
Cytosine Methylation Changes the Preferred Cis-Regulatory Configuration of Arabidopsis WUSCHEL-Related Homeobox 14

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
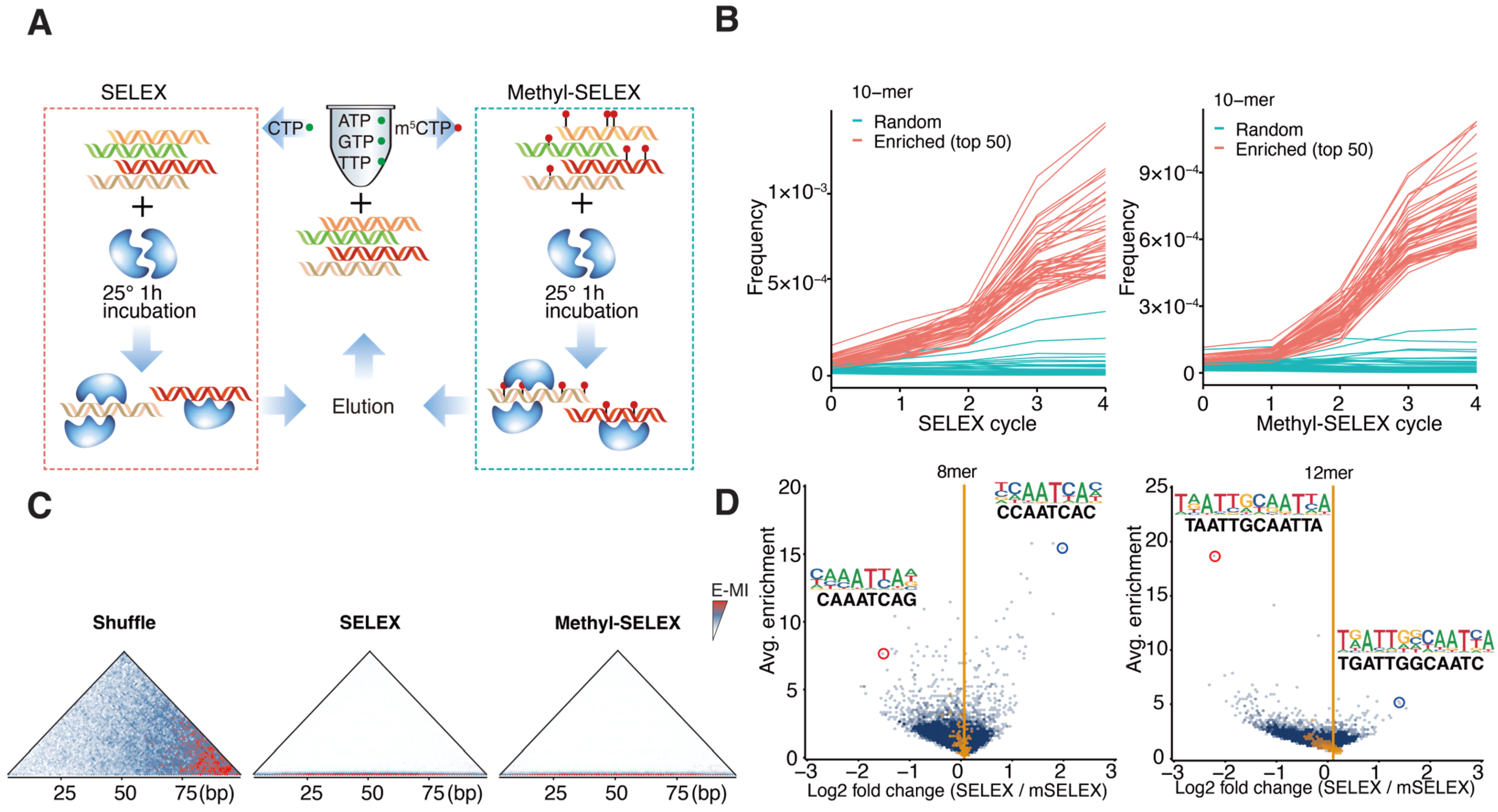
2.1. Methylation Inhibits Monomeric Binding but Enhances Dimeric Binding of AtWOX14
