
Enhancing HDAC Inhibitor Screening: Addressing Zinc Parameterization and Ligand Protonation in Docking Studies

 ,
,  ,
,  , , and
, , and 
Abstract

1. Introduction
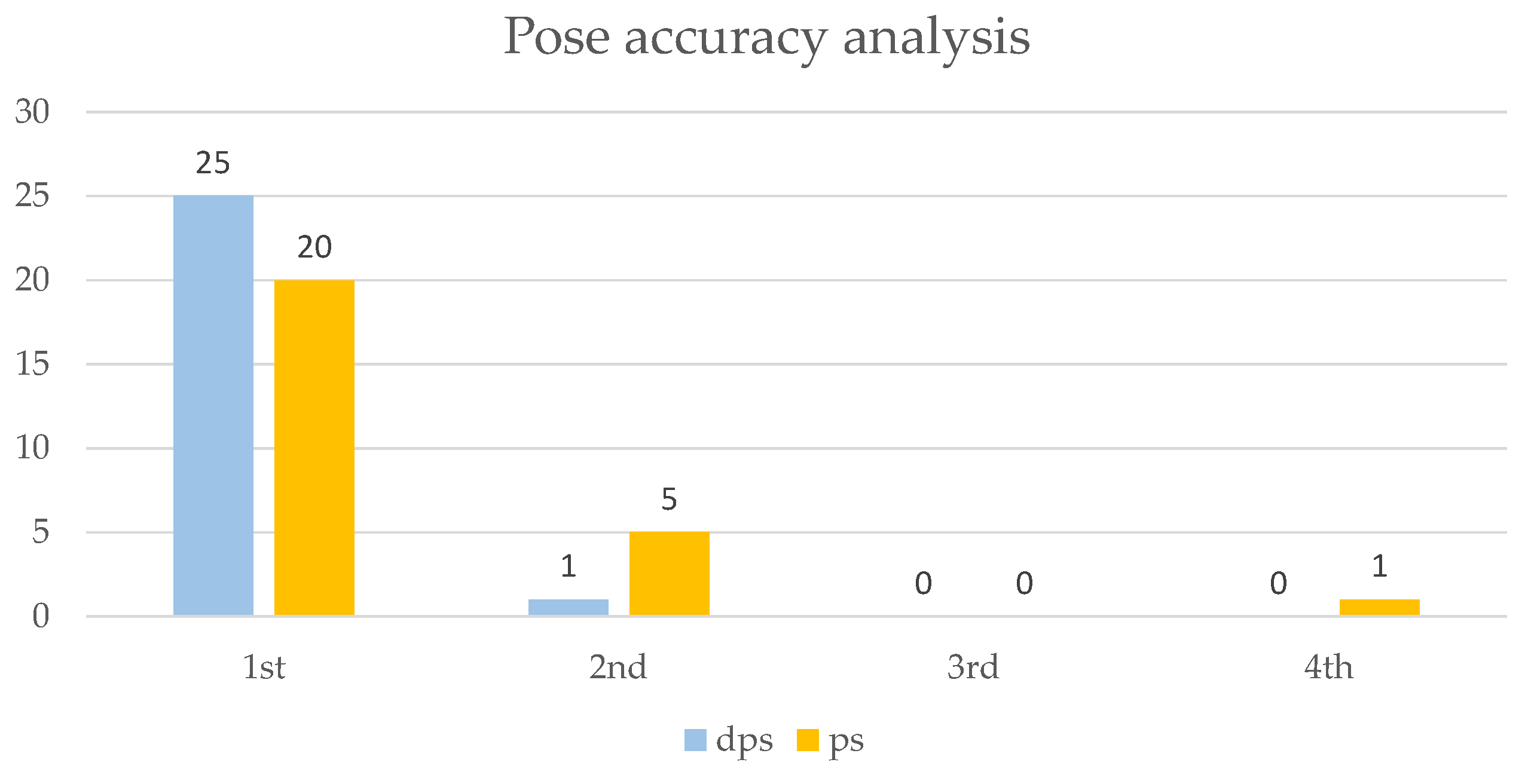
2. Results and Discussion
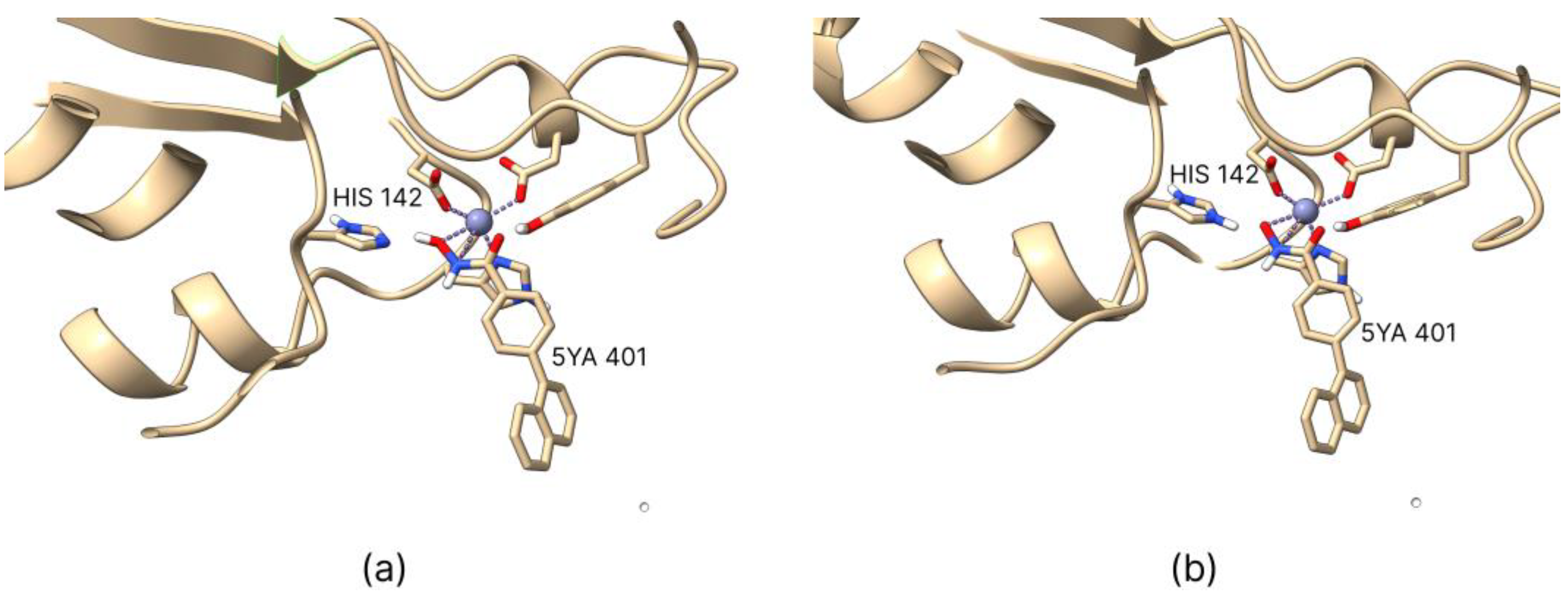
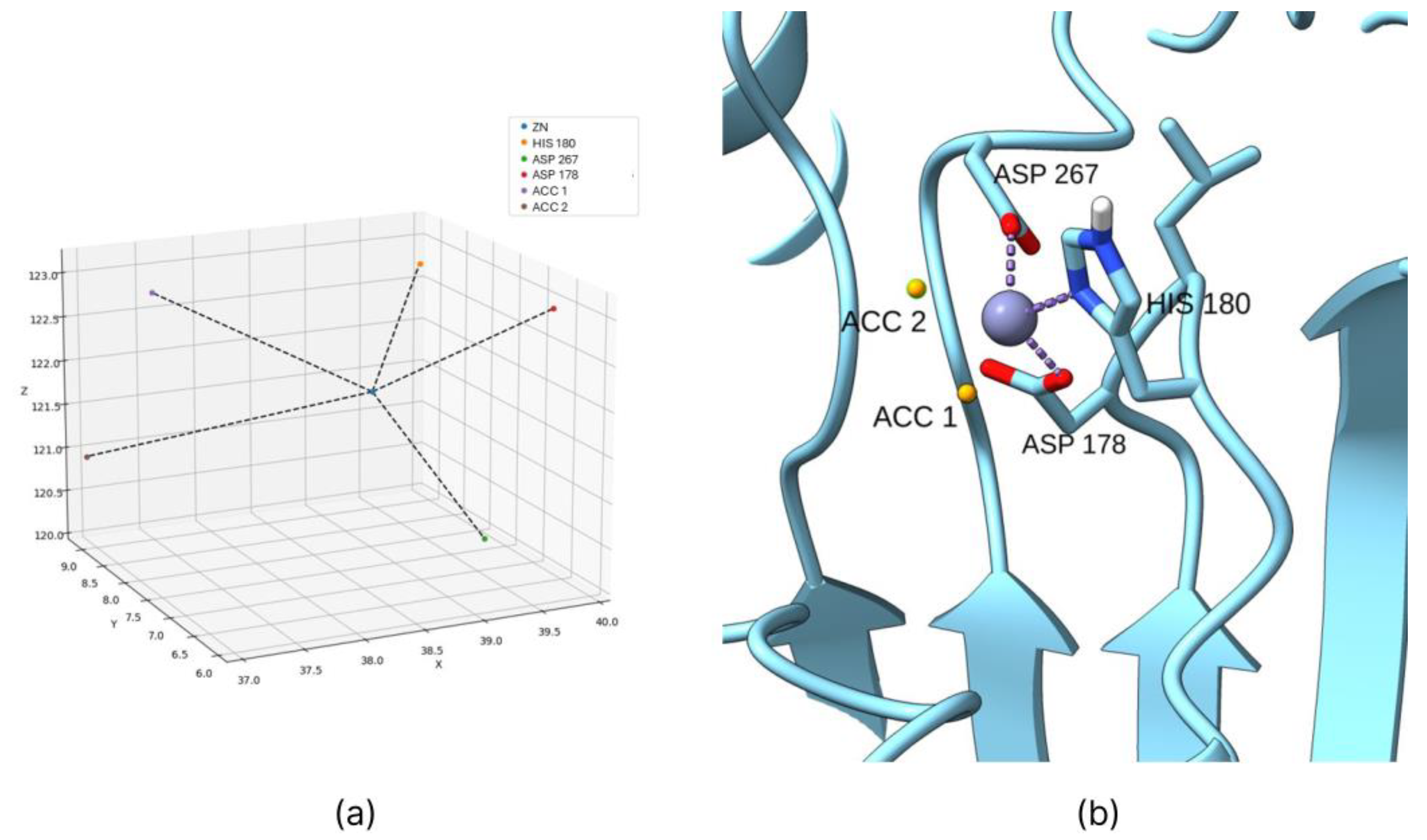
2.1. Coordination Geometry of the Zinc Ion
2.2. Active Ligands Selection
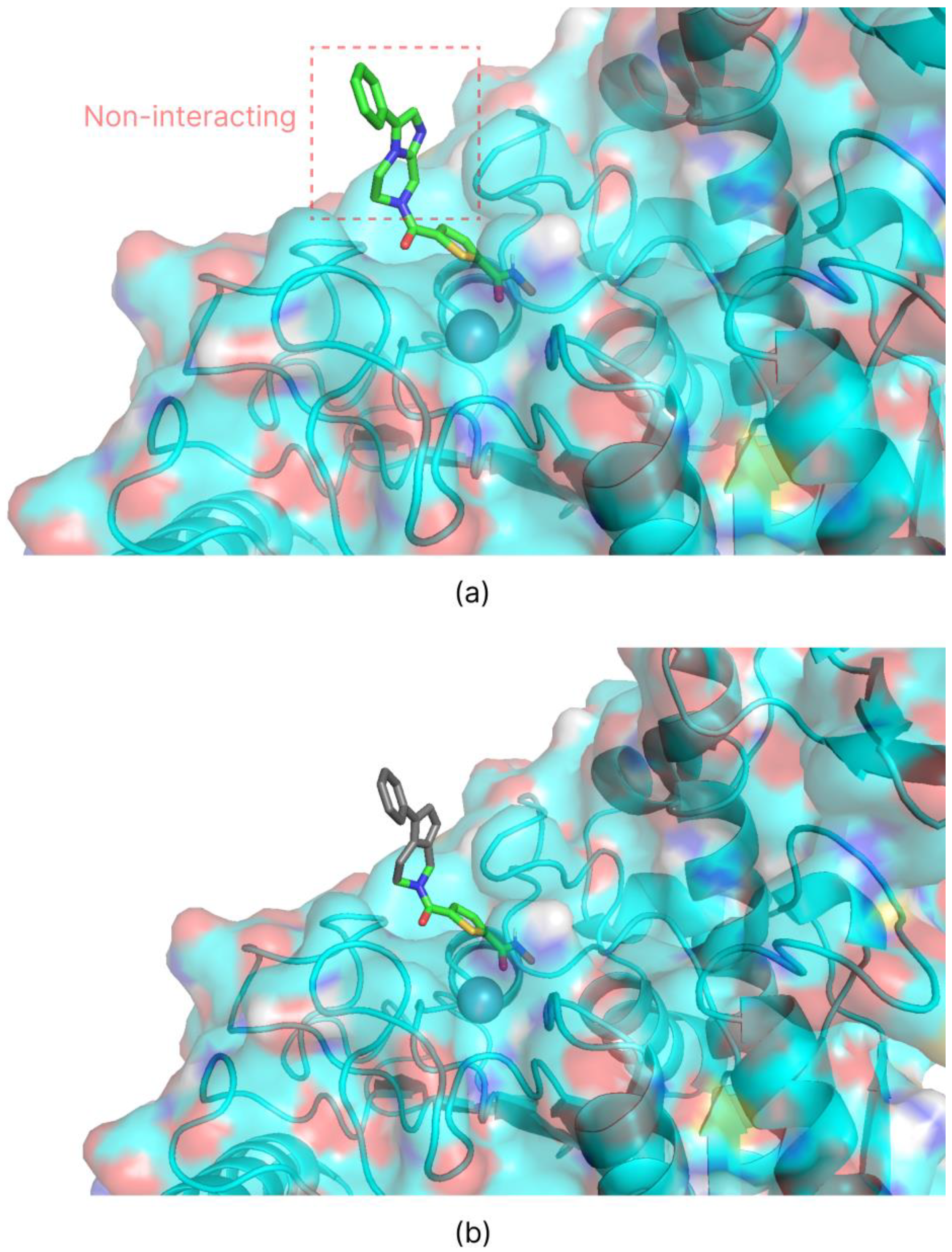
2.3. Introduction of Biases
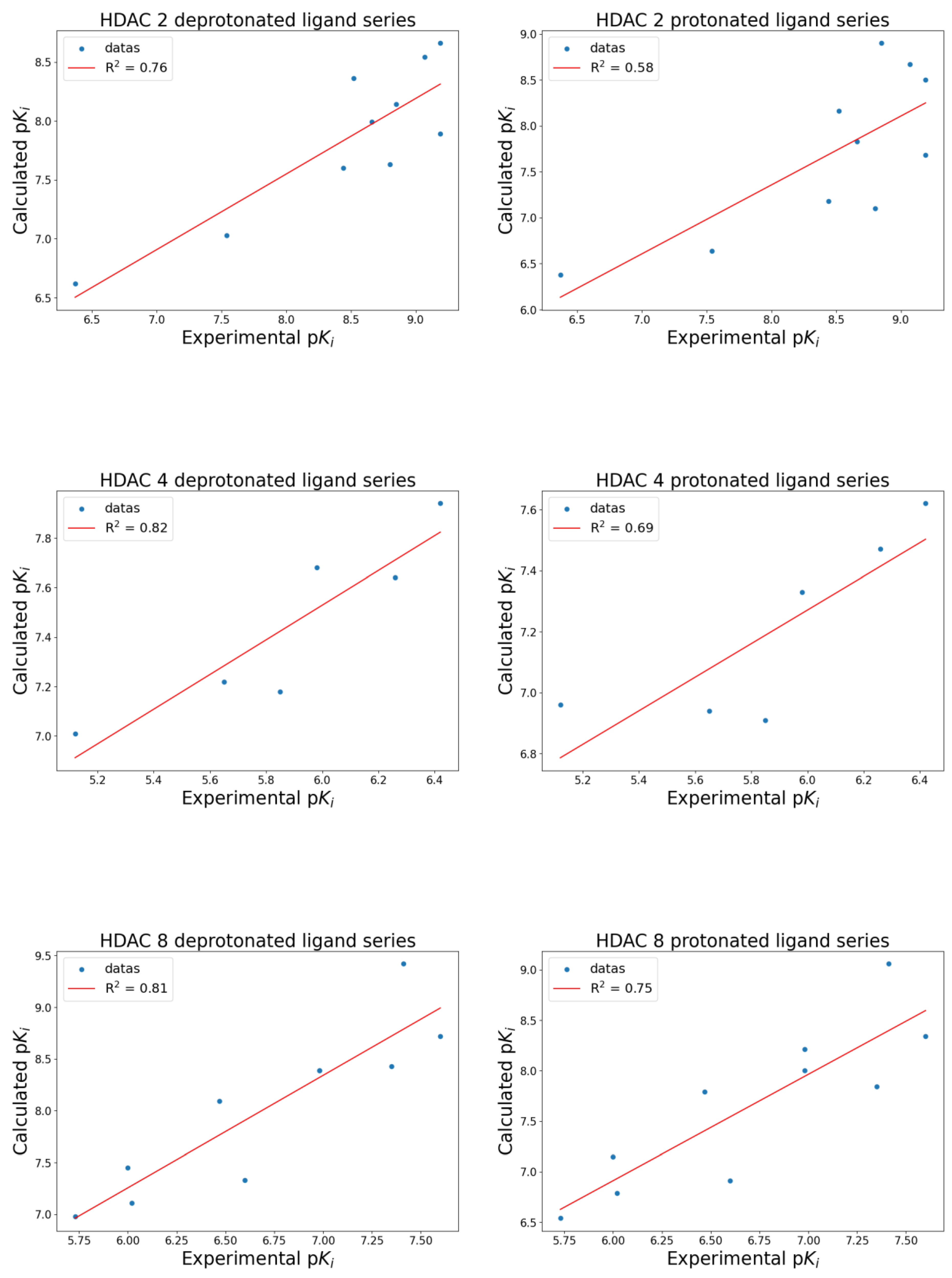
2.4. Virtual Screening
3. Materials and Methods
3.1. Biological Data and 3D Structures Generation
3.2. Protein Preparation
3.3. Molecular Docking Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, R.; Wang, G.; Zhang, Y. The Development Prospection of HDAC Inhibitors as a Potential Therapeutic Direction in Alzheimer’s Disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic Modulation and Understanding of HDAC Inhibitors in Cancer Therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.; Rocca, R.; Alcaro, S.; Artese, A. The Histone Deacetylase Family: Structural Features and Application of Combined Computational Methods. Pharmaceuticals 2024, 17, 620. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method Using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Kubinyi, H. Success stories of computer-aided design. In Computer Applications in Pharmaceutical Research and Development; Ekins, S., Ed.; Wiley: Hoboken, NJ, USA, 2006; pp. 377–424. ISBN 978-0-471-73779-7. [Google Scholar]
- Gastreich, M.; Lilienthal, M.; Briem, H.; Claussen, H. Ultrafast de Novo Docking Combining Pharmacophores and Combinatorics. J. Comput. Aided Mol. Des. 2007, 20, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4 Zn: An Improved AutoDock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Liao, S.; Gu, J.; Jiang, H.; Wang, X.; Li, H. An Accurate Metalloprotein-Specific Scoring Function and Molecular Docking Program Devised by a Dynamic Sampling and Iteration Optimization Strategy. J. Chem. Inf. Model. 2015, 55, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lyu, N.; Diao, H.; Jin, S.; Zeng, T.; Zhou, Y.; Wu, R. GM-DockZn: A Geometry Matching-Based Docking Algorithm for Zinc Proteins. Bioinformatics 2020, 36, 4004–4011. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. GPDOCK: Highly Accurate Docking Strategy for Metalloproteins Based on Geometric Probability. Brief. Bioinform. 2023, 24, bbac620. [Google Scholar] [CrossRef] [PubMed]
