
scRNA-seq Can Identify Different Cell Populations in Ovarian Cancer Bulk RNA-seq Experiments
 ,
,
Abstract
1. Introduction
2. Results
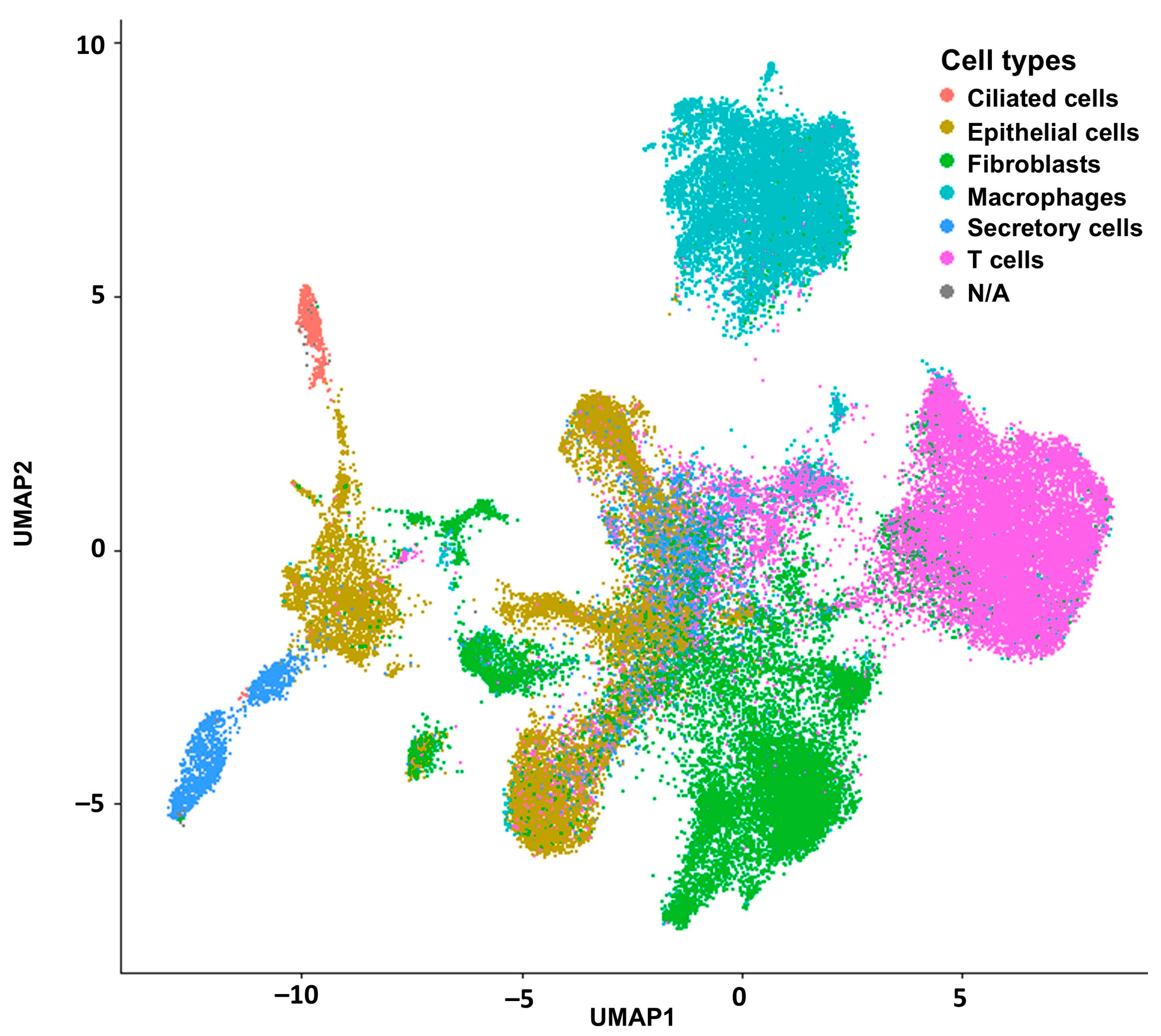
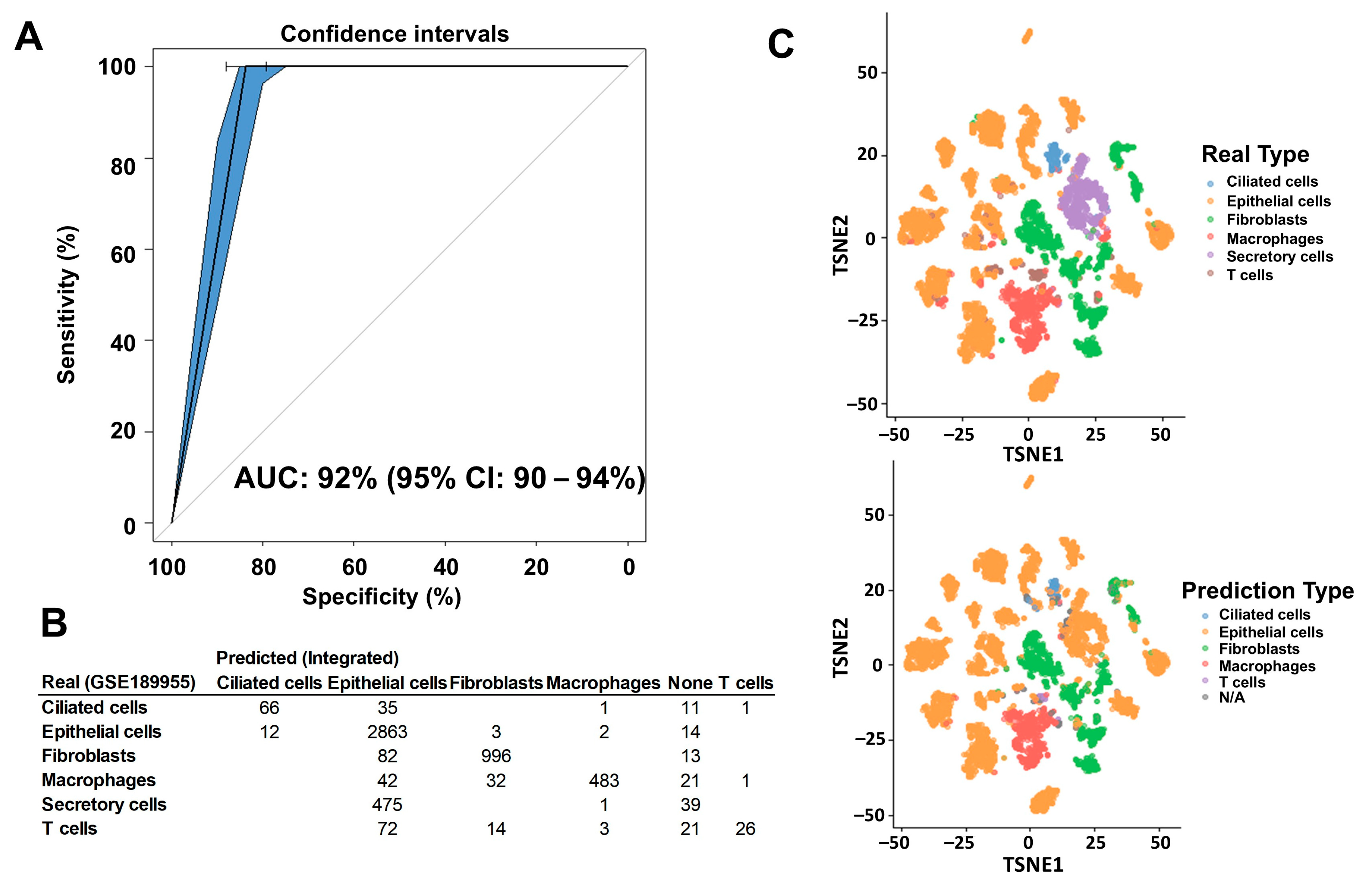
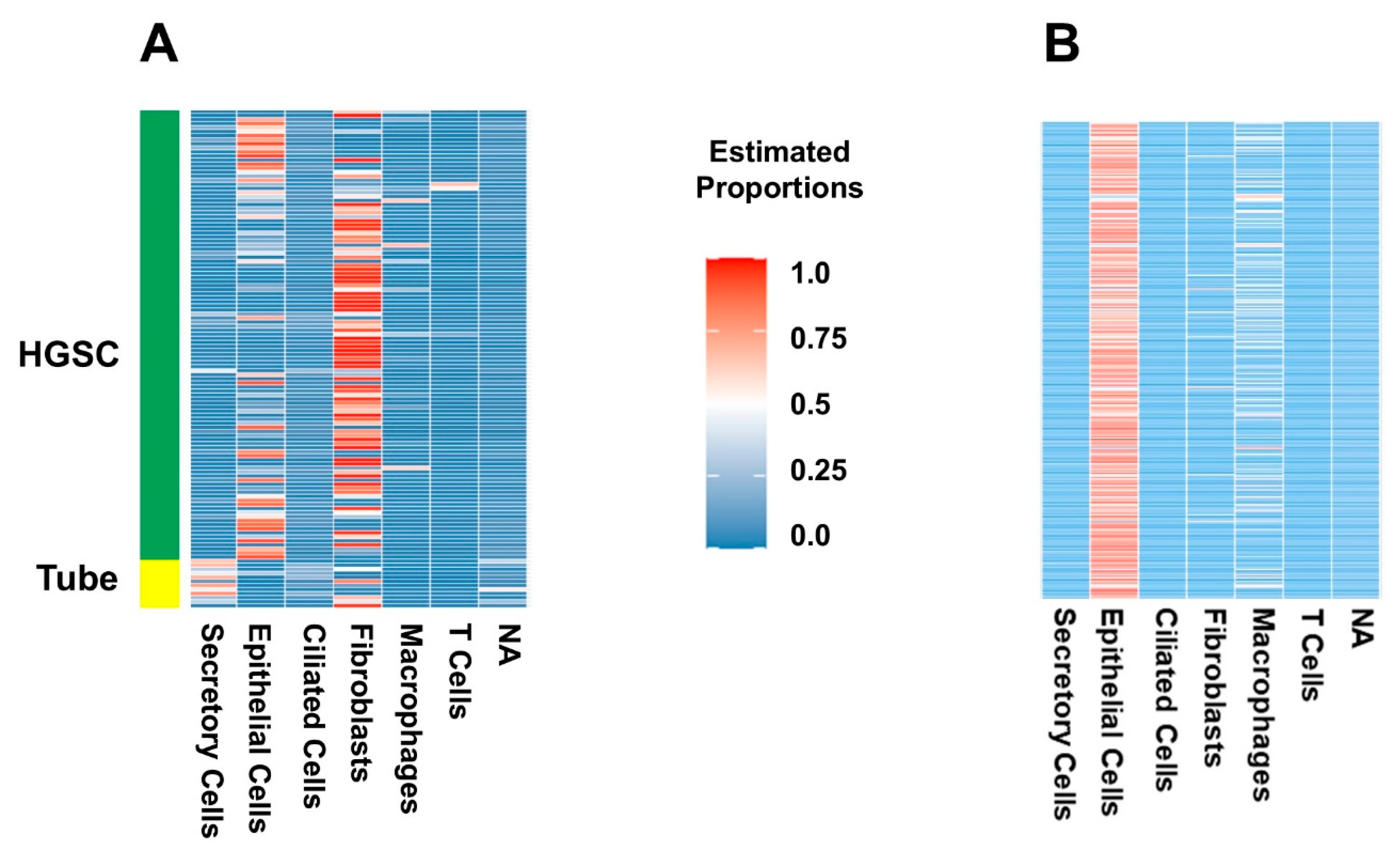
2.1. Deconvolution of Bulk RNAseq Revealed Fibroblasts, Immune Cells and Epithelial Cells in the UI Dataset Which Was Similar in the TCGA Dataset
2.2. Certain Cell Types Were Associated with Worse Patient Outcomes Depending on the Dataset and Analysis Used
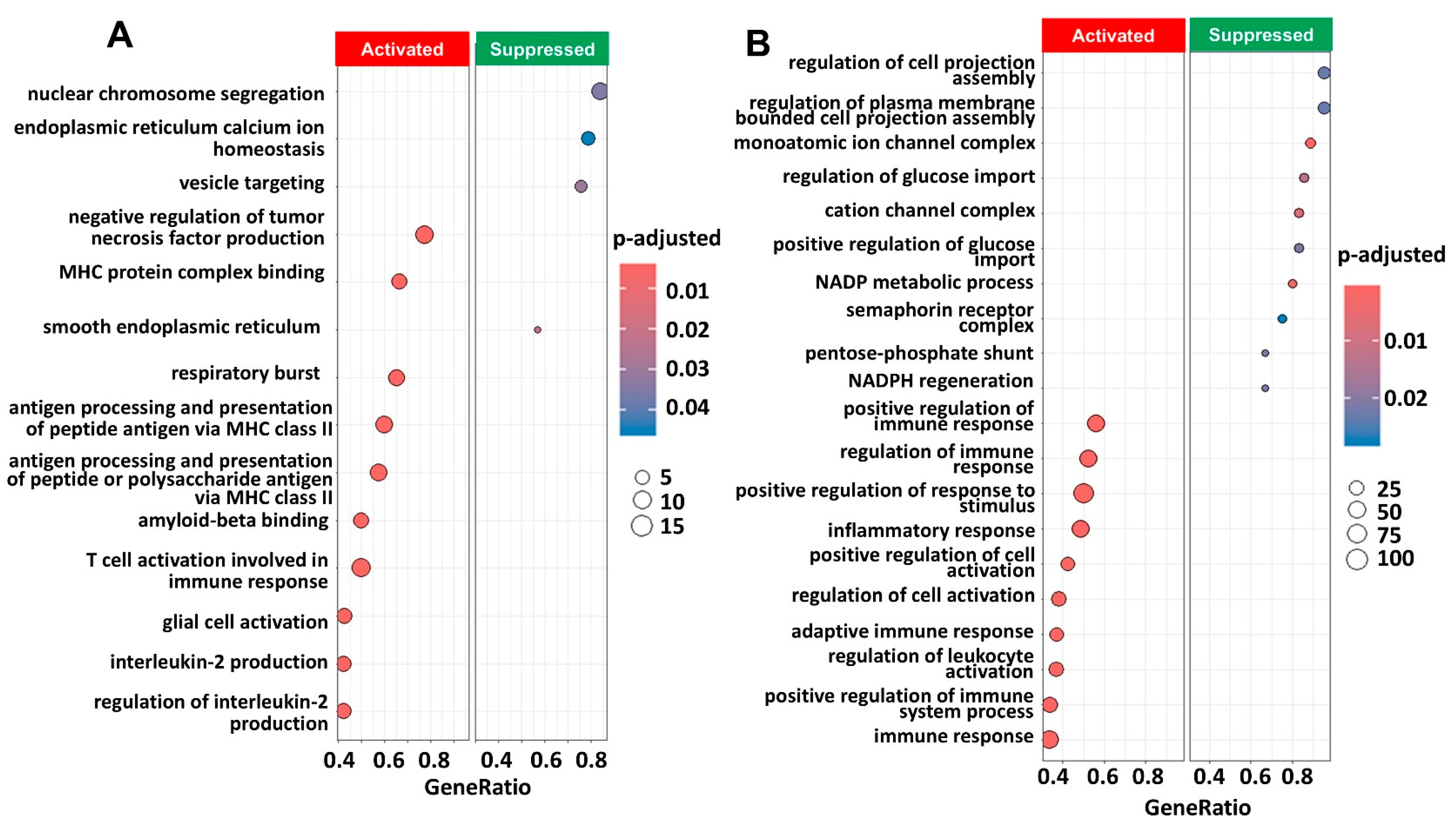
2.3. Pathway Enrichment Analysis Showed Immune Pathways Were Activated
3. Discussion
4. Methods
4.1. Data and Specimen Procurement
4.2. Data Preprocessing
4.3. Data Analysis
4.4. Deconvolution of Bulk RNA-seq from HGSC Datasets
4.5. Comparison of Patient Outcomes to Clustered Cell Proportions
4.6. Pathway Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | artificial intelligence |
| AUC | area under the curve |
| Biobank | Department of Obstetrics and Gynecology Gynecologic Oncology Biobank |
| EOC | epithelial ovarian |
| FT | fallopian tube |
| HGSC | High-grade serous cancer |
| HPCA | Human Primary Cell Atlas |
| IRB | Institutional review board |
| MuSiC | MUlti-Subject SIngle Cell deconvolution |
| RNA-seq | RNA sequencing |
| ROC | receiver operating characteristic |
| scRNA-seq | single-cell RNA-seq |
| spRNAseq | spatial RNA sequencing |
| TAM | tumor-associated macrophages |
| TCGA | The Cancer Genome Atlas |
| UI | University of Iowa |
| UIHG | University of Iowa Institute of Human Genetics |
| UMAP | Uniform manifold approximation and projection |
| WHTR | Women’s Health Tissue Repository |
References
