
Unraveling NETs in Sepsis: From Cellular Mechanisms to Clinical Relevance
 ,
,  ,
, 
Abstract
1. Introduction
2. Neutrophil Extracellular Traps (NETs): Formation and Function
2.1. NETosis
2.1.1. Types of NETosis
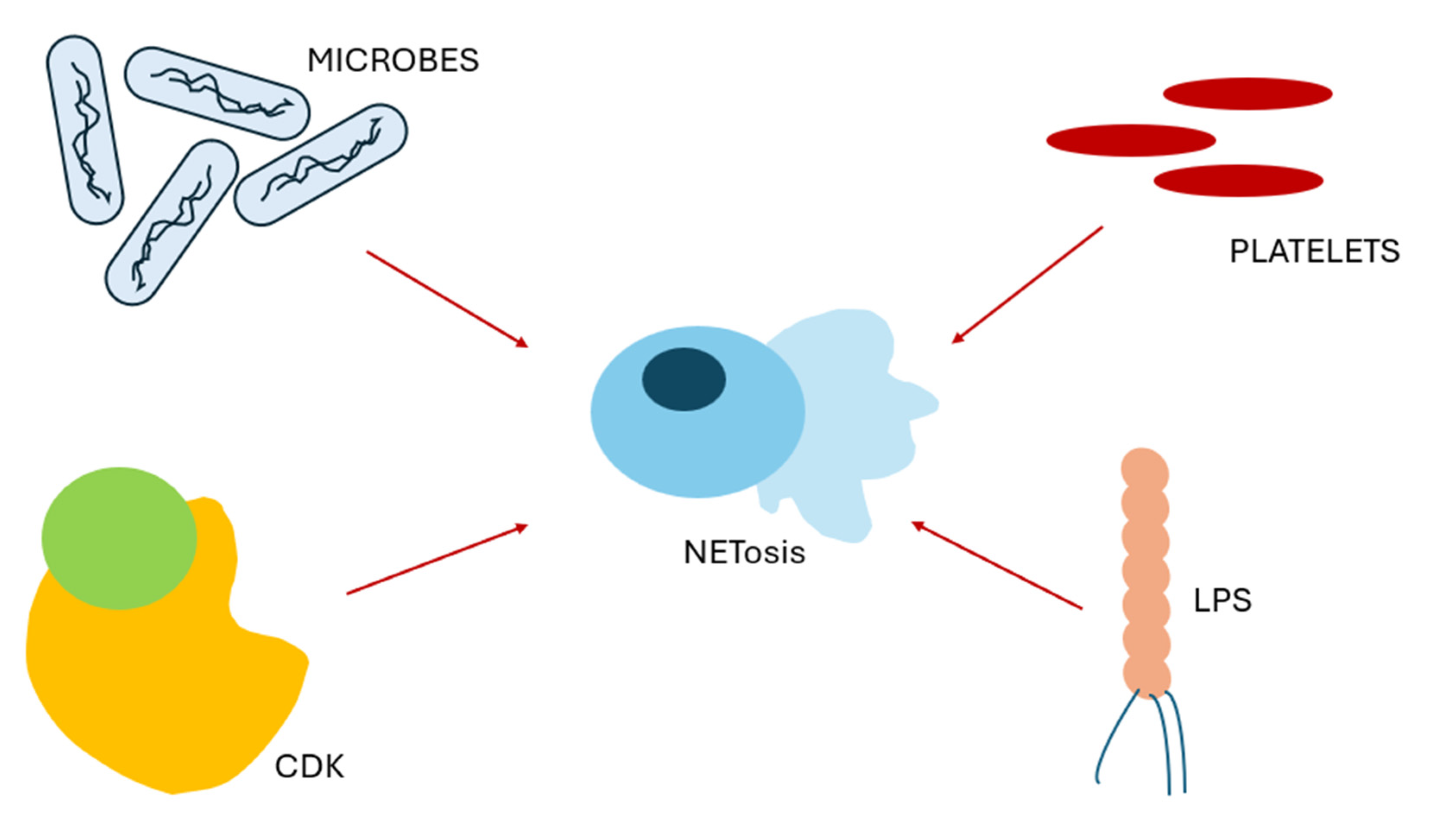
2.1.2. Stimuli for NETosis
2.1.3. Influences of Neutrophil Phenotype on NETosis
3. Role of NETs in Sepsis
3.1. Beneficial Role of NETs in Sepsis
3.2. Detrimental Roles of NETs in Sepsis
3.2.1. Endothelial Injury and Barrier Disruption
3.2.2. Sepsis-Induced Thrombosis
3.2.3. Pathological Angiogenesis in Sepsis
4. Biomarkers of NET Activation in Inflammation and Sepsis
4.1. Nucleosomes
4.2. MPO and MPO-DNA
4.3. CfDNA
4.4. Nitric Oxide (NO)
4.5. Others
5. Therapeutic Strategies Targeting NETs
5.1. Inhibition of NETosis
5.1.1. PAD4
5.1.2. ROS
5.1.3. MPO
5.2. Degradation of Already Formed NETs
DNASe
5.3. Blocking the Pathogenic Effects of NETs
5.3.1. Heparin
5.3.2. Aspirin
5.4. Novel Approaches
5.5. Extracorporeal Sorption Strategies for NET Clearance
6. Conclusions
7. Methods
Funding
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Gaieski, D.F.; Edwards, J.M.; Kallan, M.J.; Carr, B.G. Benchmarking the Incidence and Mortality of Severe Sepsis in the United States. Crit. Care Med. 2013, 41, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cicchinelli, S.; Pignataro, G.; Gemma, S.; Piccioni, A.; Picozzi, D.; Ojetti, V.; Franceschi, F.; Candelli, M. PAMPs and DAMPs in Sepsis: A Review of Their Molecular Features and Potential Clinical Implications. Int. J. Mol. Sci. 2024, 25, 962. [Google Scholar] [CrossRef] [PubMed]
- Milot, E.; Fotouhi-Ardakani, N.; Filep, J.G. Myeloid Nuclear Differentiation Antigen, Neutrophil Apoptosis and Sepsis. Front. Immunol. 2012, 3, 34306. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, X.; Chatterjee, V.; Meegan, J.E.; Beard, R.S., Jr.; Yuan, S.Y. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front. Immunol. 2019, 10, 1037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Qu, M.; Li, W.; Wu, D.; Cata, J.P.; Miao, C. Neutrophil, Neutrophil Extracellular Traps and Endothelial Cell Dysfunction in Sepsis. Clin. Transl. Med. 2023, 13, e1170. [Google Scholar] [CrossRef]
- Borregaard, N.; Sørensen, O.E.; Theilgaard-Mönch, K. Neutrophil Granules: A Library of Innate Immunity Proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil Function: From Mechanisms to Disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil Extracellular Traps Capture and Kill Candida Albicans Yeast and Hyphal Forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils Sense Microbe Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef]
- Manda, A.; Pruchniak, M.P.; Araźna, M.; Demkow, U.A. Neutrophil Extracellular Traps in Physiology and Pathology. Central Eur. J. Immunol. 2014, 1, 116–121. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase Is Required for Neutrophil Extracellular Trap Formation: Implications for Innate Immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 Is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking In Vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gómez, R.M.; Schattner, M. Mediators and Molecular Pathways Involved in the Regulation of Neutrophil Extracellular Trap Formation Mediated by Activated Platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462.e5. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Ode, Y.; Aziz, M.; Wang, P. CIRP Increases ICAM-1+ Phenotype of Neutrophils Exhibiting Elevated iNOS and NETs in Sepsis. J. Leukoc. Biol. 2018, 103, 693–707. [Google Scholar] [CrossRef]
- Zhao, Y.; Yi, W.; Wan, X.; Wang, J.; Tao, T.; Li, J.; Wang, J.; Deng, X. Blockade of ICAM-1 Improves the Outcome of Polymicrobial Sepsis via Modulating Neutrophil Migration and Reversing Immunosuppression. Mediat. Inflamm. 2014, 2014, 195290. [Google Scholar] [CrossRef]
