
Improving ALS Molecular Diagnosis Through Functional Assays: Reassessment of a SOD1 Variant of Uncertain Significance

 , ,
, ,  , ,
, , 


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Case Presentation
2.1. Identification of a Genetic Variant in an ALS Patient
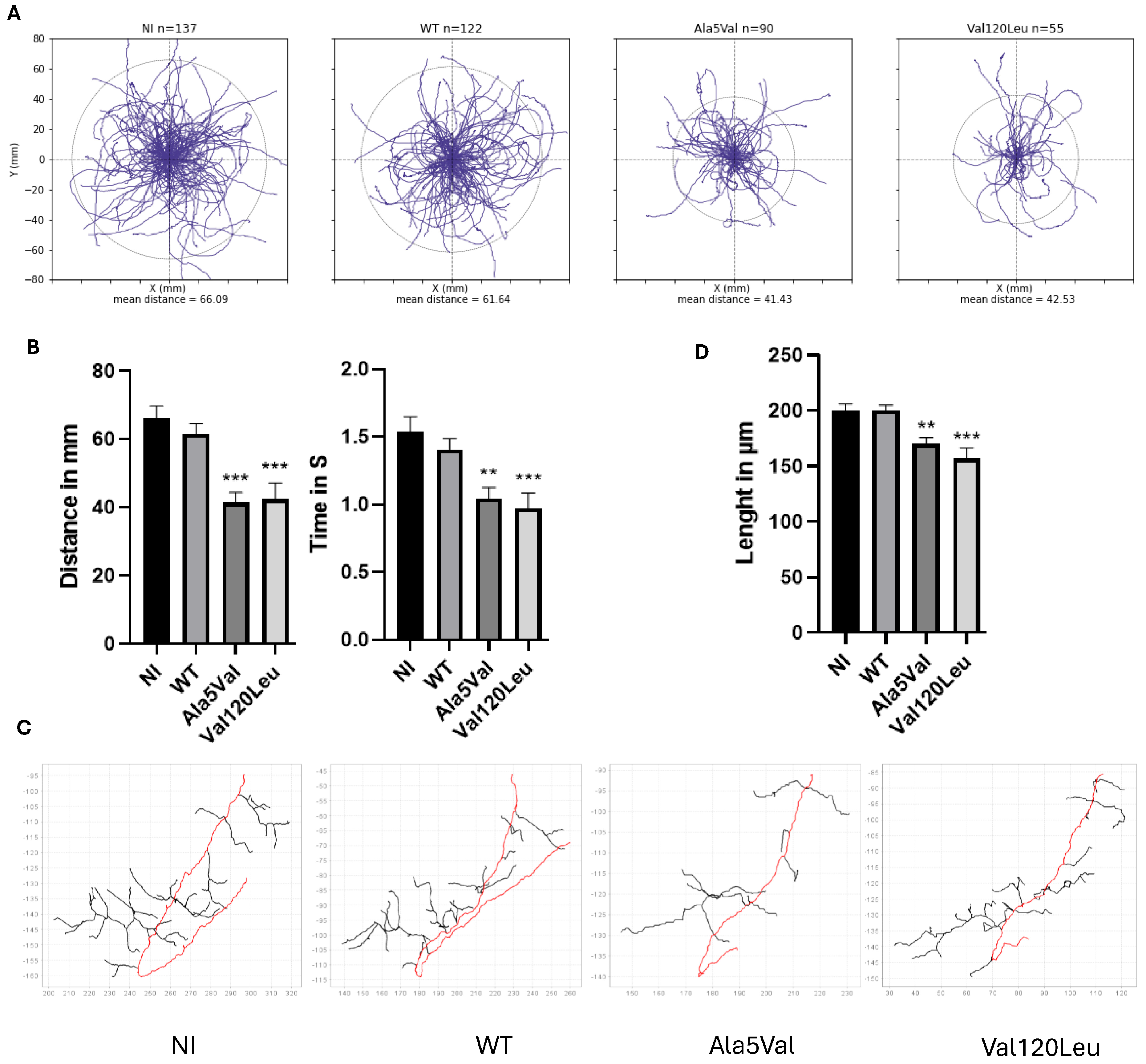
2.2. Functional Analysis of the Genetic Variant
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pottinger, T.D.; Motelow, J.E.; Povysil, G.; Moreno, C.A.M.; Ren, Z.; Phatnani, H.; New York Genome Center ALS Sequencing Consortium; Aitman, T.J.; Santoyo-Lopez, J.; Scottish Genomes Partnership; et al. Rare Variant Analyses Validate Known ALS Genes in a Multi-Ethnic Population and Identifies ANTXR2 as a Candidate in PLS. MedRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Yang, Y.; Guo, Y.; Deng, H. Genetic architecture of amyotrophic lateral sclerosis: A comprehensive review. J. Genet. Genom. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in Silico Algorithms for Use with ACMG/AMP Clinical Variant Interpretation Guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, R.J. The functional genomics laboratory: Functional validation of genetic variants. J. Inherit. Metab. Dis. 2018, 41, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Camu, W.; Brulard, C.; Marouillat, S.; Couratier, P.; Camdessanché, J.-P.; Cintas, P.; Verschueren, A.; Soriani, M.-H.; Desnuelle, C.; et al. Effect of Familial Clustering in the Genetic Screening of 235 French ALS Families. J. Neurol. Neurosurg. Psychiatry 2021, 92, 479–484. [Google Scholar] [CrossRef] [PubMed]
- de Fuenmayor-Fernández de la Hoz, C.P.; Hernández-Laín, A.; Olivé, M.; Arteche López, A.; Esteban, J.; Domínguez-González, C. SOD1 Mutations in Adult-Onset Distal Spinal Muscular Atrophy. Eur. J. Neurol. 2020, 27, e75–e76. [Google Scholar] [CrossRef] [PubMed]
- Nunes Gonçalves, J.P.; Leoni, T.B.; Martins, M.P.; Peluzzo, T.M.; Dourado, M.E.T.; Saute, J.A.M.; Paranhos Miranda Covaleski, A.P.; Bulle de Oliveira, A.S.; Claudino, R.; Marques, W.; et al. Genetic Epidemiology of Familial ALS in Brazil. Neurobiol. Aging 2021, 102, 227.e1–227.e4. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhou, Q.; Chen, Y.; Ou, R.; Cao, B.; Xu, Y.; Yang, J.; Shang, H.-F. Analysis of SOD1 Mutations in a Chinese Population with Amyotrophic Lateral Sclerosis: A Case-Control Study and Literature Review. Sci. Rep. 2017, 7, 44606. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, N.A.S.; Pinho, B.R.; Oliveira, J.M.A. Swimming against ALS: How to Model Disease in Zebrafish for Pathophysiological and Behavioral Studies. Neurosci. Biobehav. Rev. 2023, 148, 105138. [Google Scholar] [CrossRef] [PubMed]