- Arcon, J.P.; Modenutti, C.P.; Avendaño, D.; Lopez, E.D.; Defelipe, L.A.; Ambrosio, F.A.; Turjanski, A.G.; Forli, S.; Marti, M.A. AutoDock Bias: Improving Binding Mode Prediction and Virtual Screening Using Known Protein–Ligand Interactions. Bioinformatics 2019, 35, 3836–3838. [Google Scholar] [CrossRef]
- Hakkennes, M.L.A.; Buda, F.; Bonnet, S. MetalDock: An Open Access Docking Tool for Easy and Reproducible Docking of Metal Complexes. J. Chem. Inf. Model. 2023, 63, 7816–7825. [Google Scholar] [CrossRef]
- Clemente, C.M.; Prieto, J.M.; Martí, M. Unlocking Precision Docking for Metalloproteins. J. Chem. Inf. Model. 2024, 64, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Drakontaeidi, A.; Pontiki, E. A Review on Molecular Docking on HDAC Isoforms: Novel Tool for Designing Selective Inhibitors. Pharmaceuticals 2023, 16, 1639. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical Phylogenetics of Histone Deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Wang, D.; Helquist, P.; Wiest, O. Zinc Binding in HDAC Inhibitors: A DFT Study. J. Org. Chem. 2007, 72, 5446–5449. [Google Scholar] [CrossRef] [PubMed]
- Ruibo, W.; Zhenyu, L.; Zexing, C.; Yingkai, Z. Zinc Chelation with Hydroxamate in Histone Deacetylases Modulated by Water Access to the Linker Binding Channel. J. Am. Chem. Soc. 2021, 133, 6110–6113. [Google Scholar] [CrossRef]
