
tRNA Modifications: A Tale of Two Viruses—SARS-CoV-2 and ZIKV



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
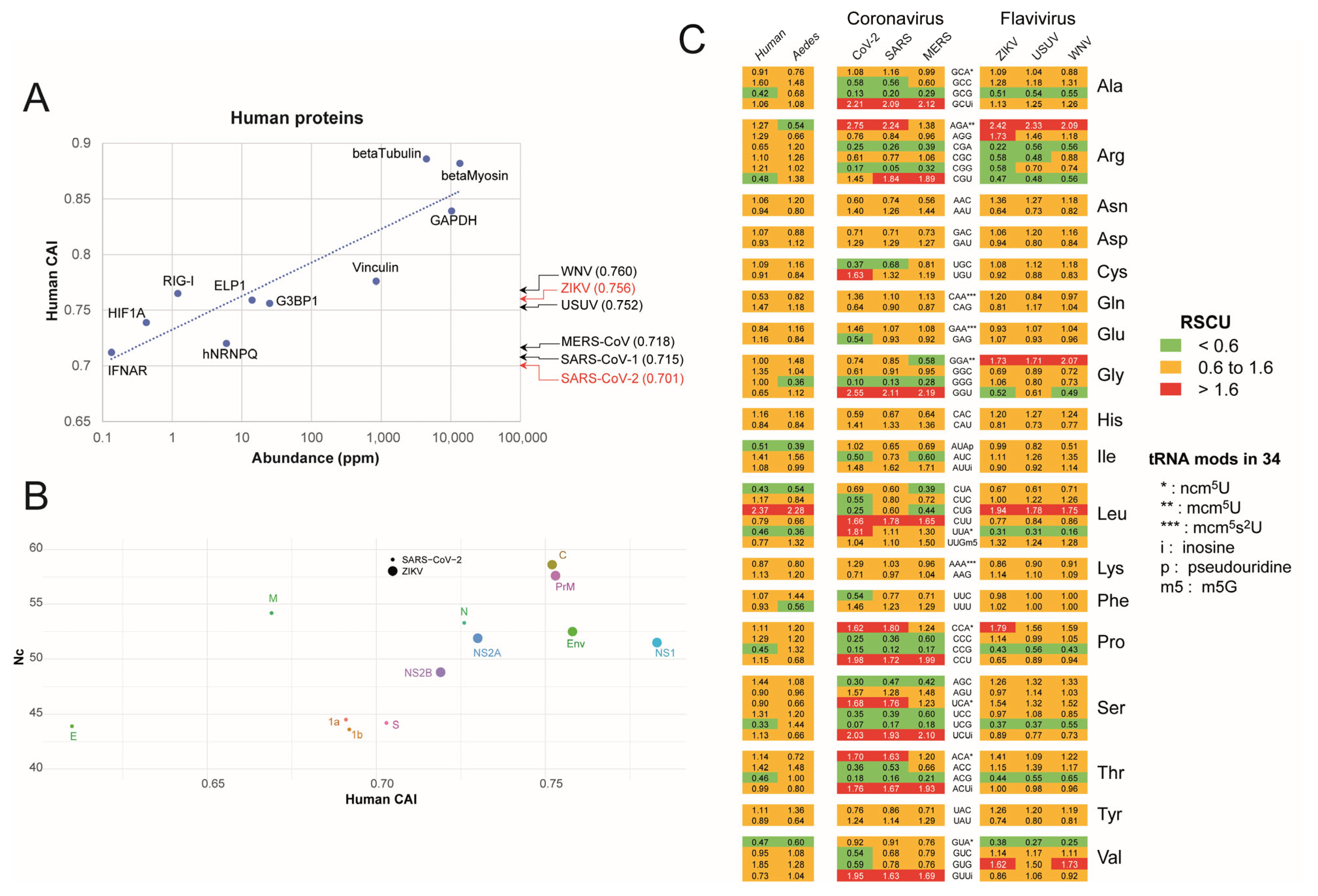
2.1. Analysis of SARS-CoV-2 and ZIKV Codon Bias
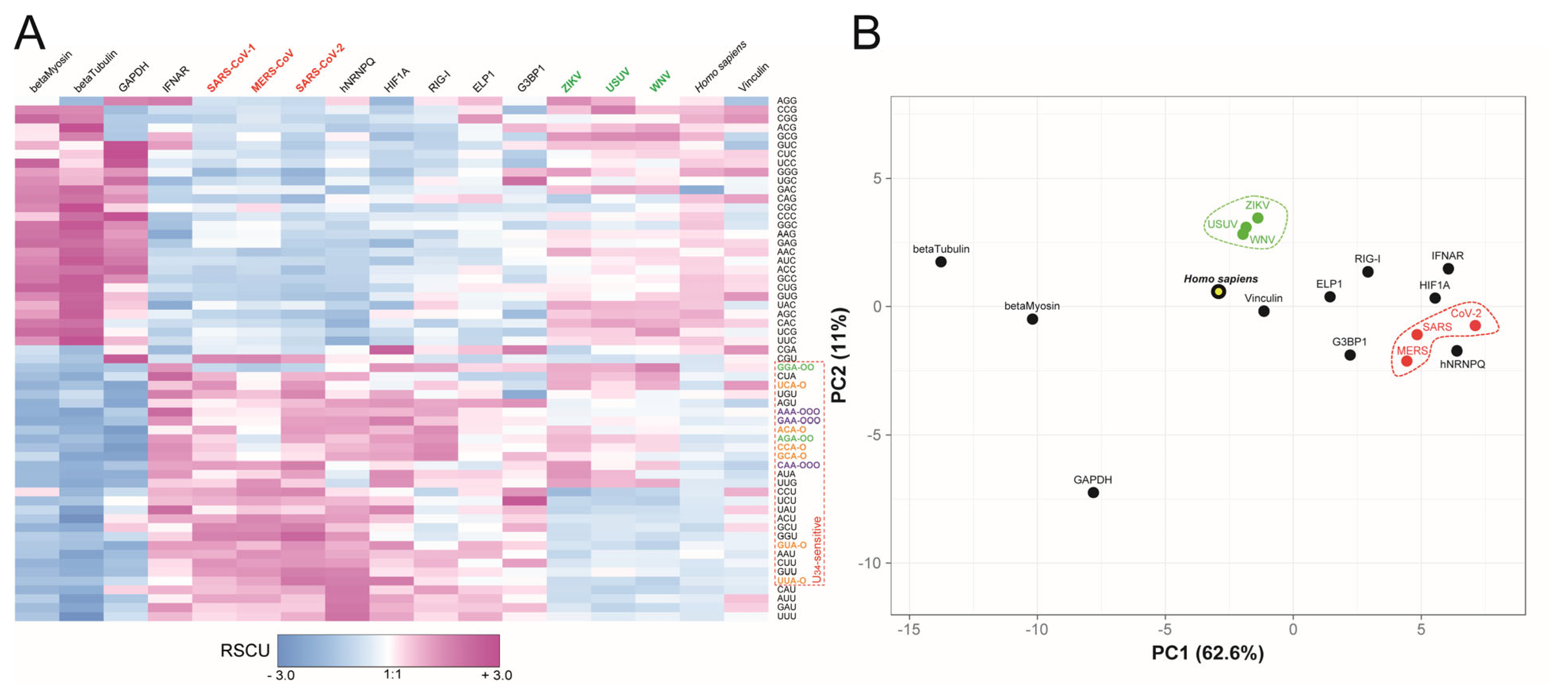
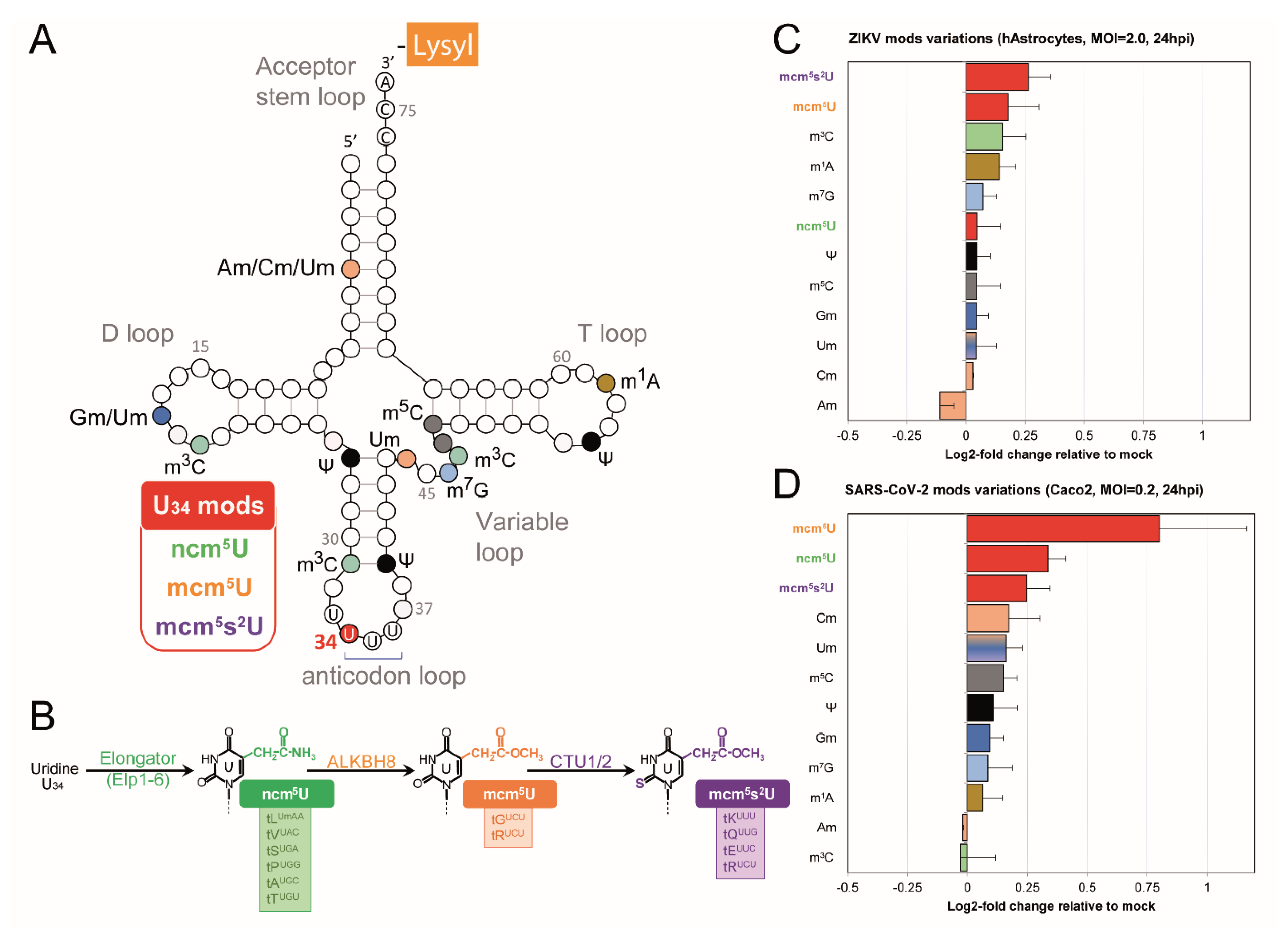
2.2. tRNA Modification Remodeling During ZIKV and SARS-CoV-2 Infection
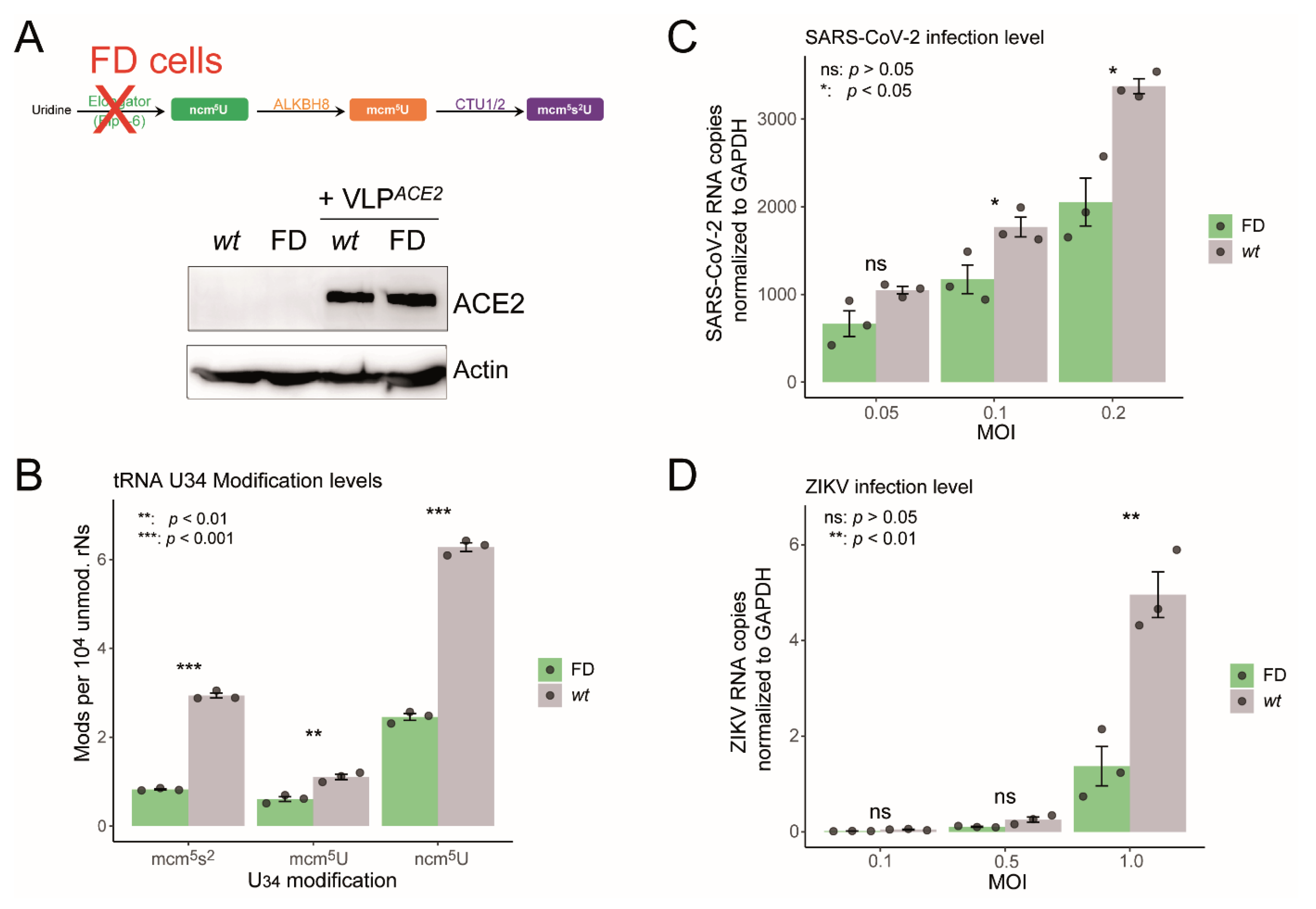
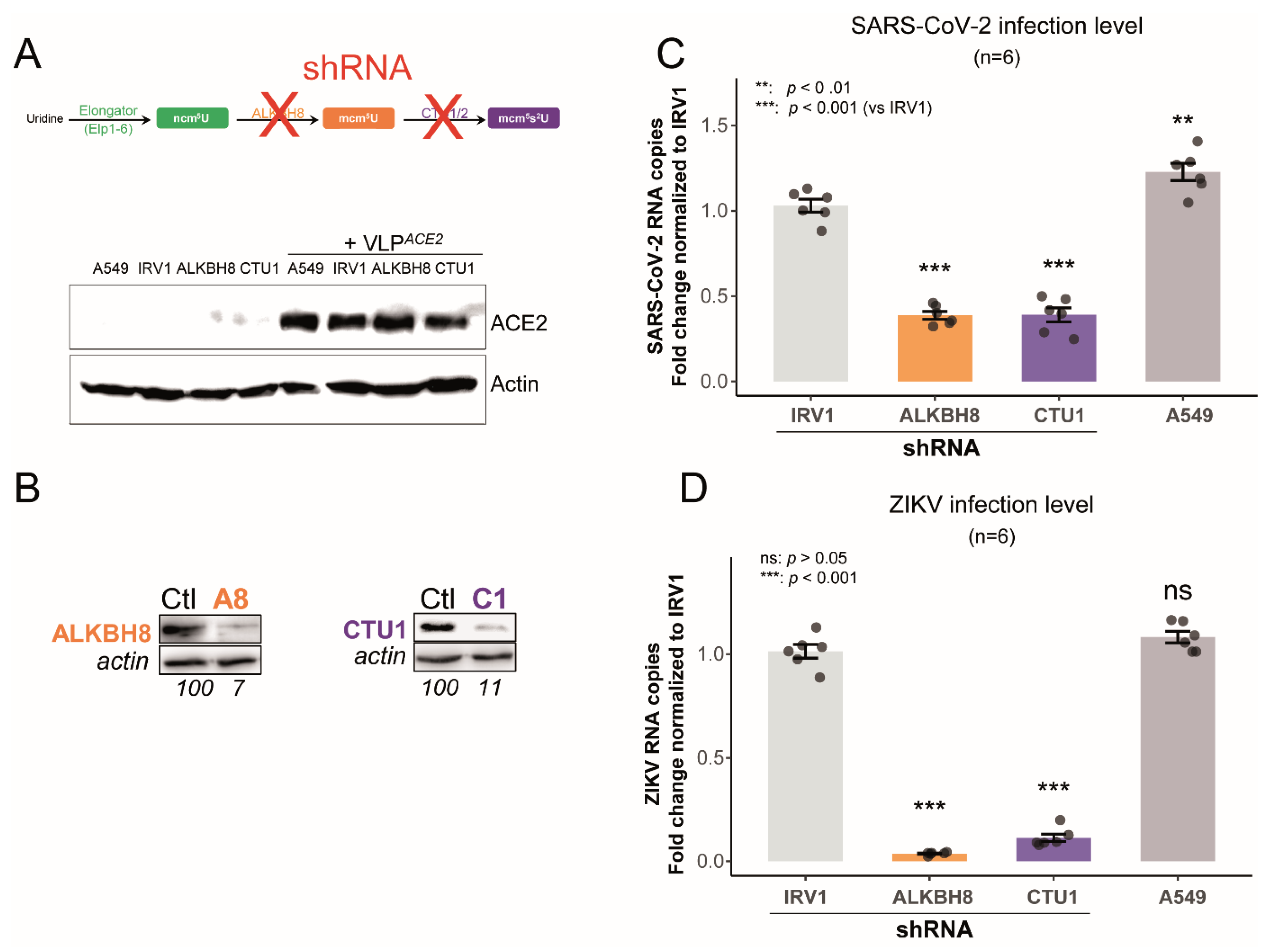
2.3. Impaired U34 tRNA Generation Decreases Translation and Limits SARS-CoV-2 and ZIKV Replication
3. Discussion
4. Material and Methods
4.1. Bio-Informatics—Codon Analysis
4.2. Cells and Viruses
4.3. Quantification of tRNA Modifications by Mass Spectrometry (LC-MS/MS)
4.4. Gene Invalidation (CRISPR/Cas9 and shRNA)
4.5. Assays for Viral Replication
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schultz, S.K.; Kothe, U. RNA Modifying Enzymes Shape tRNA Biogenesis and Function. J. Biol. Chem. 2024, 300, 107488. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Nedialkova, D.D.; Leidel, S.A. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, N.; Rodnina, M.V. tRNA Wobble Modifications and Protein Homeostasis. Translation 2016, 4, e1143076. [Google Scholar] [CrossRef]
- Tuorto, F.; Lyko, F. Genome Recoding by tRNA Modifications. Open Biol. 2016, 6, 160287. [Google Scholar] [CrossRef]
