
Assessing Determinants of Response to PARP Inhibition in Germline ATM Mutant Melanoma

 , , , , , ,
, , , , , ,  ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Melanoma ATM-Deficient Cell Lines Are HR Deficient
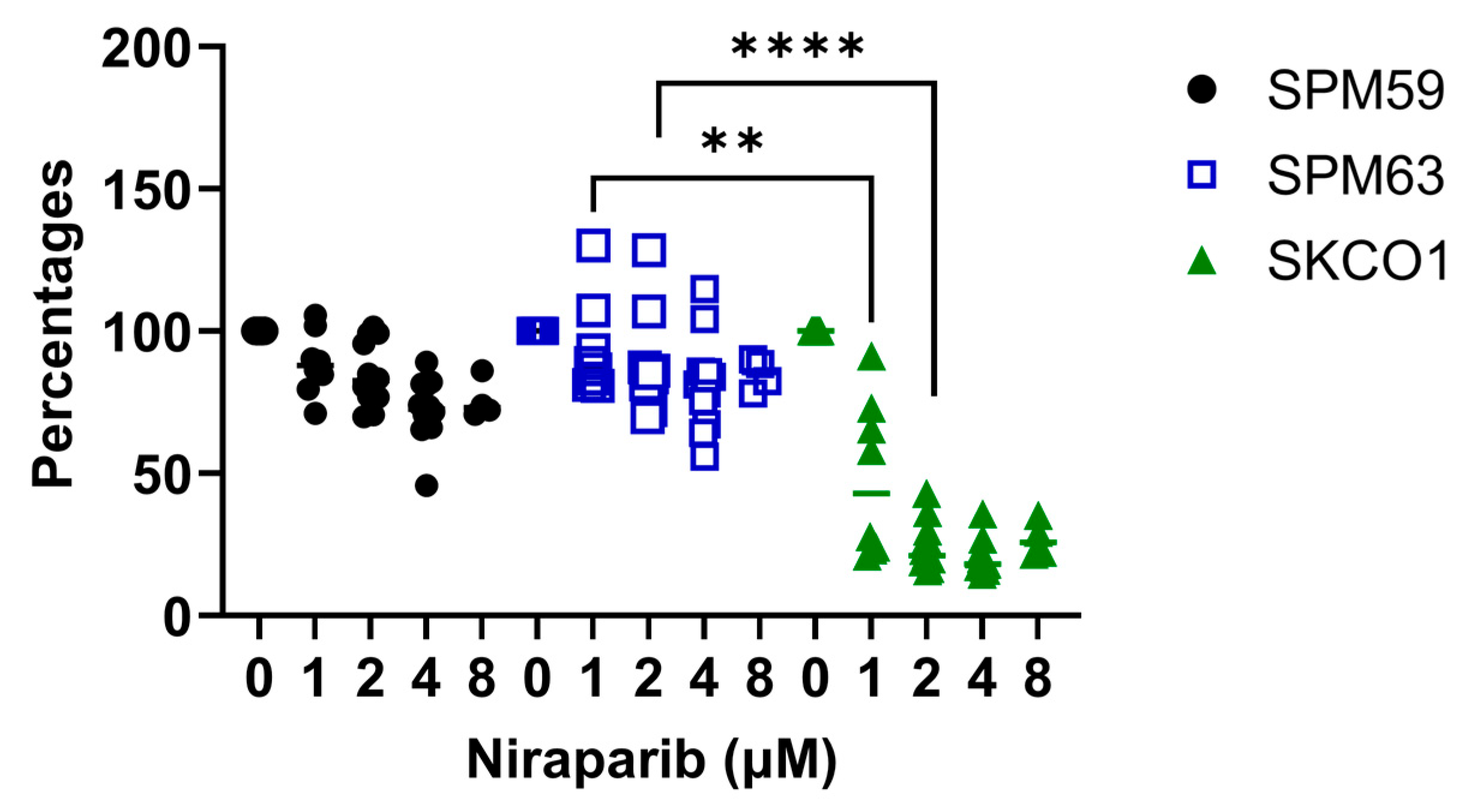
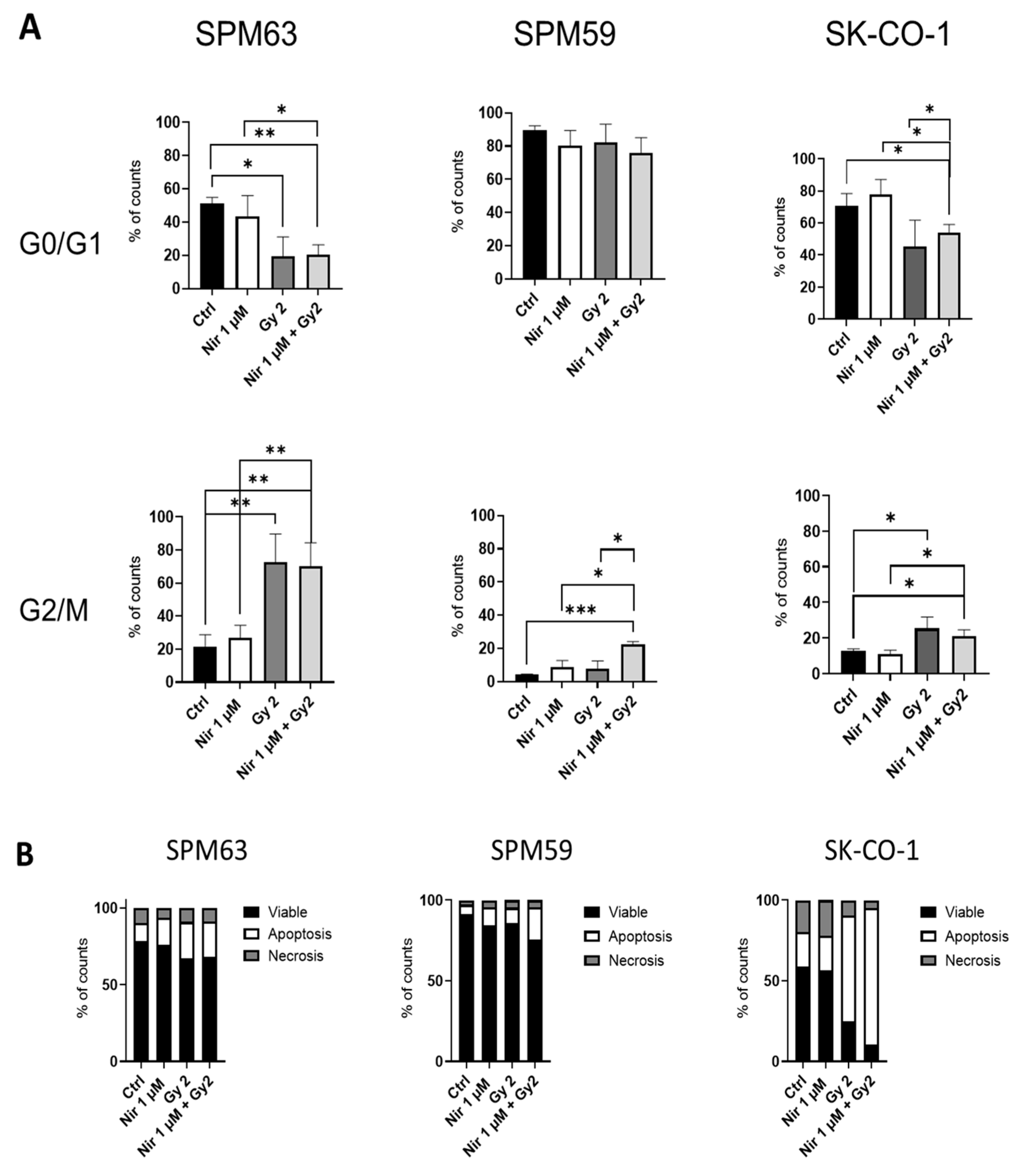
2.2. ATM-Deficient Melanoma Cells Do Not Respond to PARPi Despite HRD
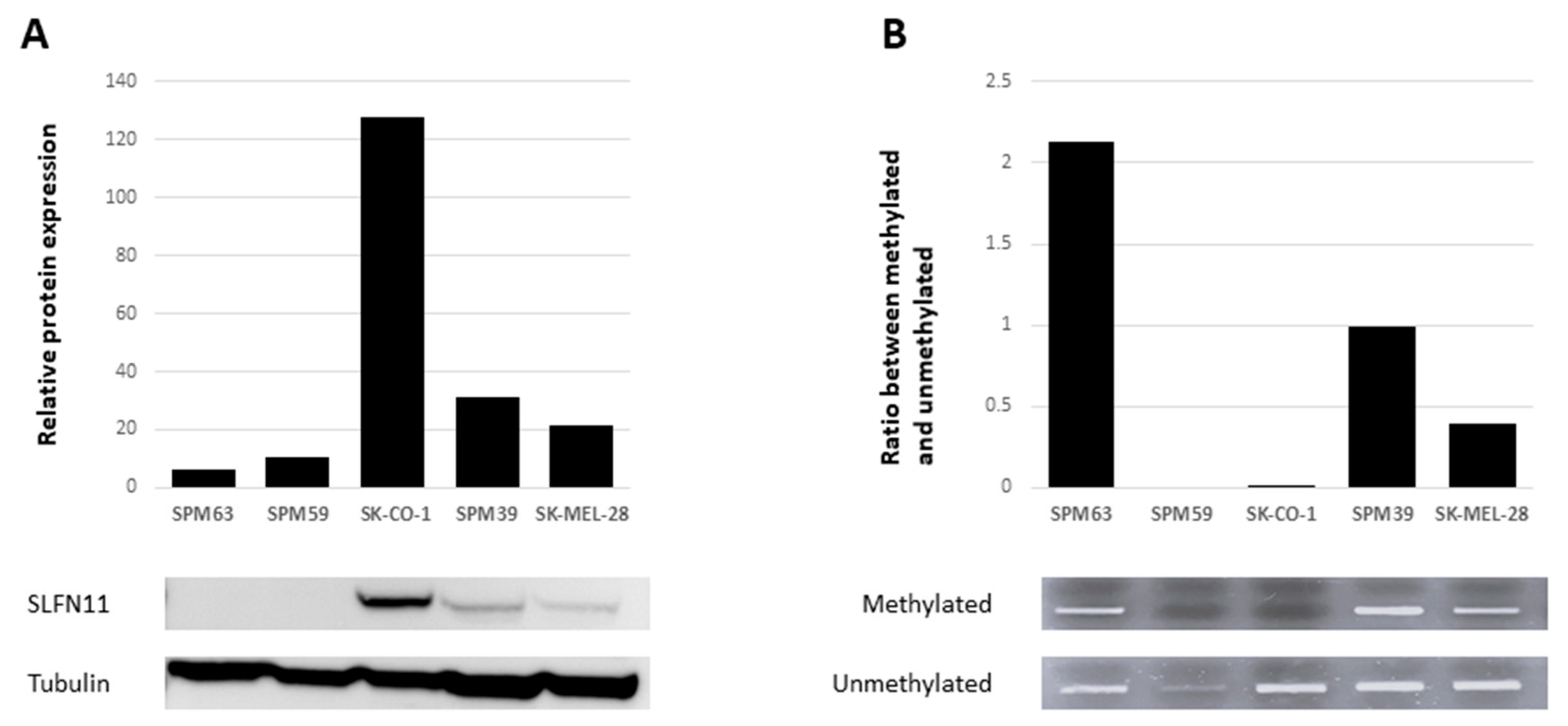
2.3. SLFN11 Inactivation Correlates with Resistance to PARPi
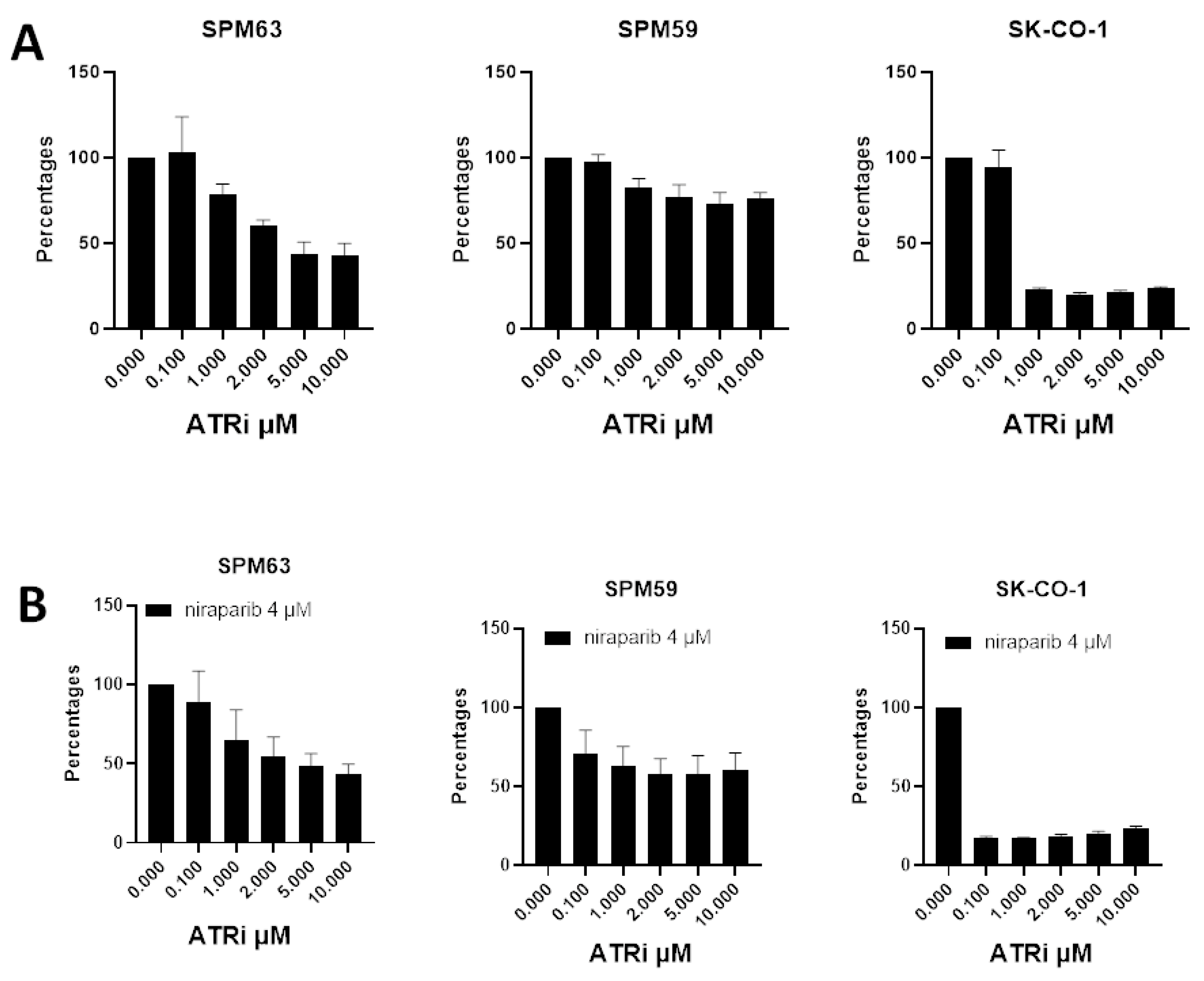
2.4. ATM-Deficient Melanoma Cells Show Sensitivity to ATRi
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Immunostaining of RAD51 Foci
4.3. Proliferation Assay
4.4. Apoptosis and Cell Cycle Analysis
4.5. Western Blot
4.6. DNA Extraction and Methylation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AT | Ataxia–telangiectasia |
| ATM | Ataxia–telangiectasia-mutated |
| ATRi | ATR inhibitor |
| DDR | DNA damage response |
| DSB | DNA double-strand breaks |
| FFPE | Formalin-fixed paraffin-embedded |
| GWAS | Genome-wide association study |
| HR | Homologous recombination |
| HRD | Homologous recombination deficiency |
| HRR | Homologous recombination repair |
| LOF | Loss-of-function |
| LOH | Loss of heterozygosity |
| MSP | Methylation-specific PCR |
| PV | Pathogenic variants |
| PARPi | Poly ADP-Ribose Polymerase inhibitors |
| SLFN11 | Schlafen family member 11 |
References
- Amaral, T.; Ottaviano, M.; Arance, A.; Blank, C.; Chiarion-Sileni, V.; Donia, M.; Dummer, R.; Garbe, C.; Gershenwald, J.E.; Gogas, H.; et al. Cutaneous Melanoma: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up☆. Ann. Oncol. 2025, 36, 10–30. [Google Scholar] [CrossRef]