- Schwarz, R.F.; Ng, C.K.Y.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and Temporal Heterogeneity in High-Grade Serous Ovarian Cancer: A Phylogenetic Analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Gonzalez Bosquet, J.; Devor, E.J.; Newtson, A.M.; Smith, B.J.; Bender, D.P.; Goodheart, M.J.; McDonald, M.E.; Braun, T.A.; Thiel, K.W.; Leslie, K.K. Creation and Validation of Models to Predict Response to Primary Treatment in Serous Ovarian Cancer. Sci. Rep. 2021, 11, 5957. [Google Scholar] [CrossRef]
- Gogineni, V.; Morand, S.; Staats, H.; Royfman, R.; Devanaboyina, M.; Einloth, K.; Dever, D.; Stanbery, L.; Aaron, P.; Manning, L.; et al. Current Ovarian Cancer Maintenance Strategies and Promising New Developments. J. Cancer 2021, 12, 38–53. [Google Scholar] [CrossRef]
- Gonzalez Bosquet, J.; Newtson, A.M.; Chung, R.K.; Thiel, K.W.; Ginader, T.; Goodheart, M.J.; Leslie, K.K.; Smith, B.J. Prediction of Chemo-Response in Serous Ovarian Cancer. Mol. Cancer 2016, 15, 66. [Google Scholar] [CrossRef]
- Bi, J.; Thiel, K.W.; Litman, J.M.; Zhang, Y.; Devor, E.J.; Newtson, A.M.; Schnieders, M.J.; Bosquet, J.G.; Leslie, K.K. Characterization of a TP53 Somatic Variant of Unknown Function From an Ovarian Cancer Patient Using Organoid Culture and Computational Modeling. Clin. Obstet. Gynecol. 2020, 63, 109–119. [Google Scholar] [CrossRef]
- Newtson, A.M.; Devor, E.J.; Bosquet, J.G. Prediction of Epithelial Ovarian Cancer Outcomes With Integration of Genomic Data. Clin. Obstet. Gynecol. 2020, 63, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Bosquet, J.G.; Marchion, D.C.; Chon, H.; Lancaster, J.M.; Chanock, S. Analysis of Chemotherapeutic Response in Ovarian Cancers Using Publicly Available High-Throughput Data. Cancer Res. 2014, 74, 3902–3912. