
Novel Hydroxyl-Functional Aliphatic CO2-Based Polycarbonates: Synthesis and Properties

 , , , , , , ,
, , , , , , ,  and
and 
Abstract
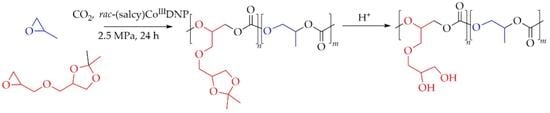
1. Introduction
2. Results and Discussion
2.1. Terpolymerization of Solketal Glycidyl Ether, Propylene Oxide, and Carbon Dioxide
2.2. Deprotection of Poly(solketal glycidyl ether carbonate-co-propylene carbonate)
2.3. Properties of Poly(solketal glycidyl ether carbonate-co-propylene carbonate) Before and After Deprotection
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Solketal Glycidyl Ether
3.3. Polymer Synthesis
3.4. Deprotection of Copolymers Containing SolGE Carbonate Units
3.5. Depolymerization
3.6. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhat, G.A.; Darensbourg, D.J. Coordination complexes as catalysts for the coupling reactions of oxiranes and carbon dioxide. Coord. Chem. Rev. 2023, 492, 215277. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, Y.; Yin, H.; Chen, X.; Wang, N.; Wang, Z. A review on catalytic copolymerization of carbon dioxide and epoxides. Mater. Today Sustain. 2025, 31, 101148. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Insertion reactions of CO2 and COS into metal-alkoxide bonds: Reactivity differences on copolymer formation with epoxides. J. Organomet. Chem. 2024, 1017, 123264. [Google Scholar] [CrossRef]
- Chen, C.; Gnanou, Y.; Feng, X. Borinane-based organoboron catalysts for alternating copolymerization of CO2 with cyclic ethers: Improved productivity and facile recovery. Polym. Chem. 2022, 13, 6312–6321. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, T.; Wang, S.; Han, D.; Huang, S.; Guo, H.; Xiao, M.; Meng, Y. CO2 derived ABA triblock all-polycarbonate thermoplastic elastomer with ultra-high elastic recovery. J. CO2 Util. 2024, 85, 102853. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Zhang, H.; Bordeianu, C.; Colson, Y.L.; Grinstaff, M.W. Synthesis of Polyethylene Glycol–Poly(glycerol carbonate) Block Copolymeric Micelles as Surfactant-Free Drug Delivery Systems. ACS Macro Lett. 2023, 12, 974–979. [Google Scholar] [CrossRef]
- Rosetto, G.; Vidal, F.; McGuire, T.M.; Kerr, R.W.F.; Williams, C.K. High Molar Mass Polycarbonates as Closed-Loop Recyclable Thermoplastics. J. Am. Chem. Soc. 2024, 146, 8381–8393. [Google Scholar] [CrossRef]
- Siragusa, F.; Detrembleur, C.; Grignard, B. The advent of recyclable CO2-based polycarbonates. Polym. Chem. 2023, 14, 1164–1183. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, X.-B. Current Challenges and Perspectives in CO2-Based Polymers. Macromolecules 2023, 56, 1759–1777. [Google Scholar] [CrossRef]
- Zheng, X.-P.; Wang, W.-Z.; Cui, Y.-K.; Jia, X.-G.; Li, H.-J.; Liu, X.-Y.; Chen, H.-P.; Mao, Z.-H. Degradable Pressure-Sensitive Adhesive Prepared From CO2 -Based Polycarbonate. J. Polym. Sci. 2025, 63, 864–875. [Google Scholar] [CrossRef]
