
Muscarinic Receptor Antagonism and TRPM3 Activation as Stimulators of Mitochondrial Function and Axonal Repair in Diabetic Sensorimotor Polyneuropathy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Diabetic Sensorimotor Polyneuropathy
3. Epidemiology—Economic and Social Burden of DSPN
4. Modeling DSPN
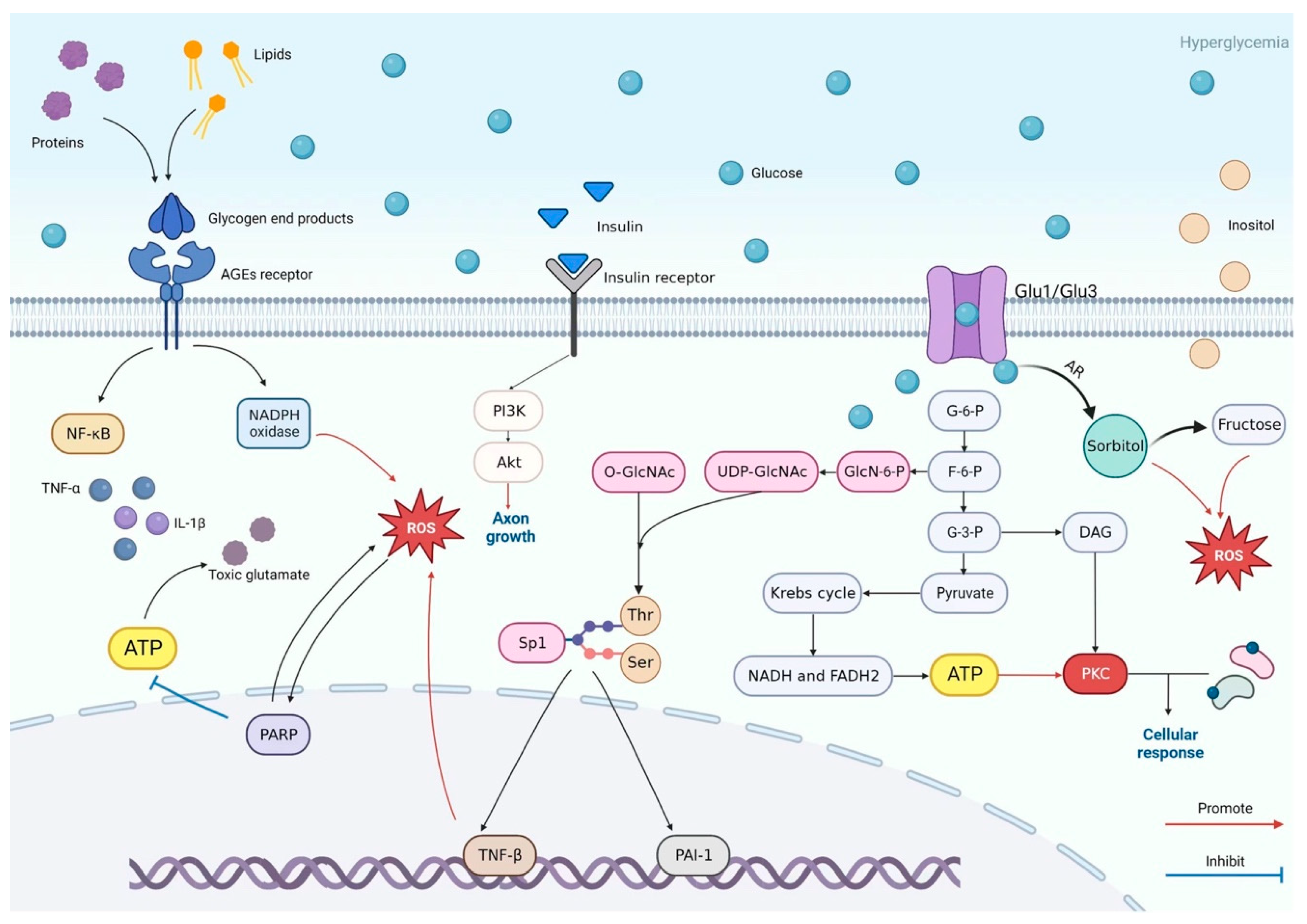
5. Pathogenesis of DSPN: Crosstalk Between Metabolic Pathways
6. Molecular and Cellular Mechanisms Driving DSPN
6.1. Mitochondrial Dysfunction in DSPN Pathology
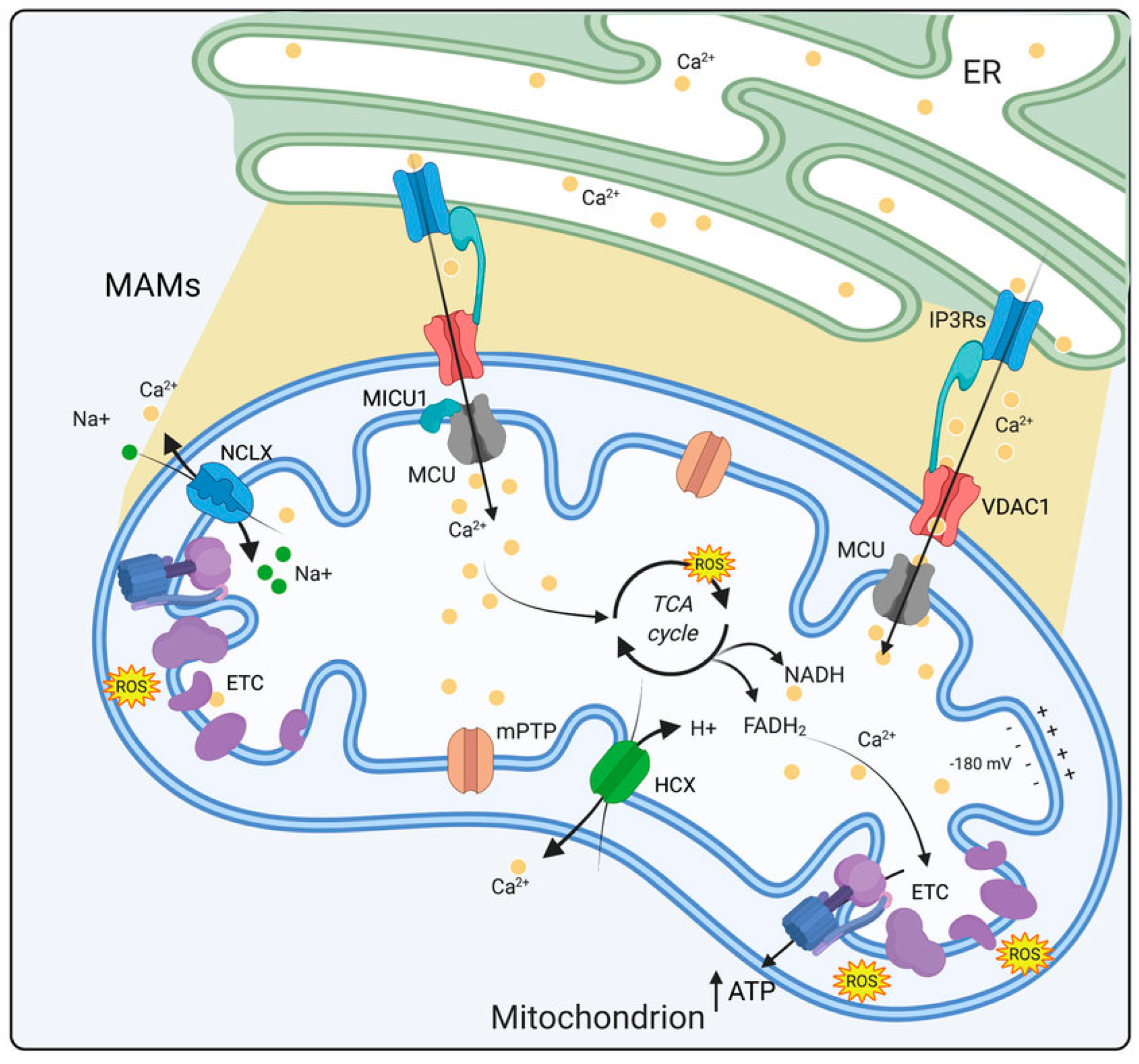
6.2. Ca2+ Imbalance in Progression of DSPN
7. AMP-Activated Protein Kinase (AMPK)
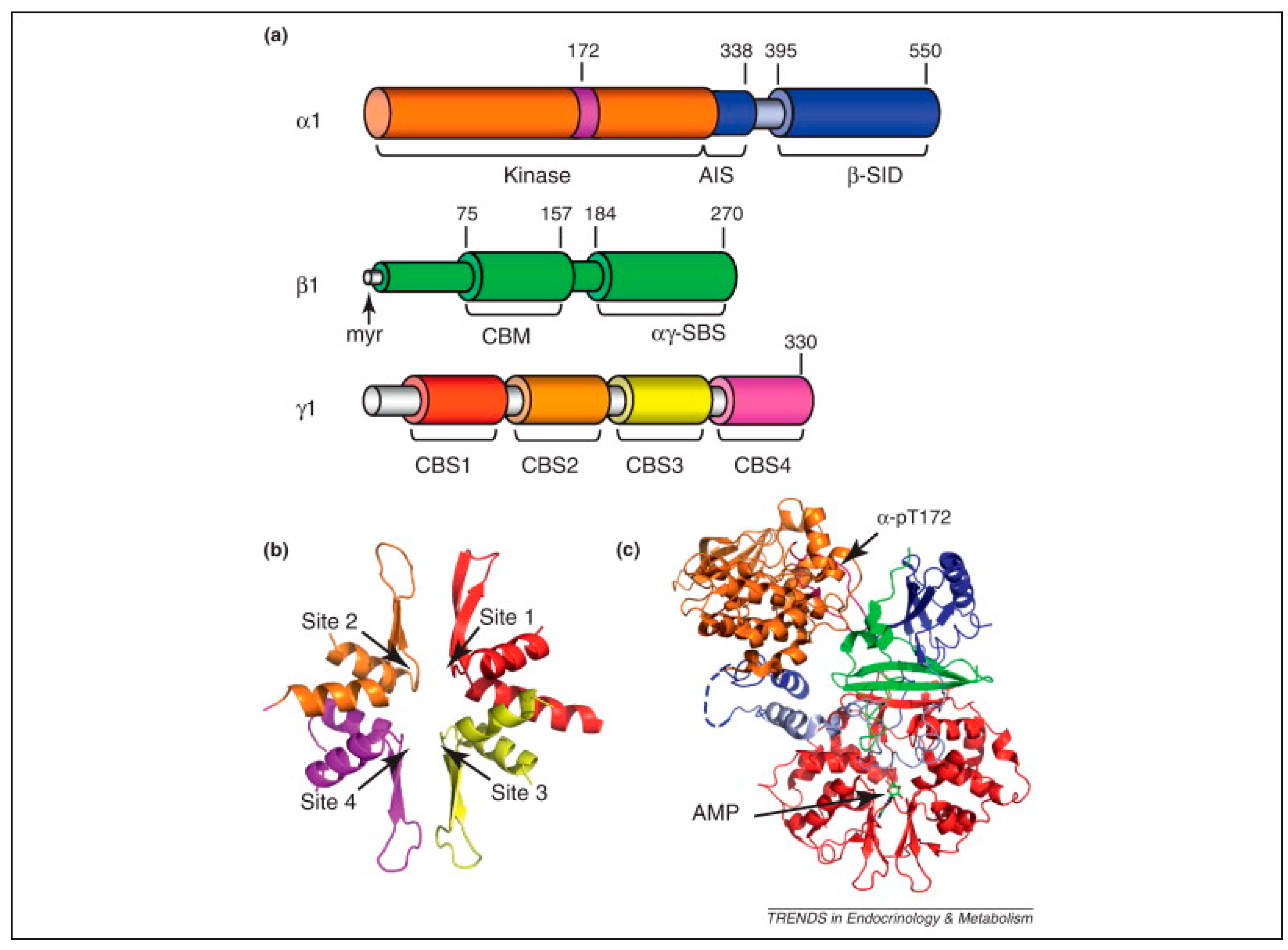
7.1. AMPK Structure and Function
7.2. AMPK Abnormalities in DSPN
8. Role of M1R in Nerve Repair
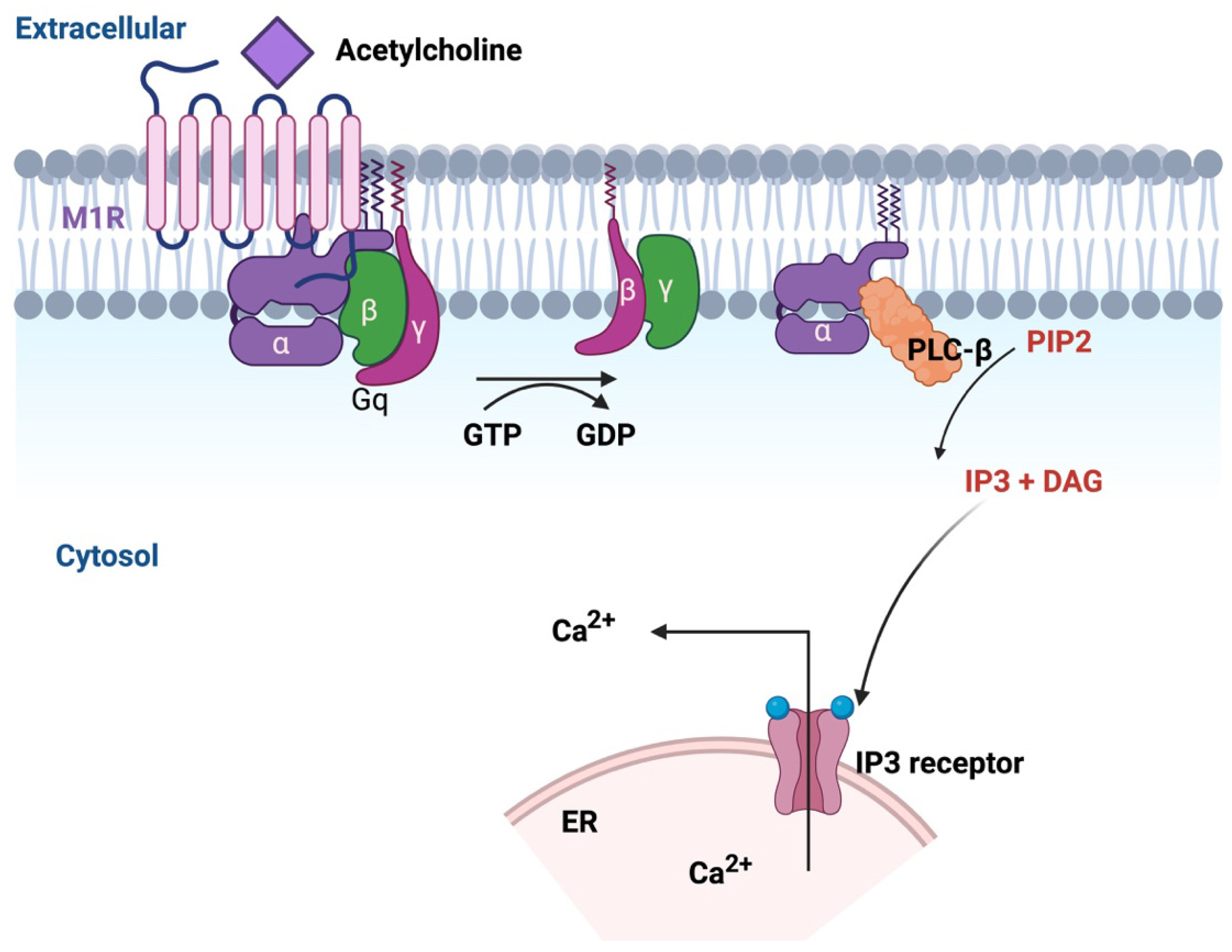
8.1. GPCR Pharmacology
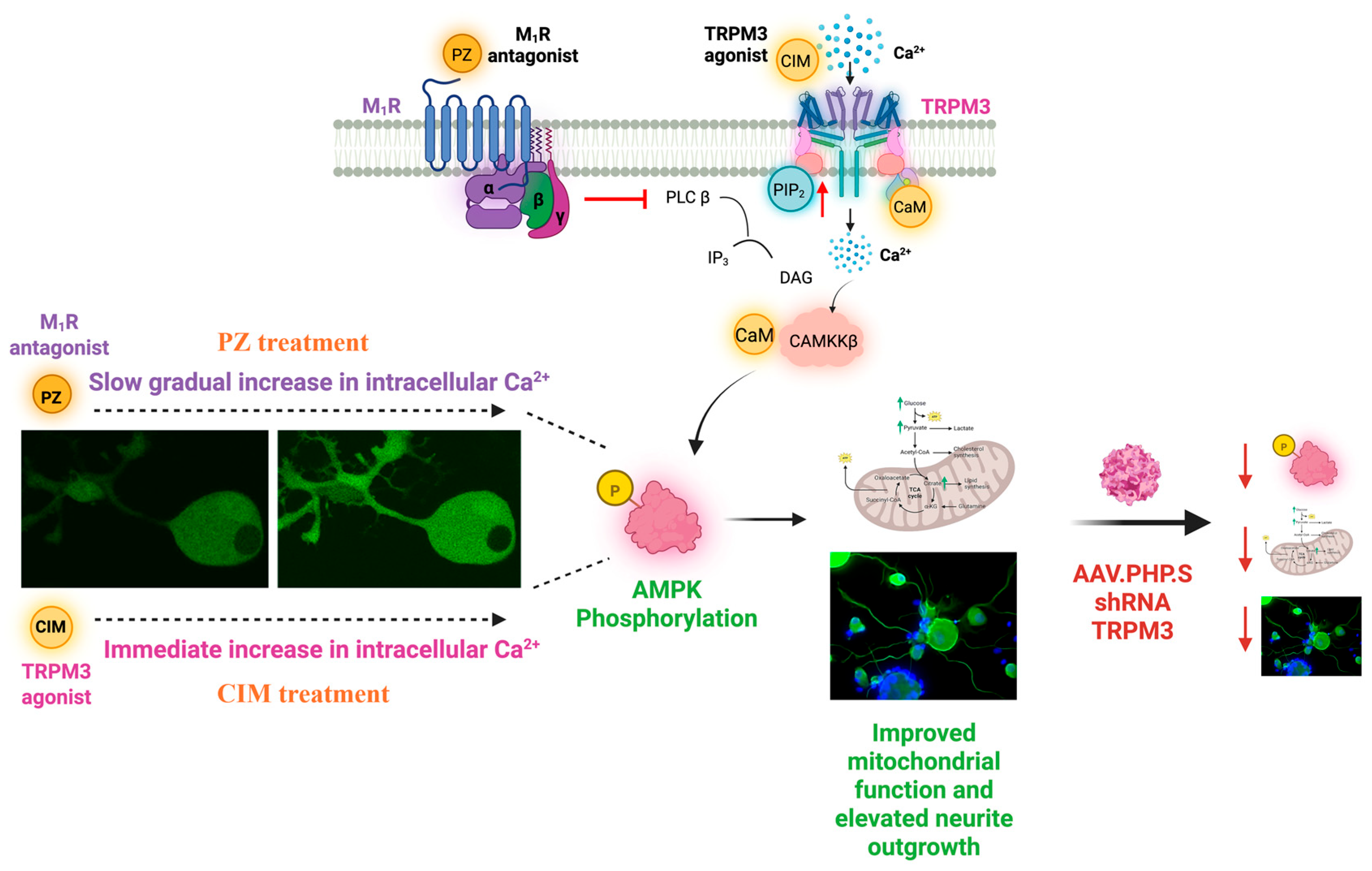
