3 Gene Repair in IPEX Patient Cells. Sci. Adv. 2020, 6, eaaz0571. [Google Scholar] [CrossRef] [PubMed]
- Haseda, F.; Imagawa, A.; Murase-Mishiba, Y.; Terasaki, J.; Hanafusa, T. CD4+CD45RA−FoxP3high Activated Regulatory T Cells Are Functionally Impaired and Related to Residual Insulin-secreting Capacity in Patients with Type 1 Diabetes. Clin. Exp. Immunol. 2013, 173, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Noori-Zadeh, A.; Mesbah-Namin, S.A.; Bistoon-beigloo, S.; Bakhtiyari, S.; Abbaszadeh, H.-A.; Darabi, S.; Rajabibazl, M.; Abdanipour, A. Regulatory T Cell Number in Multiple Sclerosis Patients: A Meta-Analysis. Mult. Scler. Relat. Disord. 2016, 5, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Grant, C.R.; Holder, B.S.; Cardone, J.; Martinez-Llordella, M.; Ma, Y.; Heneghan, M.A.; Mieli-Vergani, G.; Vergani, D.; Longhi, M.S. In Autoimmune Hepatitis Type 1 or the Autoimmune Hepatitis–Sclerosing Cholangitis Variant Defective Regulatory T-cell Responsiveness to IL-2 Results in Low IL-10 Production and Impaired Suppression. Hepatology 2015, 62, 863–875. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Rani, S.; Kumaran, M.S.; Bhatia, A.; Parsad, D. Expression of Th17- and Treg-specific Transcription Factors in Vitiligo Patients. Int. J. Dermatol. 2020, 59, 474–481. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of Regulatory T Cell in the Pathogenesis of Inflammatory Bowel Disease. World J. Gastroenterol. WJG 2016, 22, 2195–2205. [Google Scholar] [CrossRef]
- Bluestone, J.A.; McKenzie, B.S.; Beilke, J.; Ramsdell, F. Opportunities for Treg Cell Therapy for the Treatment of Human Disease. Front. Immunol. 2023, 14, 1166135. [Google Scholar] [CrossRef] [PubMed]
- Selck, C.; Dominguez-Villar, M. Antigen-Specific Regulatory T Cell Therapy in Autoimmune Diseases and Transplantation. Front. Immunol. 2021, 12, 661875. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Kawakami, R.; Mikami, N. Treg-Based Immunotherapy for Antigen-Specific Immune Suppression and Stable Tolerance Induction: A Perspective. Immunother. Adv. 2023, 3, Itad007. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, Q.; Dorfman, R.G.; Huang, X.; Fan, T.; Zhang, H.; Zhang, J.; Yu, C. Butyrate Inhibits Interleukin-17 and Generates Tregs to Ameliorate Colorectal Colitis in Rats. BMC Gastroenterol. 2016, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Shaikh, N.A.; Zhang, X.B.; Abdalla, M.I.; Baylink, D.J.; Tang, X. Enhancing Human Treg Cell Induction through Engineered Dendritic Cells and Zinc Supplementation. Crit. Rev. Immunol. 2024, 44, 37–52. [Google Scholar] [CrossRef]
- Obregon, C.; Kumar, R.; Pascual, M.A.; Vassalli, G.; Golshayan, D. Update on Dendritic Cell-Induced Immunological and Clinical Tolerance. Front. Immunol. 2017, 8, 1514. [Google Scholar] [CrossRef]
- Mansilla, M.J.; Hilkens, C.M.U.; Martínez-Cáceres, E.M. Challenges in Tolerogenic Dendritic Cell Therapy for Autoimmune Diseases: The Route of Administration. Immunother. Adv. 2023, 3, ltad012. [Google Scholar] [CrossRef]
- Morante-Palacios, O.; Fondelli, F.; Ballestar, E.; Martínez-Cáceres, E.M. Tolerogenic Dendritic Cells in Autoimmunity and Inflammatory Diseases. Trends Immunol. 2021, 42, 59–75. [Google Scholar] [CrossRef]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q. Next-Generation Regulatory T Cell Therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef] [PubMed]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juścińska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Młynarski, W.; et al. Therapy of Type 1 Diabetes with CD4+CD25highCD127-Regulatory T Cells Prolongs Survival of Pancreatic Islets—Results of One Year Follow-Up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The Effect of Low-Dose IL-2 and Treg Adoptive Cell Therapy in Patients with Type 1 Diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef]