- Jackman, J.E.; Alfonzo, J.D. Transfer RNA Modifications: Nature’s Combinatorial Chemistry Playground. WIREs RNA 2013, 4, 35–48. [Google Scholar] [CrossRef]
- Barraud, P.; Tisné, C. To Be or Not to Be Modified: Miscellaneous Aspects Influencing Nucleotide Modifications in tRNAs. IUBMB life 2019, 71, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Hopper, A. Multiple Layers of Stress-Induced Regulation in tRNA Biology. Life 2016, 6, 16. [Google Scholar] [CrossRef]
- Yared, M.-J.; Marcelot, A.; Barraud, P. Beyond the Anticodon: tRNA Core Modifications and Their Impact on Structure, Translation and Stress Adaptation. Genes 2024, 15, 374. [Google Scholar] [CrossRef]
- Nunes, A.; Ribeiro, D.R.; Marques, M.; Santos, M.A.S.; Ribeiro, D.; Soares, A.R. Emerging Roles of tRNAs in RNA Virus Infections. Trends Biochem. Sci. 2020, 45, 794–805. [Google Scholar] [CrossRef]
- Goffena, J.; Lefcort, F.; Zhang, Y.; Lehrmann, E.; Chaverra, M.; Felig, J.; Walters, J.; Buksch, R.; Becker, K.G.; George, L. Elongator and Codon Bias Regulate Protein Levels in Mammalian Peripheral Neurons. Nat. Commun. 2018, 9, 889. [Google Scholar] [CrossRef]
- Johansson, M.J.O.; Esberg, A.; Huang, B.; Björk, G.R.; Byström, A.S. Eukaryotic Wobble Uridine Modifications Promote a Functionally Redundant Decoding System. Mol. Cell. Biol. 2008, 28, 3301–3312. [Google Scholar] [CrossRef]