- Marranci, A.; Maresca, L.; Lodovichi, S.; Luserna di Rorà, A.G.; Stecca, B.; Poliseno, L. PARP1 in Melanoma: Mechanistic Insights and Implications for Basic and Clinical Research. Cancer Lett. 2025, 617, 217599. [Google Scholar] [CrossRef]
- Chan, W.Y.; Brown, L.J.; Reid, L.; Joshua, A.M. PARP Inhibitors in Melanoma—An Expanding Therapeutic Option? Cancers 2021, 13, 4520. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Desprez, P.-Y.; de Semir, D.; Woo, R.W.L.; Sharma, A.; Jones, R.; Caressi, C.; Nosrati, M.; Janiczek, E.; Rivera Penafiel, J.; et al. Phase II Study of Niraparib in Patients With Advanced Melanoma With Homologous Recombination Pathway Gene Mutations. JCO Precis. Oncol. 2025, 9, e2400658. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533, Erratum in N. Eng. J. Med. 2017, 377, 1700. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Kim, K.B.; Soroceanu, L.; de Semir, D.; Millis, S.Z.; Ross, J.; Vosoughi, E.; Dar, A.A.; Nosrati, M.; Desprez, P.Y.; Ice, R.; et al. Prevalence of Homologous Recombination Pathway Gene Mutations in Melanoma: Rationale for a New Targeted Therapeutic Approach. J. Investig. Dermatol. 2021, 141, 2028–2036.e2. [Google Scholar] [CrossRef]
- Fröhlich, L.M.; Niessner, H.; Sauer, B.; Kämereit, S.; Chatziioannou, E.; Riel, S.; Sinnberg, T.; Schittek, B. PARP Inhibitors Effectively Reduce MAPK Inhibitor Resistant Melanoma Cell Growth and Synergize with MAPK Inhibitors through a Synthetic Lethal Interaction In Vitro and In Vivo. Cancer Res. Commun. 2023, 3, 1743–1755. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef]