- Chernikova, E.V.; Beletskaya, I.P. From epoxides and carbon dioxide to polycarbonates: Synthesis, properties and applications. Russ. Chem. Rev. 2024, 93, RCR5112. [Google Scholar] [CrossRef]
- Cui, S.; Borgemenke, J.; Liu, Z.; Keener, H.M.; Li, Y. Innovative sustainable conversion from CO2 and biodiesel-based crude glycerol waste to bio-based polycarbonates. J. CO2 Util. 2019, 34, 198–206. [Google Scholar] [CrossRef]
- Li, H.; Wang, W.; Liu, S.; Xue, D.; Wang, J.; Liu, Y.; Huang, Q. Design and syntheses of functional carbon dioxide-based polycarbonates via ternary copolymerization. J. CO2 Util. 2024, 80, 102689. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, S.; Li, P.; Wang, H.; Cui, X.; Qian, B.; Shi, F. Zinc-Catalyzed Carbon Dioxide Based Biodegradable Polycarbonate Synthesis. Synthesis 2025, 57, 1791–1806. [Google Scholar] [CrossRef]
- Ye, S.; Wang, S.; Lin, L.; Xiao, M.; Meng, Y. CO2 derived biodegradable polycarbonates: Synthesis, modification and applications. Adv. Ind. Eng. Polym. Res. 2019, 2, 143–160. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Q.; Ji, R.; Xie, E.; Sun, A.; Xiao, B.; Huang, C.; Ma, S.; Wei, J.; Yang, X.; et al. Biodegradable composites of poly(propylene carbonate) mixed with silicon nitride for osteogenic activity of adipose-derived stem cells and repair of bone defects. J. Mater. Chem. B 2023, 11, 6922–6933. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Comments on the depolymerization of polycarbonates derived from epoxides and carbon dioxide: A mini review. Polym. Degrad. Stab. 2018, 149, 45–51. [Google Scholar] [CrossRef]
- Phillips, O.; Schwartz, J.M.; Kohl, P.A. Thermal decomposition of poly(propylene carbonate): End-capping, additives, and solvent effects. Polym. Degrad. Stab. 2016, 125, 129–139. [Google Scholar] [CrossRef]
- Gallin, C.F.; Lee, W.-W.; Byers, J.A. A Simple, Selective, and General Catalyst for Ring Closing Depolymerization of Polyesters and Polycarbonates for Chemical Recycling. Angew. Chem. Int. Ed. 2023, 62, e202303762. [Google Scholar] [CrossRef]
- Liu, W.-N.; Wang, M.; Ding, Z.; Li, Y.; Wang, B. Modular Access to Aliphatic Polycarbonates with Tunable Properties and Dual Closed-Loop Recyclability by Polycondensation–Depolymerization–Repolymerization Strategy. Angew. Chem. Int. Ed. 2025, 64, e202505333. [Google Scholar] [CrossRef]
- Ekladious, I.; Liu, R.; Zhang, H.; Foil, D.H.; Todd, D.A.; Graf, T.N.; Padera, R.F.; Oberlies, N.H.; Colson, Y.L.; Grinstaff, M.W. Synthesis of poly(1,2-glycerol carbonate)–paclitaxel conjugates and their utility as a single high-dose replacement for multi-dose treatment regimens in peritoneal cancer. Chem. Sci. 2017, 8, 8443–8450. [Google Scholar] [CrossRef]
- Thomas, A.; Müller, S.S.; Frey, H. Beyond Poly(ethylene glycol): Linear Polyglycerol as a Multifunctional Polyether for Biomedical and Pharmaceutical Applications. Biomacromolecules 2014, 15, 1935–1954. [Google Scholar] [CrossRef]