- Dall’Era, M.; Pauli, M.L.; Remedios, K.; Taravati, K.; Sandova, P.M.; Putnam, A.L.; Lares, A.; Haemel, A.; Tang, Q.; Hellerstein, M.; et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 431–440. [Google Scholar] [CrossRef]
- Voskens, C.; Stoica, D.; Rosenberg, M.; Vitali, F.; Zundler, S.; Ganslmayer, M.; Knott, H.; Wiesinger, M.; Wunder, J.; Kummer, M.; et al. Autologous Regulatory T-Cell Transfer in Refractory Ulcerative Colitis with Concomitant Primary Sclerosing Cholangitis. Gut 2023, 72, 49–53. [Google Scholar] [CrossRef]
- Chwojnicki, K.; Iwaszkiewicz-Grześ, D.; Jankowska, A.; Zieliński, M.; Łowiec, P.; Gliwiński, M.; Grzywińska, M.; Kowalczyk, K.; Konarzewska, A.; Glasner, P.; et al. Administration of CD4+CD25highCD127−FoxP3+ Regulatory T Cells for Relapsing-Remitting Multiple Sclerosis: A Phase 1 Study. BioDrugs 2021, 35, 47–60. [Google Scholar] [CrossRef]
- Motwani, K.; Peters, L.D.; Vliegen, W.H.; El-sayed, A.G.; Seay, H.R.; Lopez, M.C.; Baker, H.V.; Posgai, A.L.; Brusko, M.A.; Perry, D.J.; et al. Human Regulatory T Cells From Umbilical Cord Blood Display Increased Repertoire Diversity and Lineage Stability Relative to Adult Peripheral Blood. Front. Immunol. 2020, 11, 611. [Google Scholar] [CrossRef]
- Dijke, I.E.; Hoeppli, R.E.; Ellis, T.; Pearcey, J.; Huang, Q.; McMurchy, A.N.; Boer, K.; Peeters, A.M.A.; Aubert, G.; Larsen, I.; et al. Discarded Human Thymus Is a Novel Source of Stable and Long-Lived Therapeutic Regulatory T Cells. Am. J. Transplant. 2016, 16, 58–71. [Google Scholar] [CrossRef]
- Di Ianni, M.; Del Papa, B.; Cecchini, D.; Bonifacio, E.; Moretti, L.; Zei, T.; Iacucci Ostini, R.; Falzetti, F.; Fontana, L.; Tagliapietra, G.; et al. Immunomagnetic Isolation of CD4+CD25+FoxP3+ Natural T Regulatory Lymphocytes for Clinical Applications. Clin. Exp. Immunol. 2009, 156, 246–253. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-Man Clinical Results of the Treatment of Patients with Graft versus Host Disease with Human Ex Vivo Expanded CD4+CD25+CD127− T Regulatory Cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, K.; Levings, M.K. Genetic Engineering of Regulatory T Cells for Treatment of Autoimmune Disorders Including Type 1 Diabetes. Diabetologia 2024, 67, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Eder, R.; Boeld, T.J.; Doser, K.; Piseshka, B.; Andreesen, R.; Edinger, M. Only the CD45RA+ Subpopulation of CD4+CD25high T Cells Gives Rise to Homogeneous Regulatory T-Cell Lines upon in Vitro Expansion. Blood 2006, 108, 4260–4267. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Fischer, A.; Roessner, S.; Lorenz, C.; Hirschmann, S.; Atreya, R.; Neufert, C.; Atreya, I.; Neurath, M.F.; Schuler, G. Characterization and Expansion of Autologous GMP-Ready Regulatory T Cells for TREG-Based Cell Therapy in Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1348–1359. [Google Scholar] [CrossRef]
- Levings, M.K.; Sangregorio, R.; Roncarolo, M.-G. Human Cd25+Cd4+ T Regulatory Cells Suppress Naive and Memory T Cell Proliferation and Can Be Expanded in Vitro without Loss of Function. J. Exp. Med. 2001, 193, 1295–1302. [Google Scholar] [CrossRef]
- Putnam, A.L.; Brusko, T.M.; Lee, M.R.; Liu, W.; Szot, G.L.; Ghosh, T.; Atkinson, M.A.; Bluestone, J.A. Expansion of Human Regulatory T-Cells From Patients With Type 1 Diabetes. Diabetes 2009, 58, 652–662. [Google Scholar] [CrossRef]
- Ulbar, F.; Villanova, I.; Giancola, R.; Baldoni, S.; Guardalupi, F.; Fabi, B.; Olioso, P.; Capone, A.; Sola, R.; Ciardelli, S.; et al. Clinical-Grade Expanded Regulatory T Cells Are Enriched with Highly Suppressive Cells Producing IL-10, Granzyme B, and IL-35. Biol. Blood Marrow Transplant. 2020, 26, 2204–2210. [Google Scholar] [CrossRef]