- Alarcón, P.; Manosalva, C.; Conejeros, I.; Carretta, M.D.; Muñoz-Caro, T.; Silva, L.M.R.; Taubert, A.; Hermosilla, C.; Hidalgo, M.A.; Burgos, R.A. D(−) Lactic Acid-Induced Adhesion of Bovine Neutrophils onto Endothelial Cells Is Dependent on Neutrophils Extracellular Traps Formation and CD11b Expression. Front. Immunol. 2017, 8, 975. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Voels, S.; Granot, Z. Isolation and Characterization of Low- vs. High-Density Neutrophils in Cancer. In The Tumor Microenvironment; Ursini-Siegel, J., Beauchemin, N., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1458, pp. 179–193. ISBN 978-1-4939-3799-8. [Google Scholar]
- Kanamaru, R.; Ohzawa, H.; Miyato, H.; Matsumoto, S.; Haruta, H.; Kurashina, K.; Saito, S.; Hosoya, Y.; Yamaguchi, H.; Yamashita, H.; et al. Low Density Neutrophils (LDN) in Postoperative Abdominal Cavity Assist the Peritoneal Recurrence through the Production of Neutrophil Extracellular Traps (NETs). Sci. Rep. 2018, 8, 632. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.-E.; Scheiermann, C.; et al. Neutrophil Ageing Is Regulated by the Microbiome. Nature 2015, 525, 528–532. [Google Scholar] [CrossRef]
- Buchanan, J.T.; Simpson, A.J.; Aziz, R.K.; Liu, G.Y.; Kristian, S.A.; Kotb, M.; Feramisco, J.; Nizet, V. DNase Expression Allows the Pathogen Group A Streptococcus to Escape Killing in Neutrophil Extracellular Traps. Curr. Biol. 2006, 16, 396–400. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular Neutrophil Extracellular Traps Capture Bacteria from the Bloodstream during Sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An Endonuclease Allows Streptococcus Pneumoniae to Escape from Neutrophil Extracellular Traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef]
- Wartha, F.; Beiter, K.; Albiger, B.; Fernebro, J.; Zychlinsky, A.; Normark, S.; Henriques-Normark, B. Capsule and D-Alanylated Lipoteichoic Acids Protect Streptococcus Pneumoniae against Neutrophil Extracellular Traps. Cell. Microbiol. 2007, 9, 1162–1171. [Google Scholar] [CrossRef]
- Zhong, H.; Lu, R.-Y.; Wang, Y. Neutrophil Extracellular Traps in Fungal Infections: A Seesaw Battle in Hosts. Front. Immunol. 2022, 13, 977493. [Google Scholar] [CrossRef]
- Dömer, D.; Walther, T.; Möller, S.; Behnen, M.; Laskay, T. Neutrophil Extracellular Traps Activate Proinflammatory Functions of Human Neutrophils. Front. Immunol. 2021, 12, 636954. [Google Scholar] [CrossRef] [PubMed]
- Retter, A.; Singer, M.; Annane, D. “The NET Effect”: Neutrophil Extracellular Traps—A Potential Key Component of the Dysregulated Host Immune Response in Sepsis. Crit. Care 2025, 59. [Google Scholar] [CrossRef]
- Zhu, S.; Yu, Y.; Qu, M.; Qiu, Z.; Zhang, H.; Miao, C.; Guo, K. Neutrophil Extracellular Traps Contribute to Immunothrombosis Formation via the STING Pathway in Sepsis-Associated Lung Injury. Cell Death Discov. 2023, 9, 315. [Google Scholar] [CrossRef]
- Hellenthal, K.E.M.; Brabenec, L.; Wagner, N.-M. Regulation and Dysregulation of Endothelial Permeability during Systemic Inflammation. Cells 2022, 11, 1935. [Google Scholar] [CrossRef]
- Allingham, M.J.; Van Buul, J.D.; Burridge, K. ICAM-1-Mediated, Src- and Pyk2-Dependent Vascular Endothelial Cadherin Tyrosine Phosphorylation Is Required for Leukocyte Transendothelial Migration. J. Immunol. 2007, 179, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Radeva, M.Y.; Waschke, J. Mind the Gap: Mechanisms Regulating the Endothelial Barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhu, P.; Jiang, L.; Hu, Y.; Huang, L.; He, Y.; Zhang, H. Neutrophil Extracellular Traps Induced by IL-1β Promote Endothelial Dysfunction and Aggravate Limb Ischemia. Hypertens. Res. 2024, 47, 1654–1667. [Google Scholar] [CrossRef]
- Ramos, A.; Youssef, L.; Molina, P.; Torramadé-Moix, S.; Martinez-Sanchez, J.; Moreno-Castaño, A.B.; Blasco, M.; Guillén-Olmos, E.; De Moner, B.; Pino, M.; et al. Circulating Extracellular Vesicles and Neutrophil Extracellular Traps Contribute to Endothelial Dysfunction in Preeclampsia. Front. Immunol. 2024, 15, 1488127. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Mo, L.; Zeng, G.; Ma, P.; Zhang, X.; Peng, Y.; Zhang, X.; Zhou, Y.; Jiang, Y.; Huang, N.; et al. Inhibition of Neutrophil Extracellular Traps Alleviates Blood–Brain Barrier Disruption and Cognitive Dysfunction via Wnt3/β-Catenin/TCF4 Signaling in Sepsis-Associated Encephalopathy. J. Neuroinflamm. 2025, 22, 87. [Google Scholar] [CrossRef] [PubMed]