- Ray, W.C.; Grinstaff, M.W. Polycarbonate and Poly(carbonate−ester)s Synthesized from Biocompatible Building Blocks of Glycerol and Lactic Acid. Macromolecules 2003, 36, 3557–3562. [Google Scholar] [CrossRef]
- Wolinsky, J.B.; Ray, W.C.; Colson, Y.L.; Grinstaff, M.W. Poly(carbonate ester)s Based on Units of 6-Hydroxyhexanoic Acid and Glycerol. Macromolecules 2007, 40, 7065–7068. [Google Scholar] [CrossRef]
- Parzuchowski, P.G.; Świderska, A.; Roguszewska, M.; Frączkowski, T.; Tryznowski, M. Amine functionalized polyglycerols obtained by copolymerization of cyclic carbonate monomers. Polymer 2018, 151, 250–260. [Google Scholar] [CrossRef]
- Holmiere, S.; Valentin, R.; Maréchal, P.; Mouloungui, Z. Esters of oligo-(glycerol carbonate-glycerol): New biobased oligomeric surfactants. J. Colloid Interface Sci. 2017, 487, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Rokicki, G.; Rakoczy, P.; Parzuchowski, P.; Sobiecki, M. Hyperbranched aliphatic polyethers obtained from environmentally benign monomer: Glycerol carbonate. Green Chem. 2005, 7, 529–539. [Google Scholar] [CrossRef]
- Sonnati, M.O.; Amigoni, S.; Taffin de Givenchy, E.P.; Darmanin, T.; Choulet, O.; Guittard, F. Glycerol carbonate as a versatile building block for tomorrow: Synthesis, reactivity, properties and applications. Green Chem. 2013, 15, 283–306. [Google Scholar] [CrossRef]
- Helou, M.; Miserque, O.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Organocatalysts for the Controlled “Immortal” Ring-Opening Polymerization of Six-Membered-Ring Cyclic Carbonates: A Metal-Free, Green Process. Chem. Eur. J. 2010, 16, 13805–13813. [Google Scholar] [CrossRef]
- Simon, J.; Olsson, J.V.; Kim, H.; Tenney, I.F.; Waymouth, R.M. Semicrystalline Dihydroxyacetone Copolymers Derived from Glycerol. Macromolecules 2012, 45, 9275–9281. [Google Scholar] [CrossRef]
- Inoue, S. Copolymerization of Carbon Dioxide and Epoxide: Functionality of the Copolymer. J. Macromol. Sci. Chem. A. 1979, 13, 651–664. [Google Scholar] [CrossRef]
- Zhang, H.; Grinstaff, M.W. Synthesis of Atactic and Isotactic Poly(1,2-glycerol carbonate)s: Degradable Polymers for Biomedical and Pharmaceutical Applications. J. Am. Chem. Soc. 2013, 135, 6806–6809. [Google Scholar] [CrossRef]
- Scharfenberg, M.; Hilf, J.; Frey, H. Functional Polycarbonates from Carbon Dioxide and Tailored Epoxide Monomers: Degradable Materials and Their Application Potential. Adv. Funct. Mater. 2018, 28, 1704302. [Google Scholar] [CrossRef]
- Geschwind, J.; Frey, H. Stable, Hydroxyl Functional Polycarbonates with Glycerol Side Chains Synthesized From CO2 and Isopropylidene(glyceryl glycidyl ether). Macromol. Rapid Commun. 2013, 34, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Łukaszczyk, J.; Jaszcz, K.; Kuran, W.; Listoś, T. Synthesis and Modification of Functional Polycarbonates with Pendant Allyl Groups. Macromol. Biosci. 2001, 1, 282–289. [Google Scholar] [CrossRef]