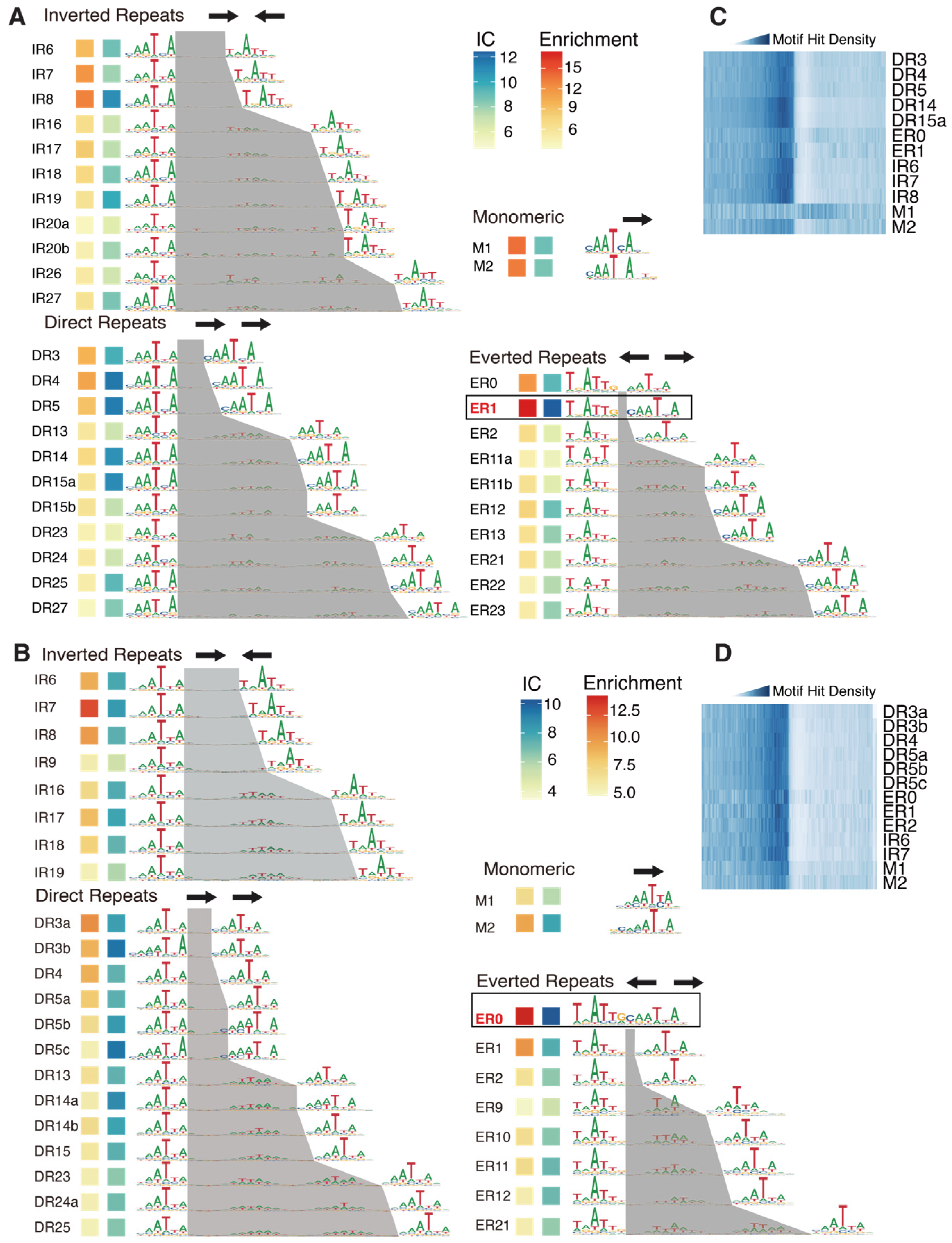
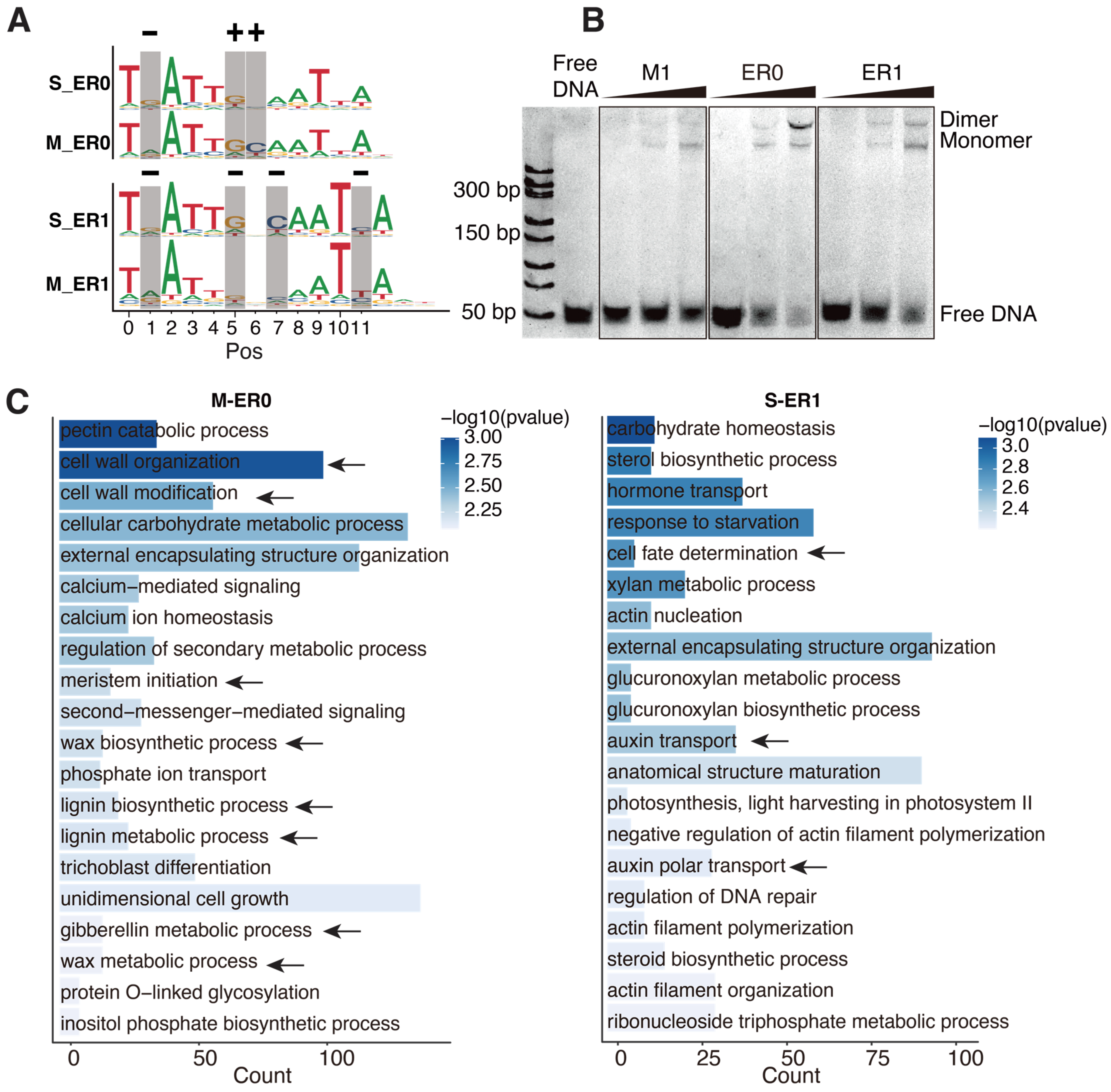
2.2. Methylation Changes the Preferred Homodimeric Configuration of AtWOX14
2.3. The Targets of ER0 and ER1 Are of Different Physiological Functions