- Meegan, J.E.; Yang, X.; Beard, R.S.; Jannaway, M.; Chatterjee, V.; Taylor-Clark, T.E.; Yuan, S.Y. Citrullinated Histone 3 Causes Endothelial Barrier Dysfunction. Biochem. Biophys. Res. Commun. 2018, 503, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Weng, J. Endothelial Dysfunction in COVID-19: An Overview of Evidence, Biomarkers, Mechanisms and Potential Therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef]
- DiStasi, M.R.; Ley, K. Opening the Flood-Gates: How Neutrophil-Endothelial Interactions Regulate Permeability. Trends Immunol. 2009, 30, 547–556. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef]
- Barkaway, A.; Rolas, L.; Joulia, R.; Bodkin, J.; Lenn, T.; Owen-Woods, C.; Reglero-Real, N.; Stein, M.; Vázquez-Martínez, L.; Girbl, T.; et al. Age-Related Changes in the Local Milieu of Inflamed Tissues Cause Aberrant Neutrophil Trafficking and Subsequent Remote Organ Damage. Immunity 2021, 54, 1494–1510.e7. [Google Scholar] [CrossRef]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated Endothelial Cells Induce Neutrophil Extracellular Traps and Are Susceptible to NETosis-mediated Cell Death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef]
- Ali, M.M.; Mahmoud, A.M.; Le Master, E.; Levitan, I.; Phillips, S.A. Role of Matrix Metalloproteinases and Histone Deacetylase in Oxidative Stress-Induced Degradation of the Endothelial Glycocalyx. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H647–H663. [Google Scholar] [CrossRef]
- Gäddnäs, F.P.; Sutinen, M.M.; Koskela, M.; Tervahartiala, T.; Sorsa, T.; Salo, T.A.; Laurila, J.J.; Koivukangas, V.; Ala-Kokko, T.I.; Oikarinen, A. Matrix-Metalloproteinase-2, -8 and -9 in Serum and Skin Blister Fluid in Patients with Severe Sepsis. Crit. Care 2010, 14, R49. [Google Scholar] [CrossRef]
- Giustozzi, M.; Ehrlinder, H.; Bongiovanni, D.; Borovac, J.A.; Guerreiro, R.A.; Gąsecka, A.; Papakonstantinou, P.E.; Parker, W.A.E. Coagulopathy and Sepsis: Pathophysiology, Clinical Manifestations and Treatment. Blood Rev. 2021, 50, 100864. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Helms, J.; Connors, J.M.; Levy, J.H. The Pathophysiology, Diagnosis, and Management of Sepsis-Associated Disseminated Intravascular Coagulation. J. Intensive Care 2023, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- De Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, Complement, and Coagulation: A Triangular Relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular Histones Increase Plasma Thrombin Generation by Impairing Thrombomodulin-dependent Protein C Activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Kim, J.E.; Yoo, H.J.; Gu, J.Y.; Kim, H.K. Histones Induce the Procoagulant Phenotype of Endothelial Cells through Tissue Factor Up-Regulation and Thrombomodulin Down-Regulation. PLoS ONE 2016, 11, e0156763. [Google Scholar] [CrossRef]
- Haubitz, M.; Gerlach, M.; Kruse, H.J.; Brunkhorst, R. Endothelial Tissue Factor Stimulation by Proteinase 3 and Elastase. Clin. Exp. Immunol. 2002, 126, 584–588. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.C.; Weitz, J.I.; Liaw, P.C. Neutrophil Extracellular Traps Promote Thrombin Generation Through Platelet-Dependent and Platelet-Independent Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Toltl, L.J.; Swystun, L.L.; Pogue, J.; Liaw, K.-L.; Weitz, J.I.; Cook, D.J.; Fox-Robichaud, A.E.; Liaw, P.C.; the Canadian Critical Care Translational Biology Group. Prognostic Utility and Characterization of Cell-Free DNA in Patients with Severe Sepsis. Crit. Care 2012, 16, R151. [Google Scholar] [CrossRef]
- Margraf, S.; Lögters, T.; Reipen, J.; Altrichter, J.; Scholz, M.; Windolf, J. Neutrophil-Derived Circulating Free Dna (Cf-Dna/Nets): A Potential Prognostic Marker for Posttraumatic Development of Inflammatory Second Hit and Sepsis. Shock 2008, 30, 352–358. [Google Scholar] [CrossRef]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence From Vascular Pathology in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef]