- Agris, P.F.; Narendran, A.; Sarachan, K.; Väre, V.Y.P.; Eruysal, E. The Importance of Being Modified: The Role of RNA Modifications in Translational Fidelity. Enzymes 2017, 41, 1–50. [Google Scholar] [CrossRef]
- Komar, A.A. The Yin and Yang of Codon Usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; de Crécy-Lagard, V.; Marck, C. Deciphering Synonymous Codons in the Three Domains of Life: Co-Evolution with Specific tRNA Modification Enzymes. FEBS Lett. 2010, 584, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The Adaptation of Codon Usage of +ssRNA Viruses to Their Hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. Canine Parvovirus Type 2 (CPV-2) and Feline Panleukopenia Virus (FPV) Codon Bias Analysis Reveals a Progressive Adaptation to the New Niche after the Host Jump. Mol. Phylogenet. Evol. 2017, 114, 82–92. [Google Scholar] [CrossRef]
- Si, F.; Jiang, L.; Yu, R.; Wei, W.; Li, Z. Study on the Characteristic Codon Usage Pattern in Porcine Epidemic Diarrhea Virus Genomes and Its Host Adaptation Phenotype. Front. Microbiol. 2021, 12, 738082. [Google Scholar] [CrossRef]
- Luo, W.; Tian, L.; Gan, Y.; Chen, E.; Shen, X.; Pan, J.; Irwin, D.M.; Chen, R.-A.; Shen, Y. The Fit of Codon Usage of Human-Isolated Avian Influenza A Viruses to Human. Infect. Genet. Evol. 2020, 81, 104181. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the Rate of Poliovirus Protein Synthesis through Large-Scale Codon Deoptimization Causes Attenuation of Viral Virulence by Lowering Specific Infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef]
- Zhou, J.-H.; Gao, Z.-L.; Zhang, J.; Ding, Y.-Z.; Stipkovits, L.; Szathmary, S.; Pejsak, Z.; Liu, Y.-S. The Analysis of Codon Bias of Foot-and-Mouth Disease Virus and the Adaptation of This Virus to the Hosts. Infect. Genet. Evol. 2013, 14, 105–110. [Google Scholar] [CrossRef]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.R. Dissimilation of Synonymous Codon Usage Bias in Virus–Host Coevolution Due to Translational Selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Eldin, P.; David, A.; Hirtz, C.; Battini, J.-L.; Briant, L. SARS-CoV-2 Displays a Suboptimal Codon Usage Bias for Efficient Translation in Human Cells Diverted by Hijacking the tRNA Epitranscriptome. Int. J. Mol. Sci. 2024, 25, 11614. [Google Scholar] [CrossRef]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, Insights on Structure, Pathogenicity and Immunity Aspects of Pandemic Human Coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and Emergence of Zika Virus. J. Infect. Dis. 2017, 216, S860–S867. [Google