- Jarvis, L.B.; Rainbow, D.B.; Coppard, V.; Howlett, S.K.; Georgieva, Z.; Davies, J.L.; Mullay, H.K.; Hester, J.; Ashmore, T.; Van Den Bosch, A.; et al. Therapeutically Expanded Human Regulatory T-Cells Are Super-Suppressive Due to HIF1A Induced Expression of CD73. Commun. Biol 2021, 4, 1186. [Google Scholar] [CrossRef]
- Terry, L.V.; Oo, Y.H. The Next Frontier of Regulatory T Cells: Promising Immunotherapy for Autoimmune Diseases and Organ Transplantations. Front. Immunol. 2020, 11, 565518. [Google Scholar] [CrossRef]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients With Refractory Crohn’s Disease. Gastroenterology 2012, 143, 1207–1217.e2. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Techmańska, I.; Juścińska, J.; Wujtewicz, M.A.; Witkowski, P.; Młynarski, W.; Balcerska, A.; et al. Administration of CD4+CD25highCD127− Regulatory T Cells Preserves β-Cell Function in Type 1 Diabetes in Children. Diabetes Care 2012, 35, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Bender, C.; Wiedeman, A.E.; Hu, A.; Ylescupidez, A.; Sietsema, W.K.; Herold, K.C.; Griffin, K.J.; Gitelman, S.E.; Long, S.A. A Phase 2 Randomized Trial with Autologous Polyclonal Expanded Regulatory T Cells in Children with New-Onset Type 1 Diabetes. Sci. Transl. Med. 2024, 16, eadn2404. [Google Scholar] [CrossRef] [PubMed]
- Blat, D.; Zigmond, E.; Alteber, Z.; Waks, T.; Eshhar, Z. Suppression of Murine Colitis and Its Associated Cancer by Carcinoembryonic Antigen-Specific Regulatory T Cells. Mol. Ther. 2014, 22, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S. CAR/FoxP3-Engineered T Regulatory Cells Target the CNS and Suppress EAE upon Intranasal Delivery. J. Neuroinflammation 2012, 9, 112. [Google Scholar] [CrossRef]
- Veerapathran, A.; Pidala, J.; Beato, F.; Yu, X.-Z.; Anasetti, C. Ex Vivo Expansion of Human Tregs Specific for Alloantigens Presented Directly or Indirectly. Blood 2011, 118, 5671–5680. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Hara, H.; Tai, H.-C.; Long, C.; Tokita, D.; Yeh, P.; Ayares, D.; Morelli, A.E.; Cooper, D.K.C. Suppressive Efficacy and Proliferative Capacity of Human Regulatory T Cells in Allogeneic and Xenogeneic Responses. Transplantation 2008, 86, 1452–1462. [Google Scholar] [CrossRef]
- Serr, I.; Drost, F.; Schubert, B.; Daniel, C. Antigen-Specific Treg Therapy in Type 1 Diabetes—Challenges and Opportunities. Front. Immunol. 2021, 12, 712870. [Google Scholar] [CrossRef]
- Taams, L.S.; Smith, J.; Rustin, M.H.; Salmon, M.; Poulter, L.W.; Akbar, A.N. Human Anergic/Suppressive CD4+CD25+ T Cells: A Highly Differentiated and Apoptosis-prone Population. Eur. J. Immunol. 2001, 31, 1122–1131. [Google Scholar] [CrossRef]
- Koenecke, C.; Czeloth, N.; Bubke, A.; Schmitz, S.; Kissenpfennig, A.; Malissen, B.; Huehn, J.; Ganser, A.; Förster, R.; Prinz, I. Alloantigen-Specific de Novo-Induced Foxp3+ Treg Revert in Vivo and Do Not Protect from Experimental GVHD. Eur. J. Immunol. 2009, 39, 3091–3096. [Google Scholar] [CrossRef]
- Mikami, N.; Kawakami, R.; Chen, K.Y.; Sugimoto, A.; Ohkura, N.; Sakaguchi, S. Epigenetic Conversion of Conventional T Cells into Regulatory T Cells by CD28 Signal Deprivation. Proc. Natl. Acad. Sci. USA 2020, 117, 12258–12268. [Google Scholar] [CrossRef]
- Akamatsu, M.; Mikami, N.; Ohkura, N.; Kawakami, R.; Kitagawa, Y.; Sugimoto, A.; Hirota, K.; Nakamura, N.; Ujihara, S.; Kurosaki, T.; et al. Conversion of Antigen-Specific Effector/Memory T Cells into Foxp3-Expressing Treg Cells by Inhibition of CDK8/19. Sci. Immunol. 2019, 4, eaaw2707. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Song, M.H.; Oh, K.I. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. J. Immunol. 2016, 196, 2119–2131. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Trifari, S.; Äijö, T.; Tsagaratou, A.; Pastor, W.A.; Zepeda-Martínez, J.A.; Lio, C.-W.J.; Li, X.; Huang, Y.; Vijayanand, P.; et al. Control of Foxp3 Stability through Modulation of TET Activity. J. Exp. Med. 2016, 213, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Requejo Cier, C.J.; Valentini, N.; Lamarche, C. Unlocking the Potential of Tregs: Innovations in CAR Technology. Front. Mol. Biosci. 2023, 10, 1267762. [Google Scholar] [CrossRef] [PubMed]