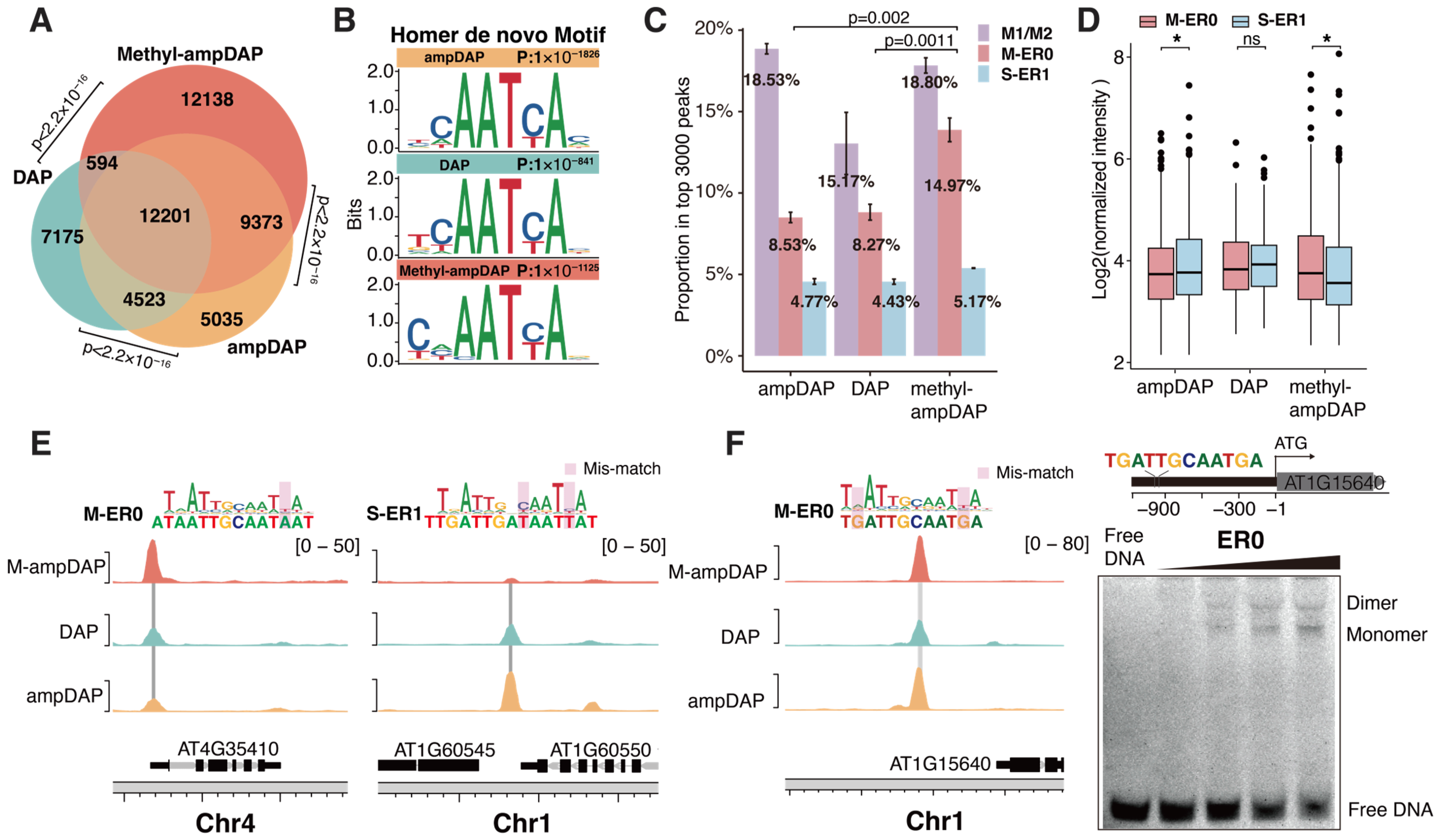
2.4. Methylation Effects on Genome-Wide Binding Targets of AtWOX14
3. Discussion
4. Materials and Methods
4.1. Cloning and Protein Expression
4.2. SELEX
4.3. Methyl-SELEX
4.4. Data Analyses of SELEX
4.5. Electrophoretic Mobility Shift Assays
- M1: ATGCTAGCTCCATCTGTATTGATTGTTTATGGCGGTGACGTACT;
- ER0: ATGCTAGCTCCATCTGTGATTGCAATCAATGGCGGTGACGTACT;
- ER1: ATGCTAGCTCCATCTGTGATTGACAATCAATGGCGGTGACGTACT;
- ER0-AT1G15640: AATAAGATGTACCCAATGATTGCAATGATGAGCCCAATGA GTGC.