Scholar] [CrossRef]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martínez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N. Zika Virus Infection as a Cause of Congenital Brain Abnormalities and Guillain-Barré Syndrome: Systematic Review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef]
- Kuno, G.; Chang, G.-J.J. Full-Length Sequencing and Genomic Characterization of Bagaza, Kedougou, and Zika Viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The Codon Adaptation Index—A Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- RoyChoudhury, S.; Mukherjee, D. A Detailed Comparative Analysis on the Overall Codon Usage Pattern in Herpesviruses. Virus Res. 2010, 148, 31–43. [Google Scholar] [CrossRef]
- Su, M.-W.; Lin, H.-M.; Yuan, H.S.; Chu, W.-C. Categorizing Host-Dependent RNA Viruses by Principal Component Analysis of Their Codon Usage Preferences. J. Comput. Biol. 2009, 16, 1539–1547. [Google Scholar] [CrossRef]
- Johansson, M.J.O.; Xu, F.; Byström, A.S. Elongator-a tRNA Modifying Complex That Promotes Efficient Translational Decoding. Biochim. Et Biophys. Acta Gene Regul. Mech. 2018, 1861, 401–408. [Google Scholar] [CrossRef]
- Donadon, I.; Pinotti, M.; Rajkowska, K.; Pianigiani, G.; Barbon, E.; Morini, E.; Motaln, H.; Rogelj, B.; Mingozzi, F.; Slaugenhaupt, S.A.; et al. Exon-Specific U1 snRNAs Improve ELP1 Exon 20 Definition and Rescue ELP1 Protein Expression in a Familial Dysautonomia Mouse Model. Hum. Mol. Genet. 