- Yajima, H.; Lee, K.J.; Zhang, S.; Kobayashi, J.; Chen, B.P.C. DNA Double Strand Break Formation upon UV-Induced Replication Stress Activates ATM and DNA-PKcs Kinases. J. Mol. Biol. 2009, 385, 800. [Google Scholar] [CrossRef]
- Putti, S.; Giovinazzo, A.; Merolle, M.; Falchetti, M.L.; Pellegrini, M. ATM Kinase Dead: From Ataxia Telangiectasia Syndrome to Cancer. Cancers 2021, 13, 5498. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, B.; Pastorino, L.; Nathan, V.; Shah, N.N.; Palmer, J.M.; Howlie, M.; Johansson, P.A.; Freedman, N.D.; Carter, B.D.; Beane-Freeman, L.; et al. Germline ATM Variants Predispose to Melanoma: A Joint Analysis across the GenoMEL and MelaNostrum Consortia. Genet. Med. 2021, 23, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.H.; Iles, M.M.; Harland, M.; Taylor, J.C.; Aitken, J.F.; Andresen, P.A.; Akslen, L.A.; Armstrong, B.K.; Avril, M.F.; Azizi, E.; et al. Genome-Wide Association Study Identifies Three New Melanoma Susceptibility Loci. Nat. Genet. 2011, 43, 1108. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Bishop, D.T.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-Wide Association Meta-Analyses Combining Multiple Risk Phenotypes Provide Insights into the Genetic Architecture of Cutaneous Melanoma Susceptibility. Nat. Genet. 2020, 52, 494–504. [Google Scholar] [CrossRef]