4.6. DAP and Data Pre-Processing
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gehring, W.J.; Müller, M.; Affolter, M.; Percival-Smith, A.; Billeter, M.; Qian, Y.Q.; Otting, G.; Wüthrich, K. The Structure of the Homeodomain and Its Functional Implications. Trends Genet. 1990, 6, 323–329. [Google Scholar] [CrossRef]
- Chandler, J.; Nardmann, J.; Werr, W. Plant Development Revolves around Axes. Trends Plant Sci. 2008, 13, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.E. Shoot Meristem Function and Leaf Polarity: The Role of Class III HD-ZIP Genes. PLoS Genet. 2006, 2, e89. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Sato, Y.; Matsuoka, M. Involvement of Homeobox Genes in Early Body Plan of Monocot. Int. Rev. Cytol. 2002, 218, 1–35, 36e. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.L.; Gago, G.M.; Palena, C.M.; Gonzalez, D.H. Homeoboxes in Plant Development. Biochim. Biophys. Acta 1998, 1442, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hake, S.; Smith, H.M.S.; Holtan, H.; Magnani, E.; Mele, G.; Ramirez, J. The Role of Knox Genes in Plant Development. Annu. Rev. Cell Dev. Biol. 2004, 20, 125–151. [Google Scholar] [CrossRef]
- Hay, A.; Craft, J.; Tsiantis, M. Plant Hormones and Homeoboxes: Bridging the Gap? Bioessays 2004, 26, 395–404. [Google Scholar] [CrossRef]
- Graaff, E. van der, E.; Laux, T.; Rensing, S.A. The WUS Homeobox-Containing (WOX) Protein Family. Genome Biol. 2009, 10, 248. [Google Scholar] [CrossRef]
- Wang, J.; Tan, M.; Wang, X.; Jia, L.; Wang, M.; Huang, A.; You, L.; Li, C.; Zhang, Y.; Zhao, Y.; et al. WUS-RELATED HOMEOBOX 14 Boosts de Novo Plant Shoot Regeneration. Plant Physiol. 2023, 192, 748. [Google Scholar] [CrossRef]
- Deveaux, Y.; Toffano-Nioche, C.; Claisse, G.; Thareau, V.; Morin, H.; Laufs, P.; Moreau, H.; Kreis, M.; Lecharny, A. Genes of the Most Conserved WOX Clade in Plants Affect Root and Flower Development in Arabidopsis. BMC Evol. Biol. 2008, 8, 291. [Google Scholar] [CrossRef] [PubMed]
- Etchells, J.P.; Provost, C.M.; Mishra, L.; Turner, S.R. WOX4 and WOX14 Act Downstream of the PXY Receptor Kinase to Regulate Plant Vascular Proliferation Independently of Any Role in Vascular Organisation. Development 2013, 140, 2224–2234. [Google Scholar] [CrossRef]
- Smit, M.E.; McGregor, S.R.; Sun, H.; Gough, C.; Bågman, A.-M.; Soyars, C.L.; Kroon, J.T.; Gaudinier, A.; Williams, C.J.; Yang, X.; et al. A PXY-Mediated Transcriptional Network Integrates Signaling Mechanisms to Control Vascular Development in Arabidopsis. Plant Cell 2019, 32, 319. [Google Scholar] [CrossRef]
- Denis, E.; Kbiri, N.; Mary, V.; Claisse, G.; Conde E Silva, N.; Kreis, M.; Deveaux, Y. WOX14 Promotes Bioactive Gibberellin Synthesis and Vascular Cell Differentiation in Arabidopsis. Plant J. 2017, 90, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Bell, O.; Tiwari, V.K.; Thomä, N.H.; Schübeler, D. Determinants and Dynamics of Genome Accessibility. Nat. Rev. Genet. 2011, 12, 554–564. [Google Scholar] [CrossRef]