2018, 27, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Karlsborn, T.; Tükenmez, H.; Chen, C.; Byström, A.S. Familial Dysautonomia (FD) Patients Have Reduced Levels of the Modified Wobble Nucleoside mcm5s2U in tRNA. Biochem. Biophys. Res. Commun. 2014, 454, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Kataoka, N.; Miyauchi, K.; Ohe, K.; Iida, K.; Yoshida, S.; Nojima, T.; Okuno, Y.; Onogi, H.; Usui, T.; et al. Rectifier of Aberrant mRNA Splicing Recovers tRNA Modification in Familial Dysautonomia. Proc. Natl. Acad. Sci. USA 2015, 112, 2764–2769. [Google Scholar] [CrossRef] [PubMed]
- Eldin, P.; Bernard, E.; Vågbø, C.B.; George, L.; Ajiro, M.; Hagiwara, M.; Slupphaug, G.; Briant, L. Zika Virus Reprograms the Host tRNA Epitranscriptome to Adapt Translation to A-Ending Codon Bias. bioRxiv 2025. [Google Scholar] [CrossRef]
- Jungfleisch, J.; Böttcher, R.; Talló-Parra, M.; Pérez-Vilaró, G.; Merits, A.; Novoa, E.M.; Díez, J. CHIKV Infection Reprograms Codon Optimality to Favor Viral RNA Translation by Altering the tRNA Epitranscriptome. Nat. Commun. 2022, 13, 4725. [Google Scholar] [CrossRef]
- Chan, C.; Kwan Sze, N.S.; Suzuki, Y.; Ohira, T.; Suzuki, T.; Begley, T.J.; Dedon, P.C. Dengue Virus Exploits the Host tRNA Epitranscriptome to Promote Viral Replication. bioRxiv 2023. [Google Scholar] [CrossRef]