- Pastorino, L.; Dalmasso, B.; Allavena, E.; Vanni, I.; Ugolini, F.; Baroni, G.; Croce, M.; Guadagno, A.; Cabiddu, F.; Andreotti, V.; et al. Ataxia-Telangiectasia Mutated Loss of Heterozygosity in Melanoma. Int. J. Mol. Sci. 2022, 23, 16027. [Google Scholar] [CrossRef]
- Smith, J.; Mun Tho, L.; Xu, N.; AGillespie, D. The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
- Raynaud, C.M.; Ahmed, E.I.; Jabeen, A.; Sanchez, A.; Sherif, S.; Carneiro-Lobo, T.C.; Awad, A.; Awartani, D.; Naik, A.; Thomas, R.; et al. Modulation of SLFN11 Induces Changes in DNA Damage Response in Breast Cancer. Cancer Cell Int. 2023, 23, 291. [Google Scholar] [CrossRef]
- Krushkal, J.; Silvers, T.; Reinhold, W.C.; Sonkin, D.; Vural, S.; Connelly, J.; Varma, S.; Meltzer, P.S.; Kunkel, M.; Rapisarda, A.; et al. Epigenome-Wide DNA Methylation Analysis of Small Cell Lung Cancer Cell Lines Suggests Potential Chemotherapy Targets. Clin. Epigenet. 2020, 12, 93. [Google Scholar] [CrossRef]
- Zhou, A.; Butt, O.; Ansstas, M.; Mauer, E.; Khaddour, K.; Ansstas, G. Determining PARP Inhibition as a Treatment Strategy in Melanoma Based on Homologous Recombination Deficiency-Related Loss of Heterozygosity. J. Natl. Compr. Cancer Netw. 2023, 21, 688–693. [Google Scholar] [CrossRef]
- Jonuscheit, S.; Jost, T.; Gajdošová, F.; Wrobel, M.; Hecht, M.; Fietkau, R.; Distel, L. Parp Inhibitors Talazoparib and Niraparib Sensitize Melanoma Cells to Ionizing Radiation. Genes 2021, 12, 849. [Google Scholar] [CrossRef]