8.2. M1R Signaling for Protection Against Dying-Back Neuropathy
8.3. Therapeutic Implications of M1R Antagonism in Diverse Peripheral Neuropathy Models
9. TRPM3: A Key Player in Sensory Perception
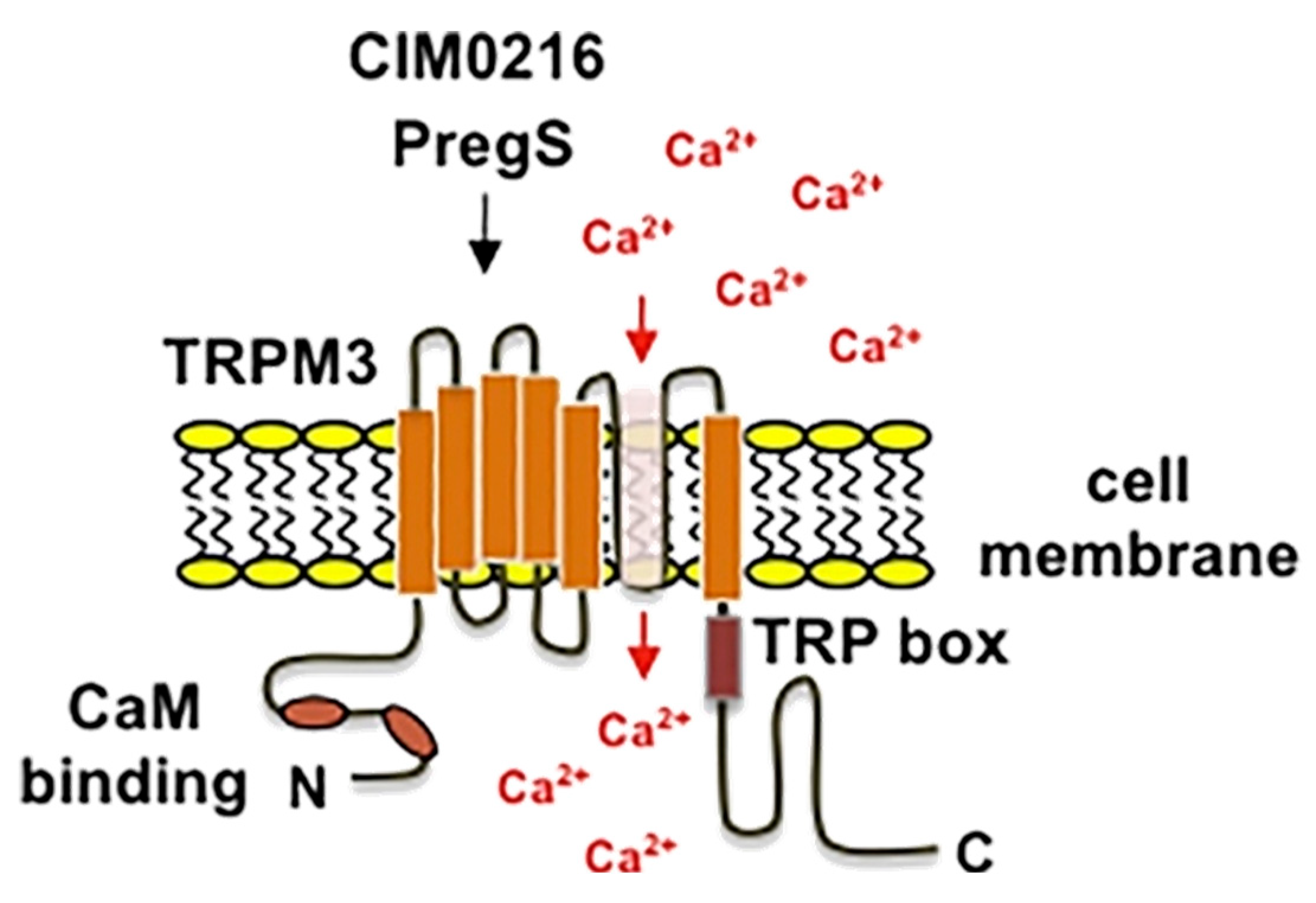
9.1. Structure and Function
9.2. TRPM3 Pharmacology
9.3. GPCR Regulation of TRPM3 Channels
9.4. Effect of TRPM3 Activation
10. The Therapeutic Potential of TRPM3 and M1R-Targeted Therapies
11. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| AGE | Advanced glycation end-product |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| CaMKKβ | Ca2+/calmodulin-dependent protein kinase kinase β |
| CBS | Cystathione β-synthase |
| CCM | Corneal confocal microscopy |
| CIPN | Chemotherapy-induced peripheral neuropathy |
| Clt | Clotrimazole |
| COX-2 | Cyclooxygenase-2 |
| DAG | Diacylglycerol |
| DEE | Developmental and epileptic encephalopathies |