- Kung, C.-T.; Su, C.-M.; Chen, C.T.; Cheng, H.-H.; Chang, M.-W.; Hung, C.-W.; Hung, S.-C.; Chang, W.-N.; Tsai, N.-W.; Wang, H.-C.; et al. Circulating Endothelial Progenitor Cells May Predict Outcomes in Adult Patients with Severe Sepsis in the Emergency Department. Clin. Chim. Acta 2016, 455, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Haem Rahimi, M.; Bidar, F.; Lukaszewicz, A.-C.; Garnier, L.; Payen-Gay, L.; Venet, F.; Monneret, G. Association of Pronounced Elevation of NET Formation and Nucleosome Biomarkers with Mortality in Patients with Septic Shock. Ann. Intensive Care 2023, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Filippini, D.F.L.; Jiang, M.; Kramer, L.; Van Der Poll, T.; Cremer, O.; Hla, T.T.W.; Retter, A.; Bos, L.D.J.; on behalf of the MARS consortium. Plasma H3.1 Nucleosomes as Biomarkers of Infection, Inflammation and Organ Failure. Crit. Care 2025, 29, 198. [Google Scholar] [CrossRef] [PubMed]
- Morimont, L.; Dechamps, M.; David, C.; Bouvy, C.; Gillot, C.; Haguet, H.; Favresse, J.; Ronvaux, L.; Candiracci, J.; Herzog, M.; et al. NETosis and Nucleosome Biomarkers in Septic Shock and Critical COVID-19 Patients: An Observational Study. Biomolecules 2022, 12, 1038. [Google Scholar] [CrossRef]
- Yaozhen, L.; Kemin, W.; Xiaoyu, J.; Yang, O.; Hongying, T.; Baihong, P. Evaluation of a Reliable Biomarker in a Cecal Ligation and Puncture-Induced Mouse Model of Sepsis. J. Vis. Exp. 2022, 63584. [Google Scholar] [CrossRef]
- Pan, B.; Li, Y.; Liu, Y.; Wang, W.; Huang, G.; Ouyang, Y. Circulating CitH3 Is a Reliable Diagnostic and Prognostic Biomarker of Septic Patients in Acute Pancreatitis. Front. Immunol. 2021, 12, 766391. [Google Scholar] [CrossRef]
- He, J.; Zheng, F.; Qiu, L.; Wang, Y.; Zhang, J.; Ye, H.; Zhang, Q. Plasma Neutrophil Extracellular Traps in Patients with Sepsis-Induced Acute Kidney Injury Serve as a New Biomarker to Predict 28-Day Survival Outcomes of Disease. Front. Med. 2024, 11, 1496966. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Kremer Hovinga, J.A.; Schatzberg, D.; Wagner, D.D.; Lämmle, B. Circulating DNA and Myeloperoxidase Indicate Disease Activity in Patients with Thrombotic Microangiopathies. Blood 2012, 120, 1157–1164. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, J.; Shi, C.; Wang, Y.; Zhang, Y.; Zhang, X.; Gong, Z. MPO-DNA Complexes and Cf-DNA in Patients with Sepsis and Their Clinical Value. Biomedicines 2024, 12, 2190. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Lin, Y.-C.; Lee, M.-J.; Chen, J.-W.; Hsu, C.-C.; Huang, T.-Y.; Chen, J.-H.; Tzeng, S.-J.; Chiu, Y.-L.; Wang, S.-R.; et al. Biomarker of Neutrophil Extracellular Traps Is Associated with Deep-Seated Infections and Predicts Mortality and Cardiovascular Morbidity in Commensal Streptococcal Bacteremia. J. Microbiol. Immunol. Infect. 2022, 55, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Lenz, M.; Maiberger, T.; Armbrust, L.; Kiwit, A.; Von Der Wense, A.; Reinshagen, K.; Elrod, J.; Boettcher, M. cfDNA and DNases: New Biomarkers of Sepsis in Preterm Neonates—A Pilot Study. Cells 2022, 11, 192. [Google Scholar] [CrossRef]
- Zhang, J.; Shao, Y.; Wu, J.; Zhang, J.; Xiong, X.; Mao, J.; Wei, Y.; Miao, C.; Zhang, H. Dysregulation of Neutrophil in Sepsis: Recent Insights and Advances. Cell Commun. Signal. 2025, 23, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zheng, Q.; Liu, X.; Ding, H.; Ma, M.; Bao, J.; Cai, Y.; Cao, C. HMGB1 Lactylation Drives Neutrophil Extracellular Trap Formation in Lactate-Induced Acute Kidney Injury. Front. Immunol. 2025, 15, 1475543. [Google Scholar] [CrossRef]
- Tang, J.; Lu, H.; Xie, Z.; Jia, X.; Su, T.; Lin, B. Identification of Potential Biomarkers for Sepsis Based on Neutrophil Extracellular Trap-Related Genes. Diagn. Microbiol. Infect. Dis. 2024, 110, 116380. [Google Scholar] [CrossRef]
- You, G.; Zhao, X.; Liu, J.; Yao, K.; Yi, X.; Chen, H.; Wei, X.; Huang, Y.; Yang, X.; Lei, Y.; et al. Machine Learning-Based Identification of CYBB and FCAR as Potential Neutrophil Extracellular Trap-Related Treatment Targets in Sepsis. Front. Immunol. 2023, 14, 1253833. [Google Scholar] [CrossRef]