- Zoppoli, G.; Regairaz, M.; Leo, E.; Reinhold, W.C.; Varma, S.; Ballestrero, A.; Doroshow, J.H.; Pommier, Y. Putative DNA/RNA Helicase Schlafen-11 (SLFN11) Sensitizes Cancer Cells to DNA-Damaging Agents. Proc. Natl. Acad. Sci. USA 2012, 109, 15030–15035. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR Inhibition Potentiates Genome Instability and Cell Death in ATM-Deficient Cancer Cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Stewart, C.A.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic Variations in Epithelial-to-Mesenchymal Transition (EMT), ATM, and SLFN11 Govern Response to PARP Inhibitors and Cisplatin in Small Cell Lung Cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y.; Murai, J.; Feng, Y.; Yu, G.K.; et al. Resistance to PARP Inhibitors by SLFN11 Inactivation Can Be Overcome by ATR Inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Pastorino, L.; Tanda, E.T.; Andreotti, V.; Dalmasso, B.; Solari, N.; Mascherini, M.; Cabiddu, F.; Guadagno, A.; Coco, S.; et al. Whole-Exome Sequencing and CfDNA Analysis Uncover Genetic Determinants of Melanoma Therapy Response in a Real-World Setting. Int. J. Mol. Sci. 2023, 24, 4302. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 Assay Feasible in Routine Tumor Samples Calls PARP Inhibitor Response beyond BRCA Mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, B.; Herencia-Roper, A.; Llop-Guevar, A.; Pedretti, F.; Moles-Fernández, A.; Viaplana, C.; Villacampa, G.; Guzmán, M.; Rodríguez, O.; Grueso, J.; et al. Preclinical In Vivo Validation of the RAD51 Test for Identification of Homologous Recombination-Deficient Tumors and Patient Stratification. Cancer Res. 2022, 82, 1646–1657. [Google Scholar] [CrossRef]