- Kalin, J.H.; Butler, K.V.; Akimova, T.; Hancock, W.W.; Kozikowski, A.P. Second-Generation Histone Deacetylase 6 Inhibitors Enhance the Immunosuppressive Effects of Foxp3+ T-Regulatory Cells. J. Med. Chem. 2012, 55, 639–651. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.S.; Jang, S.W.; Lee, G.R. Histone Deacetylase 6 Plays an Important Role in TGF-β-Induced Murine Treg Cell Differentiation by Regulating Cell Proliferation. Sci. Rep. 2022, 12, 22550. [Google Scholar] [CrossRef]
- Passerini, L.; Mel, E.R.; Sartirana, C.; Fousteri, G.; Bondanza, A.; Naldini, L.; Roncarolo, M.G.; Bacchetta, R. CD4 + T Cells from IPEX Patients Convert into Functional and Stable Regulatory T Cells by FOXP3 Gene Transfer. Sci. Transl. Med. 2013, 5, 215ra174. [Google Scholar] [CrossRef]
- Honaker, Y.; Hubbard, N.; Xiang, Y.; Fisher, L.; Hagin, D.; Sommer, K.; Song, Y.; Yang, S.J.; Lopez, C.; Tappen, T.; et al. Gene Editing to Induce FOXP3 Expression in Human CD4 + T Cells Leads to a Stable Regulatory Phenotype and Function. Sci. Transl. Med. 2020, 12, eaay6422. [Google Scholar] [CrossRef]
- Bulygin, A.S.; Khantakova, J.N.; Fisher, M.S.; Tereshchenko, V.P.; Kurilin, V.V.; Shkaruba, N.S.; Sennikov, S.V. Generation and Characterization of Stable Antigen-Specific T-Regulatory Cells Using Blockers of Cyclin-Dependent Protein Kinases. Immunologiya 2023, 44, 147–156. [Google Scholar] [CrossRef]
- Borna, S.; Lee, E.; Sato, Y.; Bacchetta, R. Towards Gene Therapy for IPEX Syndrome. Eur. J. Immunol. 2022, 52, 705–716. [Google Scholar] [CrossRef]
- Yang, S.J.; Singh, A.K.; Drow, T.; Tappen, T.; Honaker, Y.; Barahmand-pour-Whitman, F.; Linsley, P.S.; Cerosaletti, K.; Mauk, K.; Xiang, Y.; et al. Pancreatic Islet-Specific Engineered Tregs Exhibit Robust Antigen-Specific and Bystander Immune Suppression in Type 1 Diabetes Models. Sci. Transl. Med. 2022, 14, eabn1716. [Google Scholar] [CrossRef] [PubMed]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T Cells Engineered with a Novel Insulin-Specific Chimeric Antigen Receptor as a Candidate Immunotherapy for Type 1 Diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Hehlgans, T.; Feuerer, M. Engineered Treg Cells as Putative Therapeutics against Inflammatory Diseases and Beyond. Trends Immunol. 2023, 44, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef]
- Kuball, J.; Dossett, M.L.; Wolfl, M.; Ho, W.Y.; Voss, R.-H.; Fowler, C.; Greenberg, P.D. Facilitating Matched Pairing and Expression of TCR Chains Introduced into Human T Cells. Blood 2007, 109, 2331–2338. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Uckert, W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J. Immunol. 2010, 184, 6223–6231. [Google Scholar] [CrossRef]
- Bialer, G.; Horovitz-Fried, M.; Ya’acobi, S.; Morgan, R.A.; Cohen, C.J. Selected Murine Residues Endow Human TCR with Enhanced Tumor Recognition. J. Immunol. 2010, 184, 6232–6241. [Google Scholar] [CrossRef]
- Deshpande, D.; Orange, J. Reprogramming Human T Cell Function and Specificity With Non–Viral Genome Targeting. Pediatrics 2019, 144, S63–S64. [Google Scholar] [CrossRef]
- Dawson, N.A.J.; Rosado-Sánchez, I.; Novakovsky, G.E.; Fung, V.C.W.; Huang, Q.; McIver, E.; Sun, G.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Functional Effects of Chimeric Antigen Receptor Co-Receptor Signaling Domains in Human Regulatory T Cells. Sci. Transl. Med. 2020, 12, eaaz3866. [Google Scholar] [CrossRef]