- Garcia, G.; Labrouche-Colomer, S.; Duvignaud, A.; Clequin, E.; Dussiau, C.; Trégouët, D.-A.; Malvy, D.; Prevel, R.; Zouine, A.; Pellegrin, I.; et al. Impaired Balance between Neutrophil Extracellular Trap Formation and Degradation by DNases in COVID-19 Disease. J. Transl. Med. 2024, 22, 246. [Google Scholar] [CrossRef] [PubMed]
- Kuley, R.; Duvvuri, B.; Wallin, J.J.; Bui, N.; Adona, M.V.; O’Connor, N.G.; Sahi, S.K.; Stanaway, I.B.; Wurfel, M.M.; Morrell, E.D.; et al. Mitochondrial N-Formyl Methionine Peptides Contribute to Exaggerated Neutrophil Activation in Patients with COVID-19. Virulence 2023, 14, 2218077. [Google Scholar] [CrossRef]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.M.; Voorberg, J.; Nicolaes, G.A.F. PAD4 Takes Charge during Neutrophil Activation: Impact of PAD4 Mediated NET Formation on Immune-mediated Disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef]
- Liu, X.; Li, T.; Chen, H.; Yuan, L.; Ao, H. Role and Intervention of PAD4 in NETs in Acute Respiratory Distress Syndrome. Respir. Res. 2024, 25, 63. [Google Scholar] [CrossRef]
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted Delivery of Protein Arginine Deiminase-4 Inhibitors to Limit Arterial Intimal NETosis and Preserve Endothelial Integrity. Cardiovasc. Res. 2021, 117, 2652–2663. [Google Scholar] [CrossRef]
- Floyd, J.L.; Prasad, R.; Dupont, M.D.; Adu-Rutledge, Y.; Anshumali, S.; Paul, S.; Li Calzi, S.; Qi, X.; Malepati, A.; Johnson, E.; et al. Intestinal Neutrophil Extracellular Traps Promote Gut Barrier Damage Exacerbating Endotoxaemia, Systemic Inflammation and Progression of Diabetic Retinopathy in Type 2 Diabetes. Diabetologia 2025, 68, 866–889. [Google Scholar] [CrossRef]
- Denorme, F.; Rustad, J.L.; Portier, I.; Crandell, J.L.; De Araujo, C.V.; Cody, M.J.; Campbell, R.A.; Yost, C.C. Neutrophil Extracellular Trap Inhibition Improves Survival in Neonatal Mouse Infectious Peritonitis. Pediatr. Res. 2023, 93, 862–869. [Google Scholar] [CrossRef]
- He, W.; Xi, Q.; Cui, H.; Zhang, P.; Huang, R.; Wang, T.; Wang, D. Forsythiaside B Ameliorates Coagulopathies in a Rat Model of Sepsis through Inhibition of the Formation of PAD4-Dependent Neutrophil Extracellular Traps. Front. Pharmacol. 2022, 13, 1022985. [Google Scholar] [CrossRef]
- Kolarz, B.; Ciesla, M.; Dryglewska, M.; Majdan, M. Peptidyl Arginine Deiminase Type 4 Gene Promoter Hypo-Methylation in Rheumatoid Arthritis. JCM 2020, 9, 2049. [Google Scholar] [CrossRef]
- Domiciano, T.P.; Lee, Y.; Carvalho, T.T.; Wakita, D.; Martinon, D.; Jena, P.K.; Fert-Bober, J.; Borges, V.; Crother, T.R.; Chen, S.; et al. Redundant Role of PAD2 and PAD4 in the Development of Cardiovascular Lesions in a Mouse Model of Kawasaki Disease Vasculitis. Clin. Exp. Immunol. 2024, 218, 314–328. [Google Scholar] [CrossRef]
- Wang, P.; Liu, D.; Zhou, Z.; Liu, F.; Shen, Y.; You, Q.; Lu, S.; Wu, J. The Role of Protein Arginine Deiminase 4-Dependent Neutrophil Extracellular Traps Formation in Ulcerative Colitis. Front. Immunol. 2023, 14, 1144976. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Lu, Y.; Hu, B.; Pang, Y.; Liu, B.; Zhang, M.; Wang, W.; Wang, Y. Highly-Tumor-Targeted PAD4 Inhibitors with PBA Modification Inhibit Tumors In Vivo by Specifically Inhibiting the PAD4-H3cit-NETs Pathway in Neutrophils. Eur. J. Med. Chem. 2023, 258, 115619. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J.; Samstag, Y. Devilishly Radical NETwork in COVID-19: Oxidative Stress, Neutrophil Extracellular Traps (NETs), and T Cell Suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sánchez, G.; Godínez-Méndez, L.A.; Fafutis-Morris, M.; Delgado-Rizo, V. Effect of Antioxidant Supplementation on NET Formation Induced by LPS In Vitro; the Roles of Vitamins E and C, Glutathione, and N-Acetyl Cysteine. Int. J. Mol. Sci. 2023, 24, 13162. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chu, C.; Wang, X.; Yang, C.; Deng, Y.; Duan, Z.; Wang, K.; Liu, B.; Ji, W.; Ding, W. Hesperetin Attenuates Sepsis-Induced Intestinal Barrier Injury by Regulating Neutrophil Extracellular Trap Formation via the ROS/Autophagy Signaling Pathway. Food Funct. 