- Luo, L.; Lin, D.; Li, J.; Chen, H.; Qu, Q.; Zhang, L.; Luo, Y.; Chen, J.; Jiang, D.; Lü, P.; et al. EGDB: A Comprehensive Multi-Omics Database for Energy Grasses and the Epigenomic Atlas of Pearl Millet. iMeta 2024, e263. [Google Scholar] [CrossRef]
- Li, Z.; Li, D.; Li, Y.; Guo, X.; Yang, R. Deciphering the Regulatory Code of Histone Modifications in Plants. J. Genet. Genom. 2022, 49, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Lü, P.; Yu, S.; Zhu, N.; Chen, Y.-R.; Zhou, B.; Pan, Y.; Tzeng, D.; Fabi, J.P.; Argyris, J.; Garcia-Mas, J.; et al. Genome Encode Analyses Reveal the Basis of Convergent Evolution of Fleshy Fruit Ripening. Nat. Plants 2018, 4, 784–791. [Google Scholar] [CrossRef]
- Li, M.; Yao, T.; Lin, W.; Hinckley, W.E.; Galli, M.; Muchero, W.; Gallavotti, A.; Chen, J.-G.; Huang, S.C. Double DAP-Seq Uncovered Synergistic DNA Binding of Interacting bZIP Transcription Factors. Nat. Commun. 2023, 14, 2600. [Google Scholar] [CrossRef]
- Zhu, W.; Li, H.; Dong, P.; Ni, X.; Fan, M.; Yang, Y.; Xu, S.; Xu, Y.; Qian, Y.; Chen, Z.; et al. Low Temperature-Induced Regulatory Network Rewiring via WRKY Regulators during Banana Peel Browning. Plant Physiol. 2023, 193, 855–873. [Google Scholar] [CrossRef]
- Héberlé, É.; Bardet, A.F. Sensitivity of Transcription Factors to DNA Methylation. Essays Biochem. 2019, 63, 727. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Liu, J.; Zhang, Y.; Ye, L.; Peng, Y.; Wang, H.; Diao, H.; Ma, Y.; Wang, M.; et al. Transposable Elements Orchestrate Subgenome-Convergent and -Divergent Transcription in Common Wheat. Nat. Commun. 2022, 13, 6940. [Google Scholar] [CrossRef]
- Charvin, M.; Halter, T.; Blanc-Mathieu, R.; Barraud, P.; Aumont-Nicaise, M.; Parcy, F.; Navarro, L. Single-Cytosine Methylation at W-Boxes Repels Binding of WRKY Transcription Factors through Steric Hindrance. Plant Physiol. 2023, 192, 77–84. [Google Scholar] [CrossRef]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of Cytosine Methylation on DNA Binding Specificities of Human Transcription Factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]
- Hu, S.; Wan, J.; Su, Y.; Song, Q.; Zeng, Y.; Nguyen, H.N.; Shin, J.; Cox, E.; Rho, H.S.; Woodard, C.; et al. DNA Methylation Presents Distinct Binding Sites for Human Transcription Factors. eLife 2013, 2, e00726. [Google Scholar] [CrossRef]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef]
- Boer, D.R.; Freire-Rios, A.; van den Berg, W.A.M.; Saaki, T.; Manfield, I.W.; Kepinski, S.; López-Vidrieo, I.; Franco-Zorrilla, J.M.; de Vries, S.C.; Solano, R.; et al. Structural Basis for DNA Binding Specificity by the Auxin-Dependent ARF Transcription Factors. Cell 2014, 156, 577–589. [Google Scholar] [CrossRef]