| DM | Diabetes mellitus |
| DRG | Dorsal root ganglion |
| DSPN | Diabetic sensorimotor polyneuropathy |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| GLUT3 | Glucose transporter type 3 |
| GPCR | G protein-coupled receptor |
| HDL | High-density lipoprotein |
| HFD | High-fat diet |
| HIV-DSP | HIV-associated distal sensory polyneuropathy |
| HSP | Heat shock proteins |
| IDF | International Diabetes Federation |
| IDH | Isocitrate dehydrogenase |
| IENF | Intraepidermal nerve fiber |
| IL-6 | Interleukin-6 |
| IP3 | Inositol trisphosphate |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| LDL | Low-density lipoprotein |
| LKB1 | Liver kinase B1 |
| M1R | Muscarinic acetylcholine type 1 receptor |
| MAM | Mitochondria-associated membrane |
| MAPK | Mitogen-activated protein kinase |
| MCU | Mitochondrial Ca2+ uniporter |
| MDA | Malondialdehyde |
| MT7 | Muscarinic toxin 7 |
| mtTFA | Mitochondrial transcription factor A |
| NADPH | Nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NMDA | N-methyl-D-aspartate |
| NRF | Nuclear respiratory factor |
| pChAT | Peripheral form of choline acetyltransferase |
| PDH | Pyruvate dehydrogenase |
| PGC-1α | Peroxisome proliferator-activated receptor-γ coactivator-1α |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PS | Pregnenolone sulfate |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| SERCA | Sarco/endoplasmic reticulum Ca2+ ATPase |
| STZ | Streptozotocin |
| TCA | Tricarboxylic acid |
| TNF-α | Tumor necrosis factor-α |
| TNF-β | Tumor necrosis factor-β |
| TRP | Transient receptor potential |
| TRPM3 | Transient receptor potential melastatin-3 |
| UCP | Uncoupling protein |
| VDAC | Voltage-dependent anion channel |
| VSD | Voltage-sensing domain |