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel Molecular Subtypes of Serous and Endometrioid Ovarian Cancer Linked to Clinical Outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically Relevant Gene Signatures of High-Grade Serous Ovarian Carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and Therapeutic Relevance of Molecular Subtypes in High-Grade Serous Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Chen, G.M.; Kannan, L.; Geistlinger, L.; Kofia, V.; Safikhani, Z.; Gendoo, D.M.A.; Parmigiani, G.; Birrer, M.; Haibe-Kains, B.; Waldron, L. Consensus on Molecular Subtypes of High-Grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2018, 24, 5037–5047. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk Tissue Cell Type Deconvolution with Multi-Subject Single-Cell Expression Reference. Nat. Commun. 2019, 10, 380. [Google Scholar] [CrossRef]
- Avila Cobos, F.; Alquicira-Hernandez, J.; Powell, J.E.; Mestdagh, P.; De Preter, K. Benchmarking of Cell Type Deconvolution Pipelines for Transcriptomics Data. Nat. Commun. 2020, 11, 5650. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High Grade Serous Ovarian Carcinomas Originate in the Fallopian Tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.-M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular Analysis of High-Grade Serous Ovarian Carcinoma with and without Associated Serous Tubal Intra-Epithelial Carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef]
- Liang, L.; Yu, J.; Li, J.; Li, N.; Liu, J.; Xiu, L.; Zeng, J.; Wang, T.; Wu, L. Integration of ScRNA-Seq and Bulk RNA-Seq to Analyse the Heterogeneity of Ovarian Cancer Immune Cells and Establish a Molecular Risk Model. Front. Oncol. 2021, 11, 711020. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and Pharmacological Modulators of the Tumor Immune Contexture Revealed by Deconvolution of RNA-Seq Data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef]