- Geschwind, J.; Frey, H. Poly(1,2-glycerol carbonate): A Fundamental Polymer Structure Synthesized from CO2 and Glycidyl Ethers. Macromolecules 2013, 46, 3280–3287. [Google Scholar] [CrossRef]
- Nakamura, M.; Tominaga, Y. Utilization of carbon dioxide for polymer electrolytes [II]: Synthesis of alternating copolymers with glycidyl ethers as novel ion-conductive polymers. Electrochim. Acta. 2011, 57, 36–39. [Google Scholar] [CrossRef]
- Tominaga, Y.; Shimomura, T.; Nakamura, M. Alternating copolymers of carbon dioxide with glycidyl ethers for novel ion-conductive polymer electrolytes. Polymer 2010, 51, 4295–4298. [Google Scholar] [CrossRef]
- Konieczynska, M.D.; Lin, X.; Zhang, H.; Grinstaff, M.W. Synthesis of Aliphatic Poly(ether 1,2-glycerol carbonate)s via Copolymerization of CO2 with Glycidyl Ethers Using a Cobalt Salen Catalyst and Study of a Thermally Stable Solid Polymer Electrolyte. ACS Macro Lett. 2015, 4, 533–537. [Google Scholar] [CrossRef]
- Łukaszczyk, J.; Jaszcz, K.; Kuran, W.; Listos, T. Synthesis of functional polycarbonates by copolymerization of carbon dioxide with allyl glycidyl ether. Macromol. Rapid Commun. 2000, 21, 754–757. [Google Scholar] [CrossRef]
- Geschwind, J.; Wurm, F.; Frey, H. From CO2-Based Multifunctional Polycarbonates with a Controlled Number of Functional Groups to Graft Polymers. Macromol. Chem. Phys. 2013, 214, 892–901. [Google Scholar] [CrossRef]
- Deng, K.; Wang, S.; Ren, S.; Han, D.; Xiao, M.; Meng, Y. A Novel Single-Ion-Conducting Polymer Electrolyte Derived from CO2-Based Multifunctional Polycarbonate. ACS Appl. Mater. Interfaces 2016, 8, 33642–33648. [Google Scholar] [CrossRef]
- Yang, G.-W.; Zhang, Y.-Y.; Wang, Y.; Wu, G.-P.; Xu, Z.-K.; Darensbourg, D.J. Construction of Autonomic Self-Healing CO2-Based Polycarbonates via One-Pot Tandem Synthetic Strategy. Macromolecules 2018, 51, 1308–1313. [Google Scholar] [CrossRef]
- Hilf, J.; Frey, H. Propargyl-Functional Aliphatic Polycarbonate Obtained from Carbon Dioxide and Glycidyl Propargyl Ether. Macromol. Rapid Commun. 2013, 34, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Yi, M.J.; Byun, S.H.; Park, D.W.; Kim, B.U.; Ha, C.S. Biodegradable Polycarbonate Synthesis by Copolymerization of Carbon Dioxide with Epoxides Using a Heterogeneous Zinc Complex. Macromol. Symp. 2005, 224, 181–192. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, H.; Nörnberg, B.; Theato, P.; Luinstra, G.A. Synthesis and Characterization of Hydroxyl-Functionalized Poly(propylene carbonate). Macromolecules 2014, 47, 492–497. [Google Scholar] [CrossRef]
- Inoue, S. Immortal polymerization: The outset, development, and application. J. Polym. Sci. A Polym. Chem. 2000, 38, 2861–2871. [Google Scholar] [CrossRef]
- Hilf, J.; Phillips, A.; Frey, H. Poly(carbonate) copolymers with a tailored number of hydroxyl groups from glycidyl ethers and CO2. Polym. Chem. 2014, 5, 814–818. [Google Scholar] [CrossRef]