- Baselious, F.; Hilscher, S.; Robaa, D.; Barinka, C.; Schutkowski, M.; Sippl, W. Comparative Structure-Based Virtual Screening Utilizing Optimized AlphaFold Model Identifies Selective HDAC11 Inhibitor. Int. J. Mol. Sci. 2024, 25, 1358. [Google Scholar] [CrossRef] [PubMed]
- Baselious, F.; Robaa, D.; Sippl, W. Utilization of AlphaFold Models for Drug Discovery: Feasibility and Challenges. Histone Deacetylase 11 as a Case Study. Comput. Biol. Med. 2023, 167, 107700. [Google Scholar] [CrossRef] [PubMed]
- Tretbar, M.; Schliehe-Diecks, J.; Von Bredow, L.; Tan, K.; Roatsch, M.; Tu, J.-W.; Kemkes, M.; Sönnichsen, M.; Schöler, A.; Borkhardt, A.; et al. Preferential HDAC6 Inhibitors Derived from HPOB Exhibit Synergistic Antileukemia Activity in Combination with Decitabine. Eur. J. Med. Chem. 2024, 272, 116447. [Google Scholar] [CrossRef] [PubMed]
- Ghazy, E.; Heimburg, T.; Lancelot, J.; Zeyen, P.; Schmidtkunz, K.; Truhn, A.; Darwish, S.; Simoben, C.V.; Shaik, T.B.; Erdmann, F.; et al. Synthesis, Structure-Activity Relationships, Cocrystallization and Cellular Characterization of Novel smHDAC8 Inhibitors for the Treatment of Schistosomiasis. Eur. J. Med. Chem. 2021, 225, 113745. [Google Scholar] [CrossRef] [PubMed]