- Olbrecht, S.; Busschaert, P.; Qian, J.; Vanderstichele, A.; Loverix, L.; Van Gorp, T.; Van Nieuwenhuysen, E.; Han, S.; Van den Broeck, A.; Coosemans, A.; et al. High-Grade Serous Tubo-Ovarian Cancer Refined with Single-Cell RNA Sequencing: Specific Cell Subtypes Influence Survival and Determine Molecular Subtype Classification. Genome Med. 2021, 13, 111. [Google Scholar] [CrossRef]
- Greppi, M.; Tabellini, G.; Patrizi, O.; Candiani, S.; Decensi, A.; Parolini, S.; Sivori, S.; Pesce, S.; Paleari, L.; Marcenaro, E. Strengthening the Antitumor NK Cell Function for the Treatment of Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 890. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Baillie, J.K.; Brown, H.; Freeman, T.C.; Hume, D.A. An Expression Atlas of Human Primary Cells: Inference of Gene Function from Coexpression Networks. BMC Genom. 2013, 14, 632. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, H.; Chang, X.; Hu, W.; Li, M.; Li, Y.; Liu, H.; Cheng, H.; Wang, S.; Zhou, L.; et al. Single-Cell Dissection of the Multiomic Landscape of High-Grade Serous Ovarian Cancer. Cancer Res. 2022, 82, 3903–3916. [Google Scholar] [CrossRef]
- Zhao, B.; Pei, L. A Macrophage Related Signature for Predicting Prognosis and Drug Sensitivity in Ovarian Cancer Based on Integrative Machine Learning. BMC Med. Genom. 2023, 16, 230. [Google Scholar] [CrossRef]
- Song, J.; Xiao, T.; Li, M.; Jia, Q. Tumor-Associated Macrophages: Potential Therapeutic Targets and Diagnostic Markers in Cancer. Pathol. Res. Pract. 2023, 249, 154739. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Coleman, R.L.; Sood, A.K. Targeting the Tumour Microenvironment in Ovarian Cancer. Eur. J. Cancer 2016, 56, 131–143. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International Validation of the Consensus Immunoscore for the Classification of Colon Cancer: A Prognostic and Accuracy Study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Mezheyeuski, A.; Backman, M.; Mattsson, J.; Martín-Bernabé, A.; Larsson, C.; Hrynchyk, I.; Hammarström, K.; Ström, S.; Ekström, J.; Mauchanski, S.; et al. An Immune Score Reflecting Pro- and Anti-Tumoural Balance of Tumour Microenvironment Has Major Prognostic Impact and Predicts Immunotherapy Response in Solid Cancers. EBioMedicine 2023, 88, 104452. [Google Scholar] [CrossRef]
- Mirza, M.R.; Chase, D.M.; Slomovitz, B.M.; Christensen, R.D.; Novák, Z.; Black, D.; Gilbert, L.; Sharma, S.; Valabrega, G.; Landrum, L.M.; et al. Dostarlimab for Primary Advanced or Recurrent Endometrial Cancer. N. Engl. J. Med. 2023, 388, 2145–2158. [Google Scholar] [CrossRef]
- Eskander, R.N.; Sill, M.W.; Beffa, L.; Moore, R.G.; Hope, J.M.; Musa, F.B.; Mannel, R.; Shahin, M.S.; Cantuaria, G.H.; Girda, E.; et al. Pembrolizumab plus Chemotherapy in Advanced Endometrial Cancer. N. Engl. J. Med. 2023, 388, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, K.; Roy, A.; Mandal, S. Pharmacological Inhibition of Protein Kinase D Suppresses Epithelial Ovarian Cancer via MAPK/ERK1/2/Runx2 Signalling Axis. Cell. Signal. 2023, 110, 110849. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.H.; Choi, D.; Chun, Y.J.; Noh, M. Keratinocyte-Derived IL-24 Plays a Role in the Positive Feedback Regulation of Epidermal Inflammation in Response to Environmental and Endogenous Toxic Stressors. Toxicol. Appl. Pharmacol. 2014, 280, 199–206. [Google Scholar] [CrossRef]