- Sanders, J.M.; Jeyamogan, S.; Mathew, J.M.; Leventhal, J.R. Foxp3+ Regulatory T Cell Therapy for Tolerance in Autoimmunity and Solid Organ Transplantation. Front. Immunol. 2022, 13, 1055466. [Google Scholar] [CrossRef]
- Lee, J.C.; Hayman, E.; Pegram, H.J.; Santos, E.; Heller, G.; Sadelain, M.; Brentjens, R. In Vivo Inhibition of Human CD19-Targeted Effector T Cells by Natural T Regulatory Cells in a Xenotransplant Murine Model of B Cell Malignancy. Cancer Res. 2011, 71, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-Cell Therapy in Autoimmune Disease—A Case Series with Follow-Up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef]
- Hagen, M.; Müller, F.; Wirsching, A.; Kharboutli, S.; Spoerl, S.; Düsing, C.; Krickau, T.; Metzler, M.; Völkl, S.; Aigner, M.; et al. Local Immune Effector Cell-Associated Toxicity Syndrome in CAR T-Cell Treated Patients with Autoimmune Disease: An Observational Study. Lancet Rheumatol 2025, 7, e424–e433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xiao, Z.X.; Zheng, X.; Wang, G.; Yang, L.; Shi, L.; Xiang, N.; Wang, X.; Zha, G.-F.; Schett, G.; et al. In Vivo CD19 CAR T-Cell Therapy for Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2025, 393, 1542–1544. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.G.; Hoeppli, R.E.; Huang, Q.; Gillies, J.; Luciani, D.S.; Orban, P.C.; Broady, R.; Levings, M.K. Alloantigen-Specific Regulatory T Cells Generated with a Chimeric Antigen Receptor. J. Clin. Investig. 2016, 126, 1413–1424. [Google Scholar] [CrossRef]
- Bastian, H.; Lounnas-Mourey, N.; Heimendinger, P.; Hsu, B.L.; Schreeb, K.H.; Chapman, C.; Culme-Seymour, E.; Atkinson, G.F.; Cantarovich, D. Feasibility of Manufacture of Chimeric Antigen Receptor-Regulatory T Cells from Patients with End-Stage Renal Disease. Transl. Med. Commun. 2023, 8, 15. [Google Scholar] [CrossRef]
- Cui, Y.; David, M.; Bouchareychas, L.; Rouquier, S.; Sajuthi, S.; Ayrault, M.; Navarin, C.; Lara, G.; Lafon, A.; Saviane, G.; et al. IL23R-Specific CAR Tregs for the Treatment of Crohn’s Disease. J. Crohns Colitis 2024, 19, jjae135. [Google Scholar] [CrossRef]
- Elinav, E.; Adam, N.; Waks, T.; Eshhar, Z. Amelioration of Colitis by Genetically Engineered Murine Regulatory T Cells Redirected by Antigen-Specific Chimeric Receptor. Gastroenterology 2009, 129, 1721–1731. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.-H.; Yoon, J.; Culp, W.E.; Lees, J.R.; Wucherpfennig, K.W.; Scott, D.W. Engineered MBP-Specific Human Tregs Ameliorate MOG-Induced EAE through IL-2-Triggered Inhibition of Effector T Cells. J. Autoimmun. 2018, 92, 77–86. [Google Scholar] [CrossRef]
- De Paula Pohl, A.; Schmidt, A.; Zhang, A.-H.; Maldonado, T.; Königs, C.; Scott, D.W. Engineered Regulatory T Cells Expressing Myelin-Specific Chimeric Antigen Receptors Suppress EAE Progression. Cell Immunol. 2020, 358, 104222. [Google Scholar] [CrossRef]
- Mukhatayev, Z.; Dellacecca, E.R.; Cosgrove, C.; Shivde, R.; Jaishankar, D.; Pontarolo-Maag, K.; Eby, J.M.; Henning, S.W.; Ostapchuk, Y.O.; Cedercreutz, K.; et al. Antigen Specificity Enhances Disease Control by Tregs in Vitiligo. Front. Immunol. 2020, 11, 581433. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.P.; Notley, C.A.; Xue, S.-A.; Bendle, G.M.; Holler, A.; Schumacher, T.N.; Ehrenstein, M.R.; Stauss, H.J. Adoptive Therapy with Redirected Primary Regulatory T Cells Results in Antigen-Specific Suppression of Arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 19078–19083. [Google Scholar] [CrossRef]
- Raffin, C.; Zhou, Y.; Piccoli, L.; Lanzavecchia, A.; Sadelain, M.; Tareen, S.U.; Fontenot, J.D.; Bluestone, J.A. Development of Citrullinated-Vimentin-Specific CAR for Targeting Tregs to Treat Autoimmune Rheumatoid Arthritis. J. Immunol. 2018, 200, 176.17. [Google Scholar] [CrossRef]
- Imam, S.; Jaume, J. MON-LB033 Unleashing the Anti-Inflammatory Potential of Treg Cells Against Type I Diabetes Using Advanced Chimeric Antigen Receptor Technology. J. Endocr. Soc. 2019, 3, MON-LB033. [Google Scholar] [CrossRef]