- Roggenbuck, J.; Palettas, M.; Vicini, L.; Patel, R.; Quick, A.; Kolb, S.J. Incidence of pathogenic, likely pathogenic, and uncertain ALS variants in a clinic cohort. Neurol. Genet. 2020, 6, e390. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, M.; Ramírez-Expósito, M.J.; Martínez-Martos, J.M. Pathophysiology, Clinical Heterogeneity, and Therapeutic Advances in Amyotrophic Lateral Sclerosis: A Comprehensive Review of Molecular Mechanisms, Diagnostic Challenges, and Multidisciplinary Management Strategies. Life 2025, 15, 647. [Google Scholar] [CrossRef] [PubMed]
- Opie-Martin, S.; Iacoangeli, A.; Topp, S.D.; Abel, O.; Mayl, K.; Mehta, P.R.; Shatunov, A.; Fogh, I.; Bowles, H.; Limbachiya, N.; et al. The SOD1-mediated ALS phenotype shows a decoupling between age of symptom onset and disease duration. Nat. Commun. 2022, 13, 6901, Erratum in Nat. Commun. 2024, 15, 5560. https://doi.org/10.1038/s41467-024-49938-y. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.-Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic Lateral Ssclerosis and Structural Defects in Cu,Zn Superoxide Dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedja-Iacona, L.; Forget, A.; Boisseau, C.; Marouillat, S.; Chudinova, A.; Veyrat-Durebex, C.; Guissart, C.; Lumbroso, S.; Raoul, C.; Andres, C.R.; et al. Improving ALS Molecular Diagnosis Through Functional Assays: Reassessment of a SOD1 Variant of Uncertain Significance. Int. J. Mol. Sci. 2025, 26, 7414. https://doi.org/10.3390/ijms26157414
Bedja-Iacona L, Forget A, Boisseau C, Marouillat S, Chudinova A, Veyrat-Durebex C, Guissart C, Lumbroso S, Raoul C, Andres CR, et al. Improving ALS Molecular Diagnosis Through Functional Assays: Reassessment of a SOD1 Variant of Uncertain Significance. International Journal of Molecular Sciences. 2025; 26(15):7414. https://doi.org/10.3390/ijms26157414
Chicago/Turabian StyleBedja-Iacona, Léa, Arthur Forget, Chloé Boisseau, Sylviane Marouillat, Aleksandra Chudinova, Charlotte Veyrat-Durebex, Claire Guissart, Serge Lumbroso, Cédric Raoul, Christian R. Andres, and et al. 2025. "Improving ALS Molecular Diagnosis Through Functional Assays: Reassessment of a SOD1 Variant of Uncertain Significance" International Journal of Molecular Sciences 26, no. 15: 7414. https://doi.org/10.3390/ijms26157414
APA StyleBedja-Iacona, L., Forget, A., Boisseau, C., Marouillat, S., Chudinova, A., Veyrat-Durebex, C., Guissart, C., Lumbroso, S., Raoul, C., Andres, C. R., Blasco, H., Couratier, P., Corcia, P., Verschueren, A., Mouzat, K., & Vourc’h, P. (2025). Improving ALS Molecular Diagnosis Through Functional Assays: Reassessment of a SOD1 Variant of Uncertain Significance. International Journal of Molecular Sciences, 26(15), 7414. https://doi.org/10.3390/ijms26157414