- Ghazy, E.; Zeyen, P.; Herp, D.; Hügle, M.; Schmidtkunz, K.; Erdmann, F.; Robaa, D.; Schmidt, M.; Morales, E.R.; Romier, C.; et al. Design, Synthesis, and Biological Evaluation of Dual Targeting Inhibitors of Histone Deacetylase 6/8 and Bromodomain BRPF1. Eur. J. Med. Chem. 2020, 200, 112338. [Google Scholar] [CrossRef]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Vögerl, K.; Ong, N.; Senger, J.; Herp, D.; Schmidtkunz, K.; Marek, M.; Müller, M.; Bartel, K.; Shaik, T.B.; Porter, N.J.; et al. Synthesis and Biological Investigation of Phenothiazine-Based Benzhydroxamic Acids as Selective Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2019, 62, 1138–1166. [Google Scholar] [CrossRef]
- Uba, A.I.; Zengin, G. Phenolic Compounds as Histone Deacetylase Inhibitors: Binding Propensity and Interaction Insights from Molecular Docking and Dynamics Simulations. Amino Acids 2023, 55, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Moi, D.; Pinzi, L.; Bonanni, D.; Rastelli, G. Hydroxamic Acid Derivatives: From Synthetic Strategies to Medicinal Chemistry Applications. ACS Omega 2021, 6, 21843–21849. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for Structure Building and Analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | Name 1 | Experimental Ki (nM) | Experimental pKi | Calculated pKi dps 2 | Calculated pKi ps 3 |