- Radichev, I.A.; Yoon, J.; Scott, D.W.; Griffin, K.; Savinov, A.Y. Towards Antigen-Specific Tregs for Type 1 Diabetes: Construction and Functional Assessment of Pancreatic Endocrine Marker, HPi2-Based Chimeric Antigen Receptor. Cell. Immunol. 2020, 358, 104224. [Google Scholar] [CrossRef]
- Yeh, W.-I.; Seay, H.R.; Newby, B.; Posgai, A.L.; Moniz, F.B.; Michels, A.; Mathews, C.E.; Bluestone, J.A.; Brusko, T.M. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front. Immunol. 2017, 8, 1313. [Google Scholar] [CrossRef]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I.M. Generation of Human Islet-Specific Regulatory T Cells by TCR Gene Transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.-H.; Su, Y.; Rieder, S.A.; Rossi, R.J.; Ettinger, R.A.; Pratt, K.P.; Shevach, E.M.; Scott, D.W. Engineered Antigen-Specific Human Regulatory T Cells: Immunosuppression of FVIII-Specific T- and B-Cell Responses. Blood 2015, 125, 1107–1115. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Cheong, R.M.Y.; Lo, C.; Chang, J.; Ng, B.H.; Ting, Y.T.; Monk, J.A.; Loh, K.L.; Broury, A.; Tay, E.S.V.; et al. Smith-Specific Regulatory T Cells Halt the Progression of Lupus Nephritis. Nat. Commun. 2024, 15, 899. [Google Scholar] [CrossRef]
- Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Fisher, M.S. Cellular Technologies in Immunotherapy of Autoimmune Diseases. Meditsinskaya Immunol. 2023, 44, 813–824. [Google Scholar] [CrossRef]
- Sprouse, M.L.; Shevchenko, I.; Scavuzzo, M.A.; Joseph, F.; Lee, T.; Blum, S.; Borowiak, M.; Bettini, M.L.; Bettini, M. Cutting Edge: Low-Affinity TCRs Support Regulatory T Cell Function in Autoimmunity. J. Immunol. 2018, 200, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Doglio, M.; Ugolini, A.; Bercher-Brayer, C.; Camisa, B.; Toma, C.; Norata, R.; Del Rosso, S.; Greco, R.; Ciceri, F.; Sanvito, F.; et al. Regulatory T Cells Expressing CD19-Targeted Chimeric Antigen Receptor Restore Homeostasis in Systemic Lupus Erythematosus. Nat. Commun. 2024, 15, 2542. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.R.; Maghsoudlou, P.; Copland, D.A.; Nicholson, L.B.; Dick, A.D. CAR-Treg Cell Therapies and Their Future Potential in Treating Ocular Autoimmune Conditions. Front. Ophthalmol. 2023, 3, 1184937. [Google Scholar] [CrossRef]
- Wermke, M.; Kraus, S.; Ehninger, A.; Bargou, R.C.; Goebeler, M.-E.; Middeke, J.M.; Kreissig, C.; von Bonin, M.; Koedam, J.; Pehl, M.; et al. Proof of Concept for a Rapidly Switchable Universal CAR-T Platform with UniCAR-T-CD123 in Relapsed/Refractory AML. Blood 2021, 137, 3145–3148. [Google Scholar] [CrossRef]
- Mitwasi, N.; Feldmann, A.; Bergmann, R.; Berndt, N.; Arndt, C.; Koristka, S.; Kegler, A.; Jureczek, J.; Hoffmann, A.; Ehninger, A.; et al. Development of Novel Target Modules for Retargeting of UniCAR T Cells to GD2 Positive Tumor Cells. Oncotarget 2017, 8, 108584–108603. [Google Scholar] [CrossRef]
- Kegler, A.; Koristka, S.; Bergmann, R.; Berndt, N.; Arndt, C.; Feldmann, A.; Hoffmann, A.; Bornhäuser, M.; Schmitz, M.; Bachmann, M.P. T Cells Engrafted with a UniCAR 28/z Outperform UniCAR BB/z-Transduced T Cells in the Face of Regulatory T Cell-Mediated Immunosuppression. Oncoimmunology 2019, 8, e1621676. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Monneaux, F.; Muller, S. Epitope Spreading in Systemic Lupus Erythematosus: Identification of Triggering Peptide Sequences. Arthritis Rheum. 2002, 46, 1430–1438. [Google Scholar] [CrossRef]
- Rui, X.; Calderon, F.A.; Wobma, H.; Gerdemann, U.; Albanese, A.; Cagnin, L.; Mcguckin, C.; Michaelis, K.A.; Naqvi, K.; Blazar, B.R.; et al. Human OX40L-CAR-T Regs Target Activated Antigen-Presenting Cells and Control T Cell Alloreactivity. Sci. Transl. Med. 2024, 16, eadj9331. [Google Scholar] [CrossRef]