- Talló-Parra, M.; Muscolino, E.; Díez, J. The Host tRNA Epitranscriptome: A New Player in RNA Virus Infections. Front. Virol. 2022, 2, 1073619. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A Combined Set of Tools to Assess Codon Usage Adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Athey, J.; Alexaki, A.; Osipova, E.; Rostovtsev, A.; Santana-Quintero, L.V.; Katneni, U.; Simonyan, V.; Kimchi-Sarfaty, C. A New and Updated Resource for Codon Usage Tables. BMC Bioinform. 2017, 18, 391. [Google Scholar] [CrossRef] [PubMed]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster Analysis of Microarray Data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed]
- de Hoon, M.J.L.; Imoto, S.; Nolan, J.; Miyano, S. Open Source Clustering Software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef]
- Saldanha, A.J. Java Treeview—Extensible Visualization of Microarray Data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein Abundance Data, Integrated across Model Organisms, Tissues, and Cell-Lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef]
- Mutso, M.; Saul, S.; Rausalu, K.; Susova, O.; Žusinaite, E.; Mahalingam, S.; Merits, A. Reverse Genetic System, Genetically Stable Reporter Viruses and Packaged Subgenomic Replicon Based on a Brazilian Zika Virus Isolate. J. Gen. Virol. 2017, 98, 2712–2724. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient Genome Modification by CRISPR-Cas9 Nickase with Minimal off-Target Effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Eldin, P.; Péron, S.; Galashevskaya, A.; Denis-Lagache, N.; Cogné, M.; Slupphaug, G.; Briant, L. Impact of HIV-1 Vpr Manipulation of the DNA Repair Enzyme UNG2 on B Lymphocyte Class Switch Recombination. J. Transl. Med. 2020, 18, 310. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eldin, P.; Briant, L. tRNA Modifications: A Tale of Two Viruses—SARS-CoV-2 and ZIKV. Int. J. Mol. Sci. 2025, 26, 7479. https://doi.org/10.3390/ijms26157479
Eldin P, Briant L. tRNA Modifications: A Tale of Two Viruses—SARS-CoV-2 and ZIKV. International Journal of Molecular Sciences. 2025; 26(15):7479. https://doi.org/10.3390/ijms26157479
Chicago/Turabian StyleEldin, Patrick, and Laurence Briant. 2025. "tRNA Modifications: A Tale of Two Viruses—SARS-CoV-2 and ZIKV" International Journal of Molecular Sciences 26, no. 15: 7479. https://doi.org/10.3390/ijms26157479
APA StyleEldin, P., & Briant, L. (2025). tRNA Modifications: A Tale of Two Viruses—SARS-CoV-2 and ZIKV. International Journal of Molecular Sciences, 26(15), 7479. https://doi.org/10.3390/ijms26157479