|---|---|---|---|---|---|
| HDAC 2 | LBH-589 | 0.65 | 9.19 | 8.66 | 8.50 |
| Trichostatin A | 0.65 | 9.19 | 7.89 | 7.68 | |
| PXD-101 | 0.85 | 9.07 | 8.54 | 8.67 | |
| LAQ-824 | 1.40 | 8.85 | 8.14 | 8.90 | |
| SAHA | 1.60 | 8.80 | 7.63 | 7.10 | |
| Scriptaid | 2.20 | 8.66 | 7.99 | 7.83 | |
| ITF-2357 | 3.00 | 8.52 | 8.36 | 8.16 | |
| Pyroxamide | 3.60 | 8.44 | 7.60 | 7.18 | |
| SHA | 29.00 | 7.54 | 7.03 | 6.64 | |
| 4-PBHA | 430.00 | 6.37 | 6.62 | 6.38 | |
| HDAC 4 | PXD-101 | 380.00 | 6.42 | 7.94 | 7.62 |
| LBH-589 | 550.00 | 6.26 | 7.64 | 7.47 | |
| ITF-2357 | 1050.00 | 5.98 | 7.68 | 7.33 | |
| Trichostatin A | 1400.00 | 5.85 | 7.18 | 6.91 | |
| LAQ-824 | 2250.00 | 5.65 | 7.22 | 6.94 | |
| Scriptaid | 7500.00 | 5.12 | 7.01 | 6.96 | |
| HDAC 8 | PXD-101 | 25.00 | 7.60 | 8.72 | 8.34 |
| ITF-2357 | 39.00 | 7.41 | 9.42 | 9.06 | |
| Trichostatin A | 45.00 | 7.35 | 8.43 | 7.84 | |
| LBH-589 | 105.00 | 6.98 | 8.39 | 8.00 | |
| Scriptaid | 105.00 | 6.98 | 8.39 | 8.21 | |
| SAHA | 250.00 | 6.60 | 7.33 | 6.91 | |
| LAQ-824 | 340.00 | 6.47 | 8.09 | 7.79 | |
| SHA | 950.00 | 6.02 | 7.11 | 6.79 | |
| Pyroxamide | 1000.00 | 6.00 | 7.45 | 7.15 | |
| 4-PBHA | 1850.00 | 5.73 | 6.98 | 6.54 |
| Isoform | Nonbiased RMSD (Å) | Biased RMSD (Å) |
|---|---|---|
| HDAC 2 | 8.28 | 1.98 |