- Beharaj, A.; McCaslin, E.Z.; Blessing, W.A.; Grinstaff, M.W. Sustainable polycarbonate adhesives for dry and aqueous conditions with thermoresponsive properties. Nat. Commun. 2019, 10, 5478. [Google Scholar] [CrossRef]
- Beharaj, A.; Ekladious, I.; Grinstaff, M.W. Poly(Alkyl Glycidate Carbonate)s as Degradable Pressure-Sensitive Adhesives. Angew. Chem. Int. Ed. 2019, 58, 1407–1411. [Google Scholar] [CrossRef]
- Rzhevskiy, S.A.; Shurupova, O.V.; Asachenko, A.F.; Plutalova, A.V.; Chernikova, E.V.; Beletskaya, I.P. The Role of Ligand Exchange in Salen Cobalt Complexes in the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide. Int. J. Mol. Sci. 2024, 25, 10946. [Google Scholar] [CrossRef]
- Yezrielev, A.I.; Brokhina, E.L.; Roskin, Y.S. An analytical method for calculating reactivity ratios. Polym. Sci. (USSR) (Engl. Transl.) 1969, 11, 1894–1907. [Google Scholar] [CrossRef]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Cohen, C.T.; Coates, G.W. Alternating copolymerization of propylene oxide and carbon dioxide with highly efficient and selective (salen)Co(III) catalysts: Effect of ligand and cocatalyst variation. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 5182–5191. [Google Scholar] [CrossRef]
- Rzhevskii, S.A.; Shurupova, O.V.; Topchii, M.A.; Asachenko, A.F.; Plutalova, A.V.; Beletskaya, I.P.; Chernikova, E.V. Salen cobalt complexes in polycarbonate synthesis: Bimodal versus unimodal molecular weight distribution. Russ. Chem. Bull. 2025, 74, 2156–2168. [Google Scholar] [CrossRef]
- Belov, G.P.; Lisitskaya, A.P.; Solovyeva, T.I.; Chirkov, N.M. Studies of molecular weight distribution of polyolefins. Eur. Polym. J. 1970, 6, 29–40. [Google Scholar] [CrossRef]
- Chernikova, E.V.; Mineeva, K.O. Reversible Deactivation Radical Copolymerization: Synthesis of Copolymers with Controlled Unit Sequence. Polym. Sci. Ser. C. 2022, 64, 1–25. [Google Scholar] [CrossRef]
- Li, X.; Han, L.; Zhang, R.; Li, C.; Zhang, S.; Bai, H.; Wang, X.; Wang, B.; Ma, H. Regulation from gradient to near periodic sequence during anionic copolymerization of styrene and dimethyl-[4-(1-phenyl-vinyl)phenyl]silane (DPE-SiH). Polymer 2022, 244, 124663. [Google Scholar] [CrossRef]
- Kersten, E.; Linker, O.; Blankenburg, J.; Wagner, M.; Walther, P.; Naumann, S.; Frey, H. Revealing the Monomer Gradient of Polyether Copolymers Prepared Using N-Heterocyclic Olefins: Metal-Free Anionic versus Zwitterionic Lewis Pair Polymerization. Macromol. Chem. Phys. 2023, 224, 2300097. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Perepelitsina, E.O.; Belov, G.P. The influence of reagent concentration on the kinetics of carbon dioxide-propylene oxide copolymerization in the presence of a cobalt complex. Polym. Sci. Ser. B. 2014, 56, 547–552. [Google Scholar] [CrossRef]
- Cohen, C.T.; Chu, T.; Coates, G.W. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J. Am. Chem. Soc. 2005, 127, 10869–10878. [Google Scholar] [CrossRef]