| β-SID | β-subunit interacting domain |
References
- Eid, S.A.; Rumora, A.E.; Beirowski, B.; Bennett, D.L.; Hur, J.; Savelieff, M.G.; Feldman, E.L. New perspectives in diabetic neuropathy. Neuron 2023, 111, 2623–2641. [Google Scholar] [CrossRef] [PubMed]
- Sloan, G.; Selvarajah, D.; Tesfaye, S. Pathogenesis, diagnosis and clinical management of diabetic sensorimotor peripheral neuropathy. Nat. Rev. Endocrinol. 2021, 17, 400–420. [Google Scholar] [CrossRef] [PubMed]
- Rumora, A.E.; Kim, B.; Feldman, E.L. A Role for Fatty Acids in Peripheral Neuropathy Associated with Type 2 Diabetes and Prediabetes. Antioxid. Redox Signal. 2022, 37, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Zochodne, D.W. Sensory Neurodegeneration in Diabetes: Beyond Glucotoxicity. Int. Rev. Neurobiol. 2016, 127, 151–180. [Google Scholar] [CrossRef]
- Miyashita, A.; Kobayashi, M.; Yokota, T.; Zochodne, D.W. Diabetic Polyneuropathy: New Strategies to Target Sensory Neurons in Dorsal Root Ganglia. Int. J. Mol. Sci. 2023, 24, 5977. [Google Scholar] [CrossRef]
- Ma, J.; Pan, P.; Anyika, M.; Blagg, B.S.; Dobrowsky, R.T. Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons. ACS Chem. Neurosci. 2015, 6, 1637–1648. [Google Scholar] [CrossRef]
- Smith, S.; Normahani, P.; Lane, T.; Hohenschurz-Schmidt, D.; Oliver, N.; Davies, A.H. Pathogenesis of Distal Symmetrical Polyneuropathy in Diabetes. Life 2022, 12, 1074. [Google Scholar] [CrossRef]
- McMorrow, R.; Nube, V.L.; Manski-Nankervis, J.A. Preventing diabetes-related foot ulcers through early detection of peripheral neuropathy. Aust. J. Gen. Pract. 2022, 51, 833–838. [Google Scholar] [CrossRef]
- Steinmetz, J.D.; Seeher, K.M.; Schiess, N.; Nichols, E.; Cao, B.; Servili, C.; Cavallera, V.; Cousin, E.; Hagins, H.; Moberg, M.E.; et