- Wang, T.; Liu, J.; Xiao, X.Q. Cantharidin Inhibits Angiogenesis by Suppressing VEGF-Induced JAK1/STAT3, ERK and AKT Signaling Pathways. Arch. Pharm. Res. 2015, 38, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Yang, Y.; Wang, Z.; Xu, L.; Yan, Z. Longxuetongluo Capsule Alleviates Lipopolysaccharide-Induced Neuroinflammation by Regulating Multiple Signaling Pathways in BV2 Microglia Cells. J. Chin. Med. Assoc. 2020, 83, 255–265. [Google Scholar] [CrossRef]
- Wang, C.; Sun, H.; Zhong, Y. Notoginsenoside R1 Promotes MC3T3-E1 Differentiation by up-Regulating MiR-23a via MAPK and JAK1/STAT3 Pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 602–608. [Google Scholar] [CrossRef]
- Wen, W.; Liang, W.; Wu, J.; Kowolik, C.M.; Buettner, R.; Scuto, A.; Hsieh, M.-Y.; Hong, H.; Brown, C.E.; Forman, S.J.; et al. Targeting JAK1/STAT3 Signaling Suppresses Tumor Progression and Metastasis in a Peritoneal Model of Human Ovarian Cancer. Mol. Cancer Ther. 2014, 13, 3037–3048. [Google Scholar] [CrossRef]
- Han, E.S.; Wen, W.; Dellinger, T.H.; Wu, J.; Lu, S.A.; Jove, R.; Yim, J.H. Ruxolitinib Synergistically Enhances the Anti-Tumor Activity of Paclitaxel in Human Ovarian Cancer. Oncotarget 2018, 9, 24304–24319. [Google Scholar] [CrossRef]
- Hippen, A.A.; Omran, D.K.; Weber, L.M.; Jung, E.; Drapkin, R.; Doherty, J.A.; Hicks, S.C.; Greene, C.S. Performance of Computational Algorithms to Deconvolve Heterogeneous Bulk Ovarian Tumor Tissue Depends on Experimental Factors. Genome Biol. 2023, 24, 239. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.Y. From Bulk, Single-Cell to Spatial RNA Sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- U.S. Census Bureau QuickFacts: Lowa. Available online: https://www.census.gov/quickfacts/IA (accessed on 24 November 2024).