- Qin, Z.; Thomas, C.M.; Lee, S.; Coates, G.W. Cobalt-Based Complexes for the Copolymerization of Propylene Oxide and CO2: Active and Selective Catalysts for Polycarbonate Synthesis. Angew. Chem. Int. Ed. 2003, 42, 5484–5487. [Google Scholar] [CrossRef] [PubMed]
- Elmas, S.; Subhani, M.A.; Vogt, H.; Leitner, W.; Müller, T.E. Facile insertion of CO2 into metal–phenoxide bonds. Green Chem. 2013, 15, 1356–1360. [Google Scholar] [CrossRef]
- Iaych, K.; Dumarçay, S.; Fredon, E.; Gérardin, C.; Lemor, A.; Gérardin, P. Microwave-assisted synthesis of polyglycerol from glycerol carbonate. J. Appl. Polym. Sci. 2011, 120, 2354–2360. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| fSolGE | Time, h | Conversion, % | FSolGE | Conversion, wt% | Carbonate Linkage, % | Selectivity, wt% | TOF, h−1 | ||
|---|---|---|---|---|---|---|---|---|---|
| PO | SolGE | PO | SolGE | ||||||
| 0.10 | 1 | 7.8 | 14 | 0.17 | 8.6 | >99 | 95.4 | 89.7 | 373 |
| 0.21 | 1 | 4.5 | 9.7 | 0.36 | 6.0 | >99 | 92.7 | 90.4 | 242 |
| 0.32 | 2.3 | 8.4 | 14.5 | 0.44 | 10.5 | >99 | 95.5 | 94.0 | 207 |
| 0.43 | 2.3 | 7.1 | 12.5 | 0.57 | 9.3 | >99 | 93.6 | 91.8 | 184 |
| 0.53 | 2.1 | 3.5 | 10.9 | 0.78 | 7.8 | >99 | 84.0 | 92.0 | 154 |
| 0.61 | 2 | 3.1 | 6.2 | 0.76 | 4.8 | >99 | 84.0 | 88.0 | 111 |
| 0.70 | 2 | 5.1 | 6.5 | 0.77 | 5.4 | >99 | 76.2 | 76.9 | 149 |
| fSolGE | Conversion, % | Selectivity, % | Conversion, wt% | Carbonate Linkage, % | TOF, h−1 | Mn1/Mn2 × 10−3 | ĐM1/ĐM2 | ||
|---|---|---|---|---|---|---|---|---|---|
| PO | SolGE | PO | SolGE | ||||||
| 0.21 | 91.0 | 95.0 | 98.4 | 95.8 | 84.2 | >99 | 159 | 62.2/121.9 | 1.03/1.02 |
| 0.32 | 85.1 | 90.4 | 98.0 | 96.0 | 80.3 | >99 | 143 | 59.9/93.6 | 1.04/1.16 |
| 0.43 | 81.5 | 85.2 | 96.8 | 95.0 | 73.9 | >99 | 156 | 47.8/93.4 | 1.03/1.02 |
| 0.53 | 79.5 | 82.3 | 96.2 | 95.4 | 70.9 | >99 | 152 | 39.8/77.7 | 1.03/1.02 |
| 0.81 | 32.0 | 62.4 | 94.4 | 93.3 | 50.9 | >99 | 98 | 26.5/45.3 | 1.04/1.17 |
| 0.91 | 24.3 | 45.8 | 93.1 | 93.3 | 38.0 | >99 | 76 | 18.5/24.9 | 1.05/1.31 |
| Additive | Time, h | Conversion, % | Selectivity, % | Conversion, wt% | Mn1/Mn2 × 10−3 | ĐM1/ĐM2 | FSolGE | ||
|---|---|---|---|---|---|---|---|---|---|
| PO | SolGE | PO | SolGE | ||||||
| - | 70 | 66.4 | 83.8 | 98.5 | 95.3 | 65.6 | 78.7/157.8 | 1.05/1.05 | 0.12 |
| H2O | 100 | 74.1 | 97.0 | 98.7 | 95.5 | 74.0 | 21.2 | 1.11 | 0.12 |
| No | fSolGE | Before Deprotection | CPPC, wt% | T, oC | Time, h | Deprotecting Degree, % | After Deprotection | ||
|---|---|---|---|---|---|---|---|---|---|
| Mn1/Mn2 × 10−3 | ĐM1/ĐM2 | Mn1/Mn2 × 10−3 | ĐM1/ĐM2 | ||||||
| 1 | 0.11 | 20.8/38.4 | 1.04/1.04 | 10 | 25 | 24 | 40 | 20.3/38.4 | 1.05/1.04 |
| 2 | 78.7/157.8 | 1.04/1.05 | 10 | 25 | 24 | 44 | 76.6/153.8 | 1.06/1.06 | |
| 3 | 19.1 | 1.11 | 10 | 40 | 20 | 94 | 19.4 | 1.10 | |
| 4 | 10 | 40 | 20 | >78 | 19.5 | 1.10 | |||
| 5 | 10 | 45 | 23 | 93 | 18.4 | 1.13 | |||