| HDAC 4 | 1.67 | 1.66 |
| HDAC 8 | 2.91 | 2.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buccheri, R.; Coco, A.; Pasquinucci, L.; Amata, E.; Marrazzo, A.; Rescifina, A. Enhancing HDAC Inhibitor Screening: Addressing Zinc Parameterization and Ligand Protonation in Docking Studies. Int. J. Mol. Sci. 2025, 26, 850. https://doi.org/10.3390/ijms26020850
Buccheri R, Coco A, Pasquinucci L, Amata E, Marrazzo A, Rescifina A. Enhancing HDAC Inhibitor Screening: Addressing Zinc Parameterization and Ligand Protonation in Docking Studies. International Journal of Molecular Sciences. 2025; 26(2):850. https://doi.org/10.3390/ijms26020850
Chicago/Turabian StyleBuccheri, Rocco, Alessandro Coco, Lorella Pasquinucci, Emanuele Amata, Agostino Marrazzo, and Antonio Rescifina. 2025. "Enhancing HDAC Inhibitor Screening: Addressing Zinc Parameterization and Ligand Protonation in Docking Studies" International Journal of Molecular Sciences 26, no. 2: 850. https://doi.org/10.3390/ijms26020850
APA StyleBuccheri, R., Coco, A., Pasquinucci, L., Amata, E., Marrazzo, A., & Rescifina, A. (2025). Enhancing HDAC Inhibitor Screening: Addressing Zinc Parameterization and Ligand Protonation in Docking Studies. International Journal of Molecular Sciences, 26(2), 850. https://doi.org/10.3390/ijms26020850