- Hu, Z.; Artibani, M.; Alsaadi, A.; Wietek, N.; Morotti, M.; Shi, T.; Zhong, Z.; Gonzalez, L.S.; El-Sahhar, S.; Carrami, E.M.; et al. The Repertoire of Serous Ovarian Cancer Non-Genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell 2020, 37, 226–242.e7. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.D.; Salinas, E.A.; Newtson, A.M.; Sharma, D.; Keeney, M.E.; Warrier, A.; Smith, B.J.; Bender, D.P.; Goodheart, M.J.; Thiel, K.W.; et al. An Integrated Prediction Model of Recurrence in Endometrial Endometrioid Cancers. Cancer Manag. Res. 2019, 11, 5301–5315. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread Aligner: Fast, Accurate and Scalable Read Mapping by Seed-and-Vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The SVA Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Oncology (GO) Database and Informatics Resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing, version 4.3.1; R Foundation Statistics and Computing: Vienna, Austria, 2019. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HGSC (N = 112) | Range | ||
|---|---|---|---|
| Preoperative characteristics | Age (mean) | 60 | 25–85 |
| BMI (mean) | 27.2 | 14.4–54.2 | |
| Charlson Comorbidity Index * | |||
| Low (1–3) | 8 | ||
| Medium (4–6) | 63 | ||
| High (>6) Not available | 8 33 | ||
| Preop CA-125 (mean) | 2414 | 7–32,865 | |
| Disease in Upper abdomen (Other than Omentum) by Imaging | 67 | ||
| Large bowel | 4 | ||
| Spleen | 0 | ||
| Mesenteric LN | 4 | ||
| Porta/Hepatis | 5 | ||
| Other | 28 | ||
| Disease in the Chest by Imaging | |||
| Tumor | 7 | ||
| Pleural effusion | 5 | ||
| Neoadjuvant chemotherapy | |||
| Yes | 14 | ||
| No | 91 | ||
| Operative characteristics | FIGO Stage | ||
| I–II | 4 | ||
| III | 71 | ||
| IV Not available | 28 9 | ||
| Omentectomy | 102 | ||
| Surgery large bowel | 32 | ||
| Splenectomy | 2 | ||
| Diaphragmatic stripping | 6 | ||
| Residual disease | |||
| Microscopic (R0) | 21 | ||
| <1 cm (R1) | 38 | ||
| Macroscopic (R2) Not available | 36 17 | ||
| Surgical complexity score # | |||
| Low | 55 | ||
| Intermediate | 51 | ||
| High Not available | 5 1 | ||
| Outcomes | 30-day mortality | 0 (0%) | |
| 90-day mortality | 1 (1%) | ||
| Response to chemotherapy ** | |||
| Yes | 50 | ||
| No Not available | 39 23 | ||
| Cell Proportions | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HGSC | Tube | OR | p Value | OR | p Value | |
| Ciliated cells | 0.03 | 0.13 | 2.6 × 10−15 | <0.001 * | 1.88 | 0.969 |
| Epithelial cells | 0.30 | 0.01 | 4.1 × 109 | 0.033 * | 2.3 × 108 | 0.042 * |
| Fibroblasts | 0.49 | 0.24 | 9.40 | 0.029 * | 1.68 | 0.955 |
| Macrophages | 0.04 | 0.003 | 4.3 × 107 | 0.366 | NS | NS |
| N/A | 0.05 | 0.14 | 3 × 10−23 | <0.001 * | 2.9 × 10−10 | 0.342 |
| Secretory cells | 0.09 | 0.48 | 0.006 | <0.001 * | 8.5 × 10−6 | <0.001 * |
| T cells | 0.01 | 0.00 | 1.1 × 1081 | 0.993 | NS | NS |
| Response to Chemotherapy | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| Responders | Non-Responders | OR | p Value | OR | p Value | |
| Ciliated cells | 0.15 | 0.13 | 0.42 | 0.494 | NS | |