- Karim, M.; Feng, G.; Wood, K.J.; Bushell, A.R. CD25+CD4+ Regulatory T Cells Generated by Exposure to a Model Protein Antigen Prevent Allograft Rejection: Antigen-Specific Reactivation in Vivo Is Critical for Bystander Regulation. Blood 2005, 105, 4871–4877. [Google Scholar] [CrossRef]
- Croft, M. Control of Immunity by the TNFR-Related Molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, C.; Schmitt, N.; Contin-Bordes, C.; Richez, C.; Lazaro, E.; Duffau, P.; Truchetet, M.-E.; Pascual, V.; Ueno, H.; Blanco, P. OP0009 OX40 Ligand Contributes to Human Lupus Pathogenesis by Promoting T Follicular Helper Response. Ann. Rheum. Dis. 2015, 74, 67. [Google Scholar] [CrossRef]
- Forcade, E.; Kim, H.T.; Cutler, C.; Wang, K.; Alho, A.C.; Nikiforow, S.; Ho, V.T.; Koreth, J.; Armand, P.; Alyea, E.P.; et al. Circulating T Follicular Helper Cells with Increased Function during Chronic Graft-versus-Host Disease. Blood 2016, 127, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Kim, H.T.; Nikiforow, S.; Heubeck, A.T.; Ho, V.T.; Koreth, J.; Alyea, E.P.; Armand, P.; Blazar, B.R.; Soiffer, R.J.; et al. Antibodies Targeting Surface Membrane Antigens in Patients with Chronic Graft-versus-Host Disease. Blood 2017, 130, 2889–2899. [Google Scholar] [CrossRef]
- Laustsen, J.K.; Rasmussen, T.K.; Stengaard-Pedersen, K.; Hørslev-Petersen, K.; Hetland, M.L.; Østergaard, M.; Junker, P.; Hvid, M.; Deleuran, B. Soluble OX40L Is Associated with Presence of Autoantibodies in Early Rheumatoid Arthritis. Arthritis Res. Ther. 2014, 16, 474. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, C.; Liu, M.; Shen, Y.; Hu, X.; Wang, Q.; Wu, J.; Wu, M.; Fang, Q.; Zhang, X. OX40 Signaling Is Involved in the Autoactivation of CD4+CD28− T Cells and Contributes to the Pathogenesis of Autoimmune Arthritis. Arthritis Res. Ther. 2017, 19, 67. [Google Scholar] [CrossRef]
- Duffus, K.; López-Isac, E.; Teruel, M.; Simeón, C.P.; Carreria, P.; Ortego-Centeno, N.; Vicente, E.; Worthington, J.; Herrick, A.L.; Martin, J. Association of TNFSF4 (OX40L) Polymorphisms with Systemic Sclerosis–Related Calcinosis. Rheumatology 2019, 58, 1299–1301. [Google Scholar] [CrossRef]
- Elhai, M.; Avouac, J.; Hoffmann-Vold, A.M.; Ruzehaji, N.; Amiar, O.; Ruiz, B.; Brahiti, H.; Ponsoye, M.; Fréchet, M.; Burgevin, A.; et al. OX40L Blockade Protects against Inflammation-Driven Fibrosis. Proc. Natl. Acad. Sci. USA 2016, 113, E3901–E3910. [Google Scholar] [CrossRef]
- Sitrin, J.; Suto, E.; Wuster, A.; Eastham-Anderson, J.; Kim, J.M.; Austin, C.D.; Lee, W.P.; Behrens, T.W. The Ox40/Ox40 Ligand Pathway Promotes Pathogenic Th Cell Responses, Plasmablast Accumulation, and Lupus Nephritis in NZB/W F1 Mice. J. Immunol. 2017, 199, 1238–1249. [Google Scholar] [CrossRef]
- Cortini, A.; Ellinghaus, U.; Malik, T.H.; Cunninghame Graham, D.S.; Botto, M.; Vyse, T.J. B Cell OX40L Supports T Follicular Helper Cell Development and Contributes to SLE Pathogenesis. Ann. Rheum. Dis. 2017, 76, 2095–2103. [Google Scholar] [CrossRef]
- Bittner, S.; Ruhland, B.; Hofmann, V.; Schmidleithner, L.; Schambeck, K.; Pant, A.; Stüve, P.; Delacher, M.; Echtenacher, B.; Edinger, M.; et al. Biosensors for Inflammation as a Strategy to Engineer Regulatory T Cells for Cell Therapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2208436119. [Google Scholar] [CrossRef]
- Wong, S.P.; Argyros, O.; Harbottle, R.P. Sustained Expression from DNA Vectors. Adv Genet 2015, 89, 113–152. [Google Scholar] [CrossRef]
- Wong, S.P.; Argyros, O.; Coutelle, C.; Harbottle, R.P. Non-Viral S/MAR Vectors Replicate Episomally in Vivo When Provided with a Selective Advantage. Gene Ther. 2011, 18, 82–87. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bozza, M.; De Roia, A.; Correia, M.P.; Berger, A.; Tuch, A.; Schmidt, A.; Zörnig, I.; Jäger, D.; Schmidt, P.; Harbottle, R.P. A Nonviral, Nonintegrating DNA Nanovector Platform for the Safe, Rapid, and Persistent Manufacture of Recombinant T Cells. Sci. Adv. 2021, 7, eabf1333. [Google Scholar] [CrossRef] [PubMed]