- Yan, J.; Qiu, Y.; Ribeiro dos Santos, A.M.; Yin, Y.; Li, Y.E.; Vinckier, N.; Nariai, N.; Benaglio, P.; Raman, A.; Li, X.; et al. Systematic Analysis of Transcription Factors Binding to Noncoding Variants. Nature 2021, 591, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Jolma, A.; Zhang, J.; Mondragón, E.; Morgunova, E.; Kivioja, T.; Laverty, K.U.; Yin, Y.; Zhu, F.; Bourenkov, G.; Morris, Q.; et al. Binding Specificities of Human RNA-Binding Proteins toward Structured and Linear RNA Sequences. Genome Res. 2020, 30, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Morgunova, E.; Nagy, G.; Yin, Y.; Zhu, F.; Nayak, S.P.; Xiao, T.; Sokolov, I.; Popov, A.; Laughton, C.; Grubmuller, H.; et al. Interfacial Water Confers Transcription Factors with Dinucleotide Specificity. Nat Struct Mol Biol 2025. [Google Scholar] [CrossRef]
- Zhu, F.; Farnung, L.; Kaasinen, E.; Sahu, B.; Yin, Y.; Wei, B.; Dodonova, S.O.; Nitta, K.R.; Morgunova, E.; Taipale, M.; et al. The Interaction Landscape between Transcription Factors and the Nucleosome. Nature 2018, 562, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Yuan, Z.; Zhang, X.; Chen, H.; Luo, L.; Li, W.; Li, T.; Ma, N.; Mao, F.; Lin, D.; et al. Sea-ATI Unravels Novel Vocabularies of Plant Active Cistrome. Nucleic Acids Res. 2023, 51, 11568–11583. [Google Scholar] [CrossRef]
- Nitta, K.R.; Jolma, A.; Yin, Y.; Morgunova, E.; Kivioja, T.; Akhtar, J.; Hens, K.; Toivonen, J.; Deplancke, B.; Furlong, E.E.M.; et al. Conservation of Transcription Factor Binding Specificities across 600 Million Years of Bilateria Evolution. eLife 2015, 4, e04837. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.; Lee, T.; Hung, Y.; Li, G.; KuanChieh, T.; Liu, Y.; Kuo, P.; Zheng, H.; Chang, W. PlantPAN3.0: A New and Updated Resource for Reconstructing Transcriptional Regulatory Networks from ChIP-Seq Experiments in Plants. Nucleic Acids Res. 2019, 47, D1155–D1163. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.; Meng, Y.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a Central Hub for Transcription Factors and Regulatory Interactions in Plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Bryne, J.C.; Valen, E.; Tang, M.-H.E.; Marstrand, T.; Winther, O.; da Piedade, I.; Krogh, A.; Lenhard, B.; Sandelin, A. JASPAR, the Open Access Database of Transcription Factor-Binding Profiles: New Content and Tools in the 2008 Update. Nucleic Acids Res. 2008, 36, D102–D106. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.-K. Epigenetic Gene Regulation in Plants and Its Potential Applications in Crop Improvement. Nat. Rev. Mol. Cell Biol. 2025, 26, 51–67. [Google Scholar] [CrossRef]
- Fontana, M.; Roosjen, M.; Crespo García, I.; van den Berg, W.; Malfois, M.; Boer, R.; Weijers, D.; Hohlbein, J. Cooperative Action of Separate Interaction Domains Promotes High-Affinity DNA Binding of Arabidopsis thaliana ARF Transcription Factors. Proc. Natl. Acad. Sci. USA 2023, 120, e2219916120. [Google Scholar] [CrossRef] [PubMed]
- Nanao, M.H.; Vinos-Poyo, T.; Brunoud, G.; Thévenon, E.; Mazzoleni, M.; Mast, D.; Lainé, S.; Wang, S.; Hagen, G.; Li, H.; et al. Structural Basis for Oligomerization of Auxin Transcriptional Regulators. Nat. Commun. 2014, 5, 3617. [Google Scholar] [CrossRef] [PubMed]