- Choi, S.J.; Choi, Y.I.; Kim, L.; Park, I.S.; Han, J.Y.; Kim, J.M.; Chu, Y.C. Preparation of Compact Agarose Cell Blocks from the Residues of Liquid-Based Cytology Samples. Korean J. Pathol. 2014, 48, 351–360. [Google Scholar] [CrossRef]
- Ballabeni, A.; Zamponi, R.; Moore, J.K.; Helin, K.; Kirschner, M.W. Geminin Deploys Multiple Mechanisms to Regulate Cdt1 before Cell Division Thus Ensuring the Proper Execution of DNA Replication. Proc. Natl. Acad. Sci. USA 2013, 110, E2848–E2853. [Google Scholar] [CrossRef] [PubMed]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Dey, P.; De, D.; Ghosh, U. Gamma Ray-Induced in Vitro Cell Migration via EGFR/ERK/Akt/P38 Activation Is Prevented by Olaparib Pretreatment. Int. J. Radiat. Biol. 2020, 96, 651–660. [Google Scholar] [CrossRef]
- Bridges, K.A.; Toniatti, C.; Buser, C.A.; Liu, H.; Buchholz, T.A.; Meyn, R.E.; Bridges, K.A.; Toniatti, C.; Buser, C.A.; Liu, H.; et al. Niraparib (MK-4827), a Novel Poly(ADP-Ribose) Polymerase Inhibitor, Radiosensitizes Human Lung and Breast Cancer Cells. Oncotarget 2014, 5, 5076–5086. [Google Scholar] [CrossRef]
- Amaro, A.A.; Gangemi, R.; Emionite, L.; Castagnola, P.; Filaci, G.; Jager, M.J.; Tanda, E.T.; Spagnolo, F.; Mascherini, M.; Pfeffer, U.; et al. Cerivastatin Synergizes with Trametinib and Enhances Its Efficacy in the Therapy of Uveal Melanoma. Cancers 2023, 15, 886. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, M.Y.; Gao, A.A.; Zhu, C.; He, T.; Herman, J.G.; Guo, M.Z. Epigenetic Silencing Schlafen-11 Sensitizes Esophageal Cancer to ATM Inhibitor. World J. Gastrointest. Oncol. 