- Choileáin, S.N.; Kleinewietfeld, M.; Raddassi, K.; Hafler, D.A.; Ruff, W.E.; Longbrake, E.E. CXCR3+ T Cells in Multiple Sclerosis Correlate with Reduced Diversity of the Gut Microbiome CXCR3+ T Cells in Multiple Sclerosis Correlate with Reduced Diversity of the Gut Microbiome. J. Transl. Autoimmun. 2020, 3, 100032. [Google Scholar] [CrossRef] [PubMed]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef]
- Furlan, S.N.; Singh, K.; Lopez, C.; Tkachev, V.; Hunt, D.J.; Hibbard, J.; Betz, K.M.; Blazar, B.R.; Trapnell, C.; Kean, L.S. IL-2 Enhances Ex Vivo–Expanded Regulatory T-Cell Persistence after Adoptive Transfer. Blood Adv. 2020, 4, 1594–1605. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. MTOR Signaling, Tregs and Immune Modulation. Immunotherapy 2014, 6, 1295–1311. [Google Scholar] [CrossRef]
- Li, X.; Liang, Y.; LeBlanc, M.; Benner, C.; Zheng, Y. Function of a Foxp3 Cis-Element in Protecting Regulatory T Cell Identity. Cell 2014, 158, 734–748. [Google Scholar] [CrossRef]
- He, J.; Chen, J.; Miao, M.; Zhang, R.; Cheng, G.; Wang, Y.; Feng, R.; Huang, B.; Luan, H.; Jia, Y.; et al. Efficacy and Safety of Low-Dose Interleukin 2 for Primary Sjögren Syndrome: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2241451. [Google Scholar] [CrossRef]
- Graßhoff, H.; Comdühr, S.; Monne, L.R.; Müller, A.; Lamprecht, P.; Riemekasten, G.; Humrich, J.Y. Low-Dose IL-2 Therapy in Autoimmune and Rheumatic Diseases. Front. Immunol. 2021, 12, 648408. [Google Scholar] [CrossRef]
- Le Menn, G.; Jabłońska, A.; Chen, Z. The Effects of Post-Translational Modifications on Th17/Treg Cell Differentiation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2022, 1869, 119223. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, K.; Nidhi, M.; Chandran, S.; Shevkoplyas, S.S.; Mohan, C. Manufacturing Regulatory T Cells for Adoptive Cell Therapy in Immune Diseases: A Critical Appraisal. Clin. Immunol. 2023, 251, 109328. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, A.C.; Larson, R.C.; Choi, B.D.; Bouffard, A.A.; Riley, L.S.; Schiferle, E.; Kulkarni, A.S.; Cetrulo, C.L.; Ting, D.; Blazar, B.R.; et al. Chimeric Antigen Receptor Costimulation Domains Modulate Human Regulatory T Cell Function. JCI Insight 2019, 4, e126194. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.; Klüver, A.; Fertig, L.; Menkhoff, V.D.; Subklewe, M.; Endres, S.; Kobold, S. Mechanisms and Strategies for Safe Chimeric Antigen Receptor T-cell Activity Control. Int J Cancer 2023, 153, 1706–1725. [Google Scholar] [CrossRef]
- Guercio, M.; Manni, S.; Boffa, I.; Caruso, S.; Di Cecca, S.; Sinibaldi, M.; Abbaszadeh, Z.; Camera, A.; Ciccone, R.; Polito, V.A.; et al. Inclusion of the Inducible Caspase 9 Suicide Gene in CAR Construct Increases Safety of CAR.CD19 T Cell Therapy in B-Cell Malignancies. Front. Immunol. 2021, 12, 755639. [Google Scholar] [CrossRef]
- Fritsche, E.; Volk, H.D.; Reinke, P.; Abou-El-Enein, M. Toward an Optimized Process for Clinical Manufacturing of CAR-Treg Cell Therapy. Trends Biotechnol 2020, 38, 1099–1112. [Google Scholar] [CrossRef]
- Stoops, J.; Morton, T.; Powell, J.; Pace, A.L.; Bluestone, J.A. Treg Cell Therapy Manufacturability: Current State of the Art, Challenges and New Opportunities. Front. Immunol. 2025, 16, 1604483. [Google Scholar] [CrossRef]
- Balcerek, J.; Shy, B.R.; Putnam, A.L.; Masiello, L.M.; Lares, A.; Dekovic, F.; Acevedo, L.; Lee, M.R.; Nguyen, V.; Liu, W.; et al. Polyclonal Regulatory T Cell Manufacturing Under CGMP: A Decade of Experience. Front. Immunol. 2021, 12, 744763. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).