| Epithelial cells | 0.24 | 0.22 | 0.58 | 0.658 | NS | |
| Fibroblasts | 0.43 | 0.49 | 2.35 | 0.271 | NS | |
| Macrophages | 0.08 | 0.04 | 2.2 × 10−4 | 0.039 * | 2.2 × 10−4 | 0.067 |
| Secretory cells | 0.03 | 0.06 | 12.17 | 0.263 | NS | |
| T cells | 0.07 | 0.06 | 0.42 | 0.778 | NS | |
| Clinical features | ||||||
| Age | 1.05 | 0.011 * | 1.04 | 0.055 | ||
| BMI | 1.05 | 0.160 | NS | |||
| Charlson Comorbidity Index | 0.11 | 0.068 | 0.461 | 0.564 | ||
| Preop CA125 | 1.00 | 0.874 | NS | |||
| Disease upper abdomen | 2.28 | 0.076 | 1.194 | 0.232 | ||
| Disease in chest | 2.7 × 10−8 | 0.991 | NS | |||
| Neoadjuvant chemotherapy | 8.39 | 0.009 * | 14.21 | 0.007 * | ||
| FIGO Stage | 2.67 | 0.069 | 1.849 | 0.065 | ||
| Optimal surgery (R0 + R1) | 0.46 | 0.097 | 0.406 | 0.172 | ||
| Residual micro (R0) | 0.26 | 0.048 * | 0.15 | 0.027 * | ||
| Surgical Complexity score | 2.7 × 10−8 | 0.990 | NS | |||
| Optimal surgery | Univariate | Multivariate | ||||
| Optimal | Suboptimal | OR | p value | OR | p value | |
| Ciliated cells | 0.13 | 0.16 | 2.67 | 0.406 | NS | |
| Epithelial cells | 0.22 | 0.24 | 2.01 | 0.537 | NS | |
| Fibroblasts | 0.45 | 0.43 | 0.81 | 0.762 | NS | |
| Macrophages | 0.09 | 0.05 | 0.01 | 0.108 | 0.09 | 0.279 |
| Secretory cells | 0.05 | 0.05 | 0.73 | 0.873 | NS | |
| T cells | 0.06 | 0.07 | 2.20 | 0.763 | NS | |
| Clinical features | ||||||
| Age | 0.99 | 0.587 | NS | |||
| BMI | 1.01 | 0.638 | NS | |||
| Charlson Comorbidity Index | 1.06 | 0.946 | NS | |||
| Preop CA125 | 1.00 | 0.572 | NS | |||
| Disease upper abdomen | 1.27 | 0.564 | NS | |||
| Disease in chest | 4.79 | 0.069 | 4.90 | 0.066 | ||
| Neoadjuvant chemotherapy | 8.39 | 0.558 | NS | |||
| FIGO Stage | 7.7 × 106 | 0.989 | NS | |||
| Optimal surgery (R0 + R1) | 8.4 × 10−24 | 0.999 | NS | |||
| Residual micro (R0) | 1 × 10−8 | 0.990 | NS | |||
| Surgical Complexity score | 0.53 | 0.508 | NS | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabrilovich, S.; Devor, E.; Cardillo, N.; Bender, D.; Goodheart, M.; Gonzalez-Bosquet, J. scRNA-seq Can Identify Different Cell Populations in Ovarian Cancer Bulk RNA-seq Experiments. Int. J. Mol. Sci. 2025, 26, 7512. https://doi.org/10.3390/ijms26157512
Gabrilovich S, Devor E, Cardillo N, Bender D, Goodheart M, Gonzalez-Bosquet J. scRNA-seq Can Identify Different Cell Populations in Ovarian Cancer Bulk RNA-seq Experiments. International Journal of Molecular Sciences. 2025; 26(15):7512. https://doi.org/10.3390/ijms26157512
Chicago/Turabian StyleGabrilovich, Sofia, Eric Devor, Nicholas Cardillo, David Bender, Michael Goodheart, and Jesus Gonzalez-Bosquet. 2025. "scRNA-seq Can Identify Different Cell Populations in Ovarian Cancer Bulk RNA-seq Experiments" International Journal of Molecular Sciences 26, no. 15: 7512. https://doi.org/10.3390/ijms26157512
APA StyleGabrilovich, S., Devor, E., Cardillo, N., Bender, D., Goodheart, M., & Gonzalez-Bosquet, J. (2025). scRNA-seq Can Identify Different Cell Populations in Ovarian Cancer Bulk RNA-seq Experiments. International Journal of Molecular Sciences, 26(15), 7512. https://doi.org/10.3390/ijms26157512