2023, 14, 4213–4227. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, D.; Luo, J.; Jie, J.; Liu, H.; Peng, L.; Bai, X.; Li, D. Zingerone Inhibits the Neutrophil Extracellular Trap Formation and Protects against Sepsis via Nrf2-Mediated ROS Inhibition. Oxidative Med. Cell. Longev. 2022, 2022, 3990607. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Kobayashi, E.; Ichihara, G.; Kitakata, H.; Katsumata, Y.; Sugai, K.; Hakamata, Y.; Sano, M. H2 Inhibits the Formation of Neutrophil Extracellular Traps. JACC Basic Transl. Sci. 2022, 7, 146–161. [Google Scholar] [CrossRef]
- Haute, G.V.; Luft, C.; Pedrazza, L.; Donadio, M.V.F.; De Oliveira, J.R. Octyl Gallate Decrease Lymphocyte Activation and Regulates Neutrophil Extracellular Traps Release. Mol. Biol. Rep. 2022, 49, 1593–1599. [Google Scholar] [CrossRef]
- Duan, Z.; Xie, T.; Chu, C.; Chen, F.; Wang, X.; Li, J.; Ding, W. De-Escalation Antibiotic Therapy Alleviates Organ Injury through Modulation of NETs Formation during Sepsis. Cell Death Discov. 2021, 7, 345. [Google Scholar] [CrossRef]
- Chen, Y.F.; Cheng, L.L.; Shen, Y.H.; Zhang, H.; Wang, X.J.; Xu, Y.C.; Zhang, J.; Ge, J.B. Investigate the role of neutrophil extracellular traps in immune checkpoint inhibitor-associated myocarditis with programmed death protein-1 inhibitors involvement. Zhonghua Yi Xue Za Zhi 2023, 103, 3384–3393. [Google Scholar] [CrossRef] [PubMed]
- Hemmling, H.; Hallberg, L.A.E.; Hägglund, P.; Hawkins, C.L. Histones in Neutrophil Extracellular Traps (NETs) Contain Oxidative Post-Translational Modifications Induced by the Myeloperoxidase Oxidant Hypochlorous Acid. Redox Biol. 2025, 84, 103696. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive Role of Neutrophil Extracellular Traps in Pathogen-Induced Lung Injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef]
- Thomas, G.M.; Carbo, C.; Curtis, B.R.; Martinod, K.; Mazo, I.B.; Schatzberg, D.; Cifuni, S.M.; Fuchs, T.A.; Von Andrian, U.H.; Hartwig, J.H.; et al. Extracellular DNA Traps Are Associated with the Pathogenesis of TRALI in Humans and Mice. Blood 2012, 119, 6335–6343. [Google Scholar] [CrossRef]
- Zalghout, S.; Martinod, K. Therapeutic Potential of DNases in Immunothrombosis: Promising Succor or Uncertain Future? J. Thromb. Haemost. 2025, 23, 760–778. [Google Scholar] [CrossRef]
- Zhu, C.; Xie, J.; Liu, Q.; Wang, Y.; Li, H.; Yu, C.; Li, P.; Deng, X.; Bian, J.; Wang, J. PD-L1 Promotes GSDMD-Mediated NET Release by Maintaining the Transcriptional Activity of Stat3 in Sepsis-Associated Encephalopathy. Int. J. Biol. Sci. 2023, 19, 1413–1429. [Google Scholar] [CrossRef] [PubMed]
- Sohrabipour, S.; Muniz, V.S.; Sharma, N.; Dwivedi, D.J.; Liaw, P.C. Mechanistic Studies of DNase I Activity: Impact of Heparin Variants and PAD4. Shock 2021, 56, 975–987. [Google Scholar] [CrossRef]
- Aramburu, I.V.; Hoving, D.; Vernardis, S.I.; Tin, M.C.F.; Ioannou, M.; Temkin, M.I.; De Vasconcelos, N.M.; Demichev, V.; Helbig, E.T.; Lippert, L.; et al. Functional Proteomic Profiling Links Deficient DNA Clearance with Increased Mortality in Individuals with Severe COVID-19 Pneumonia. Immunity 2022, 55, 2436–2453.e5. [Google Scholar] [CrossRef]
- Wang, D.; Hei, Y.; Sun, H.; Pan, T.; Ma, Z. Heparin and DNase I Treat Myocardial Injury in Septic Mice. Shock 2025, 63, 908–918. [Google Scholar] [CrossRef]
- Komorowicz, E.; Balázs, N.; Tanka-Salamon, A.; Varga, Z.; Szabó, L.; Bóta, A.; Longstaff, C.; Kolev, K. Size- and Charge-dependent Modulation of the Lytic Susceptibility and Mechanical Stability of Fibrin-histone Clots by Heparin and Polyphosphate Variants. J. Thromb. Haemost. 2021, 19, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Semak, V.; Lauková, L.; Fischer, M.B.; Weber, V. Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, Including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones. Int. J. Mol. Sci. 2022, 23, 1823. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.D.; Vieira-Damiani, G.; Da Silva, L.Q.; Leonardi, G.R.; Vaz, C.O.; Jacintho-Robison, B.C.; Mazetto, B.M.; De Paula, E.V.; Monica, F.Z.; Orsi, F.A. Impact of Antiplatelets, Anticoagulants and Cyclic Nucleotide Stimulators on Neutrophil Extracellular Traps (NETs) and Inflammatory Markers during COVID-19. J. Thromb. Thrombolysis 2024, 58, 199–209. [Google Scholar] [CrossRef]