2024, 16, 2060–2073. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allavena, E.; Croce, M.; Dalmasso, B.; Profumo, C.; Rigo, V.; Andreotti, V.; Vanni, I.; Pellegrino, B.; Musolino, A.; Campanini, N.; et al. Assessing Determinants of Response to PARP Inhibition in Germline ATM Mutant Melanoma. Int. J. Mol. Sci. 2025, 26, 7420. https://doi.org/10.3390/ijms26157420
Allavena E, Croce M, Dalmasso B, Profumo C, Rigo V, Andreotti V, Vanni I, Pellegrino B, Musolino A, Campanini N, et al. Assessing Determinants of Response to PARP Inhibition in Germline ATM Mutant Melanoma. International Journal of Molecular Sciences. 2025; 26(15):7420. https://doi.org/10.3390/ijms26157420
Chicago/Turabian StyleAllavena, Eleonora, Michela Croce, Bruna Dalmasso, Cecilia Profumo, Valentina Rigo, Virginia Andreotti, Irene Vanni, Benedetta Pellegrino, Antonino Musolino, Nicoletta Campanini, and et al. 2025. "Assessing Determinants of Response to PARP Inhibition in Germline ATM Mutant Melanoma" International Journal of Molecular Sciences 26, no. 15: 7420. https://doi.org/10.3390/ijms26157420
APA StyleAllavena, E., Croce, M., Dalmasso, B., Profumo, C., Rigo, V., Andreotti, V., Vanni, I., Pellegrino, B., Musolino, A., Campanini, N., Bruno, W., Mastracci, L., Zoppoli, G., Tanda, E. T., Spagnolo, F., Ghiorzo, P., & Pastorino, L. (2025). Assessing Determinants of Response to PARP Inhibition in Germline ATM Mutant Melanoma. International Journal of Molecular Sciences, 26(15), 7420. https://doi.org/10.3390/ijms26157420