- Jolma, A.; Yin, Y.; Nitta, K.R.; Dave, K.; Popov, A.; Taipale, M.; Enge, M.; Kivioja, T.; Morgunova, E.; Taipale, J. DNA-Dependent Formation of Transcription Factor Pairs Alters Their Binding Specificity. Nature 2015, 527, 384–388. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Inoue-Bungo, T.; Morii, H.; Fujikawa, A.; Sasaki, M.; Kimura, K.; Shiina, M.; Sato, K.; Kumasaka, T.; Yamamoto, M.; et al. Structural Analyses of DNA Recognition by the AML1/Runx-1 Runt Domain and Its Allosteric Control by CBFβ. Cell 2001, 104, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Brostromer, E.; Xing, D.; Jin, J.; Chong, S.; Ge, H.; Wang, S.; Gu, C.; Yang, L.; Gao, Y.Q.; et al. Probing Allostery Through DNA. Science 2013, 339, 816–819. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread Natural Variation of DNA Methylation within Angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef]
- Korhonen, J.; Martinmäki, P.; Pizzi, C.; Rastas, P.; Ukkonen, E. MOODS: Fast Search for Position Weight Matrix Matches in DNA Sequences. Bioinformatics 2009, 25, 3181–3182. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, A.; O’Malley, R.C.; Huang, S.-S.C.; Galli, M.; Nery, J.R.; Gallavotti, A.; Ecker, J.R. Mapping Genome-Wide Transcription-Factor Binding Sites Using DAP-Seq. Nat. Protoc. 2017, 12, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Z.; Zhu, W.; Ni, X.; Wang, J.; Fu, L.; Chen, J.; Li, T.; Tang, L.; Yang, Y.; et al. The MaNAP1-MaMADS1 Transcription Factor Module Mediates Ethylene-Regulated Peel Softening and Ripening in Banana. Plant Cell 2024, 37, koae282. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor Package to Plot Customizable Genomes Displaying Arbitrary Data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, D.; Zhang, X.; Luo, L.; Li, T.; Chen, H.; Ma, N.; Fu, L.; Tian, P.; Mao, F.; Lü, P.; et al. Cytosine Methylation Changes the Preferred Cis-Regulatory Configuration of Arabidopsis WUSCHEL-Related Homeobox 14. Int. J. Mol. Sci. 2025, 26, 763. https://doi.org/10.3390/ijms26020763
Jiang D, Zhang X, Luo L, Li T, Chen H, Ma N, Fu L, Tian P, Mao F, Lü P, et al. Cytosine Methylation Changes the Preferred Cis-Regulatory Configuration of Arabidopsis WUSCHEL-Related Homeobox 14. International Journal of Molecular Sciences. 2025; 26(2):763. https://doi.org/10.3390/ijms26020763
Chicago/Turabian StyleJiang, Dingkun, Xinfeng Zhang, Lin Luo, Tian Li, Hao Chen, Nana Ma, Lufeng Fu, Peng Tian, Fei Mao, Peitao Lü, and et al. 2025. "Cytosine Methylation Changes the Preferred Cis-Regulatory Configuration of Arabidopsis WUSCHEL-Related Homeobox 14" International Journal of Molecular Sciences 26, no. 2: 763. https://doi.org/10.3390/ijms26020763
APA StyleJiang, D., Zhang, X., Luo, L., Li, T., Chen, H., Ma, N., Fu, L., Tian, P., Mao, F., Lü, P., Guo, H., & Zhu, F. (2025). Cytosine Methylation Changes the Preferred Cis-Regulatory Configuration of Arabidopsis WUSCHEL-Related Homeobox 14. International Journal of Molecular Sciences, 26(2), 763. https://doi.org/10.3390/ijms26020763