- Huckriede, J.; Keulen, G.M.; Van De Poll, M.C.G.; Wichapong, K.; Reutelingsperger, C.P.M.; Nicolaes, G.A.F. Proteolytic Neutralization of Extracellular Histones by Neutrophil Elastase Is Enhanced by Heparin. J. Thromb. Haemost. 2025, 23, 2483–2493. [Google Scholar] [CrossRef]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of Neutrophil Extracellular Trap Formation by Anti-Inflammatory Drugs. J. Pharmacol. Exp. Ther. 2013, 345, 430–437. [Google Scholar] [CrossRef]
- Alsabani, M.; Abrams, S.T.; Cheng, Z.; Morton, B.; Lane, S.; Alosaimi, S.; Yu, W.; Wang, G.; Toh, C.-H. Reduction of NETosis by Targeting CXCR1/2 Reduces Thrombosis, Lung Injury, and Mortality in Experimental Human and Murine Sepsis. Br. J. Anaesth. 2022, 128, 283–293. [Google Scholar] [CrossRef]
- Zeng, T.; Liang, L.; Deng, W.; Xie, M.; Zhao, M.; Wang, S.; Liu, J.; Yang, M. BMAL1 Plays a Crucial Role in Immune Homeostasis during Sepsis-Induced Acute Lung Injury. Biochem. Pharmacol. 2024, 226, 116379. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Duan, Z.; Wang, X.; Chu, C.; Yang, C.; Chen, F.; Wang, D.; Wang, C.; Li, Q.; Ding, W. Neutrophil Extracellular Traps Impair Intestinal Barrier Functions in Sepsis by Regulating TLR9-Mediated Endoplasmic Reticulum Stress Pathway. Cell Death Dis. 2021, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Gocho, T.; Mori, H.; Islam, M.d.M.; Maruchi, Y.; Takenaka, N.; Tomino, A.; Tsuda, M.; Kano, H.; Takeyama, N. Removal of Circulating Neutrophil Extracellular Trap Components With an Immobilized Polymyxin B Filter: A Preliminary Study. Shock 2020, 54, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Jarczak, D.; Kluge, S.; Nierhaus, A. Septic Hyperinflammation—Is There a Role for Extracorporeal Blood Purification Techniques? Int. J. Mol. Sci. 2024, 25, 3120. [Google Scholar] [CrossRef]

{kind=link}
| Biomarker | Source/Mechanism | Clinical Relevance | Associated Conditions |
|---|---|---|---|
| Histone H3.1 nucleosomes | Released during cell death and neutrophil activation | Correlates with DIC, AKI, hyperinflammation, and 28-day mortality | Sepsis, COVID-19 |
| Citrullinated H3 (e.g., H3R8) | NET-specific modification by PAD4 | Indicates NET activation; associated with disease severity and prognosis; elevated in early sepsis | Sepsis, acute pancreatitis, COVID-19 |
| HMGB1 (lactylated) | DAMP, promotes release of NETs in response to lactate | Induces NETosis; contributes to AKI development | Sepsis, lactate-induced AKI |
| NET-related genes | ELANE, TLR4, MPO, PADI4, CTSG, MMP9, S100A12, CYBB, FCAR | Diagnostic and prognostic value; potential therapeutic targets; predict poor survival; targeting these reduces NET formation and lung injury | Sepsis |
| MPO | Neutrophil granule enzyme involved in ROS-dependent NETosis | Marker of NET activation and disease severity; part of MPO-DNA complexes | Sepsis, COVID-19, TMA |
| MPO-DNA complex | Marker of extracellular NET release | Correlates with inflammation, coagulation, organ damage, and bacteremia mortality | Sepsis, infective endocarditis |
| NE-DNA complex | Derived from neutrophil elastase during NETosis | Indicator of NET activation; correlates with AKI and 28-day mortality | Sepsis with AKI |
| S100A8/A9 (calprotectin) | Neutrophil-derived protein complex | Reflects neutrophil activation and NETosis; increases with disease activity | Thrombotic microangiopathies, sepsis |
| Cell-free DNA (cfDNA) | Released from NETs or damaged cells | Marker of inflammation and organ damage; effective in early sepsis detection, including in neonates | Sepsis, neonatal sepsis |
| DNases (e.g., DNase1L3) | Enzymes degrading extracellular DNA | Impaired degradation in severe disease; therapeutic interest; linked to pDC dysfunction | Severe sepsis, COVID-19 |