| 6 | 10 | 45 | 24 | 94 | 19.6 | 1.10 | |||
| 7 | 89.8/143.8 | 1.05/1.23 | 10 | 45 | 48 | 100 | 89.8/143.8 | 1.05/1.23 | |
| 8 | 0.42 | 46.5/92.2 | 1.02/1.02 | 20 | 25 | 5 | 11 | 49.3/98.7 | 1.05/1.03 |
| 9 | 47.8/93.4 | 1.03/1.02 | 20 | 40 | 4 | 16 1 | 50.1/95.2 | 1.02/1.02 | |
| 10 | 20 | 40 | 4 | 17 | 50.3/95.7 | 1.02/1.02 | |||
| 11 | 20 | 40 | 24 | 81 | – | – | |||
| 12 | 10 | 40 | 5 | 6 | 50.3/99.0 | 1.04/1.04 | |||
| 13 | 0.53 | 39.8/77.7 | 1.03/1.02 | 10 | 45 | 48 | >96 | – | – |
| 14 | 10 | 45 | 24 | 83 | – | – | |||
| No. | fSolGE | FSolGE | FGG | Mn1/Mn2 × 10−3 | Tg, °C | Tp 1, °C |
|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 88.9/194.5 | 41.2 | 241 |
| 2 | 0.11 | 0.19 | 0 | 89.8/143.8 | 35.7 | 254 |
| 3 | 0 | 0.19 | 89.8/143.8 | 21.1 | 293 | |
| 4 | 0.13 | 0 | 78.7/157.8 | 37.8 | 271 | |
| 5 | 0.14 | 0 | 20.8/38.4 | 36.0 | 268 | |
| 6 | 0.14 | 0 | 19.1 | 32.0 | 274 | |
| 7 | 0.01 | 0.13 | 19.6 | 16.5 | 290 | |
| 8 | 0.22 | 0.23 | 0 | 62.2/121.9 | 31.5 | 253 |
| 9 | 0.32 | 0.34 | 0 | 59.9/93.6 | 27.3 | 259 |
| 10 | 0.42 | 0.43 | 0 | 47.8/93.4 | 24.3 | 264 |
| 11 | 0.49 | 0 | 46.5/92.2 | 24.3 | 269 | |
| 12 | 0.36 | 0.07 | 50.1/95.2 | 21.8 | 241 | |
| 13 | 0.28 | 0.21 | 49.3/98.7 | 4.1 | 264 | |
| 14 | 0.09 | 0.34 | – | –3.1 | 230 | |
| 15 | 0.53 | 0.54 | 0 | 39.8/77.7 | 20.4 | 270 |
| 16 | 0.04 | 0.52 | – | 14.1 | 247 | |
| 17 | 0.09 | 0.45 | – | 10.5 | 250 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maximov, N.M.; Rzhevskiy, S.A.; Asachenko, A.F.; Plutalova, A.V.; Trofimchuk, E.S.; Lysenko, E.A.; Shurupova, O.V.; Tarasova, E.S.; Chernikova, E.V.; Beletskaya, I.P. Novel Hydroxyl-Functional Aliphatic CO2-Based Polycarbonates: Synthesis and Properties. Int. J. Mol. Sci. 2025, 26, 10151. https://doi.org/10.3390/ijms262010151
Maximov NM, Rzhevskiy SA, Asachenko AF, Plutalova AV, Trofimchuk ES, Lysenko EA, Shurupova OV, Tarasova ES, Chernikova EV, Beletskaya IP. Novel Hydroxyl-Functional Aliphatic CO2-Based Polycarbonates: Synthesis and Properties. International Journal of Molecular Sciences. 2025; 26(20):10151. https://doi.org/10.3390/ijms262010151
Chicago/Turabian StyleMaximov, Nikita M., Sergey A. Rzhevskiy, Andrey F. Asachenko, Anna V. Plutalova, Elena S. Trofimchuk, Evgenii A. Lysenko, Olga V. Shurupova, Ekaterina S. Tarasova, Elena V. Chernikova, and Irina P. Beletskaya. 2025. "Novel Hydroxyl-Functional Aliphatic CO2-Based Polycarbonates: Synthesis and Properties" International Journal of Molecular Sciences 26, no. 20: 10151. https://doi.org/10.3390/ijms262010151
APA StyleMaximov, N. M., Rzhevskiy, S. A., Asachenko, A. F., Plutalova, A. V., Trofimchuk, E. S., Lysenko, E. A., Shurupova, O. V., Tarasova, E. S., Chernikova, E. V., & Beletskaya, I. P. (2025). Novel Hydroxyl-Functional Aliphatic CO2-Based Polycarbonates: Synthesis and Properties. International Journal of Molecular Sciences, 26(20), 10151. https://doi.org/10.3390/ijms262010151