| fMET peptides | Mitochondrially derived DAMPs | Activate neutrophils and trigger NETosis; elevated in severe cases | COVID-19 |
| Therapeutic Strategy | Molecular Target | Mechanism of Action | Examples | Indications/Notes |
|---|---|---|---|---|
| PAD4 inhibitors | Peptidyl arginine deiminase 4 (PAD4) | Block histone citrullination → prevent DNA decondensation | Cl-amidine, GSK484 | Autoimmune diseases, sepsis |
| ROS inhibitors | Reactive oxygen species (ROS) | Reduce oxidative stress necessary for NETosis | N-acetylcysteine, apocynin | Chronic inflammation, sepsis |
| MPO inhibitors | Myeloperoxidase (MPO) | Inhibit enzyme involved in NETosis | PF-1355, AZD5904 | Autoimmune and cardiovascular diseases |
| DNases | Extracellular DNA | Degrades DNA backbone of NETs | DNase I (dornase alfa) | Cystic fibrosis, sepsis, ARDS, COVID-19 |
| Histone neutralization | Histones | Neutralizes extracellular histone toxicity | Heparin, anti-histone antibodies | Reduces endothelial damage and thrombosis |
| Heparin and derivatives | Histones, clots | Anticoagulants and neutralize NETs | Heparin, low-molecular-weight heparins | Venous thrombosis, severe COVID-19 |
| Monoclonal antibodies | Specific NET components or receptors (e.g., TLR9) | Block NET activation or effects | Under investigation | Various preclinical models |
| Upstream signaling inhibitors | Cytokines and receptors (IL-8, CXCR2, TLR) | Reduce NETosis induction | Experimental molecules | New therapeutic perspectives |
| oXiris® hemofilter, (Baxter, Pune, Maharashtra) | Inflammatory mediators, endotoxin, uremic toxins | Adsorbs cytokines and endotoxins; removes fluids and toxins | oXiris® filter (AN69 membrane) (Baxter, Pune, Maharashtra) | Sepsis, septic shock with renal failure; preliminary positive results, further RCTs needed |
| Cytosorb® (CytoSorbents Corporation, Princeton, NJ, USA) | Cytokines, inflammatory mediators | Adsorbs inflammatory mediators and toxins using polymer resin | Cytosorb® (CytoSorbents Corporation, Princeton, NJ, USA) | Sepsis, systemic inflammation; used as extracorporeal supportive therapy |
| Coupled plasma filtration adsorption (CPFA) | Plasma and inflammatory mediators | Plasma separated and purified by resin adsorption, then reinfused | CPFA | Sepsis; limited evidence, negative RCT results, no longer recommended |
| Albumin dialysis MARS® (Vantive Health LLC, Deerfield, IL, USA), Prometheus® (Fresenius Medical Care, Bad Homburg, Germany), ADVOS® (ADVITOS GmbH, Munich, Germany) | Albumin-bound toxins and inflammatory mediators | Removes albumin-bound toxins using albumin-containing dialysate | MARS® (Vantive Health LLC, Deerfield, IL, USA), Prometheus® (Fresenius Medical Care, Bad Homburg, Germany), ADVOS® (ADVITOS GmbH, Munich, Germany) | Extracorporeal liver support; high cost, complex; role in sepsis unclear |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignataro, G.; Gemma, S.; Petrucci, M.; Barone, F.; Piccioni, A.; Franceschi, F.; Candelli, M. Unraveling NETs in Sepsis: From Cellular Mechanisms to Clinical Relevance. Int. J. Mol. Sci. 2025, 26, 7464. https://doi.org/10.3390/ijms26157464
Pignataro G, Gemma S, Petrucci M, Barone F, Piccioni A, Franceschi F, Candelli M. Unraveling NETs in Sepsis: From Cellular Mechanisms to Clinical Relevance. International Journal of Molecular Sciences. 2025; 26(15):7464. https://doi.org/10.3390/ijms26157464
Chicago/Turabian StylePignataro, Giulia, Stefania Gemma, Martina Petrucci, Fabiana Barone, Andrea Piccioni, Francesco Franceschi, and Marcello Candelli. 2025. "Unraveling NETs in Sepsis: From Cellular Mechanisms to Clinical Relevance" International Journal of Molecular Sciences 26, no. 15: 7464. https://doi.org/10.3390/ijms26157464
APA StylePignataro, G., Gemma, S., Petrucci, M., Barone, F., Piccioni, A., Franceschi, F., & Candelli, M. (2025). Unraveling NETs in Sepsis: From Cellular Mechanisms to Clinical Relevance. International Journal of Molecular Sciences, 26(15), 7464. https://doi.org/10.3390/ijms26157464