al. Global, regional, and national burden of disorders affecting the nervous system, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024, 23, 344–381. [Google Scholar] [CrossRef]
- Ziegler, D.; Tesfaye, S.; Spallone, V.; Gurieva, I.; Al Kaabi, J.; Mankovsky, B.; Martinka, E.; Radulian, G.; Nguyen, K.T.; Stirban, A.O.; et al. Screening, diagnosis and management of diabetic sensorimotor polyneuropathy in clinical practice: International expert consensus recommendations. Diabetes Res. Clin. Pract. 2022, 186, 109063. [Google Scholar] [CrossRef]
- Staehelin Jensen, T. The pathogenesis of painful diabetic neuropathy and clinical presentation. Diabetes Res. Clin. Pract. 2023, 206 (Suppl. 1), 110753. [Google Scholar] [CrossRef]
- Zaino, B.; Goel, R.; Devaragudi, S.; Prakash, A.; Vaghamashi, Y.; Sethi, Y.; Patel, N.; Kaka, N. Diabetic neuropathy: Pathogenesis and evolving principles of management. Dis. Mon. 2023, 69, 101582. [Google Scholar] [CrossRef]
- Vinik, A.I.; Maser, R.E.; Mitchell, B.D.; Freeman, R. Diabetic autonomic neuropathy. Diabetes Care 2003, 26, 1553–1579. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Elafros, M.A.; Viswanathan, V.; Jensen, T.S.; Bennett, D.L.; Feldman, E.L. The global and regional burden of diabetic peripheral neuropathy. Nat. Rev. Neurol. 2025, 21, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.J.; Boyko, E.J.; IDF Diabetes Atlas 10th Edition Scientific Committee. IDF Diabetes Atlas; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- Gordois, A.; Scuffham, P.; Shearer, A.; Oglesby, A.; Tobian, J.A. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003, 26, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, E.P. Estimating the annual cost burden of diabetic peripheral neuropathy in the United States. Endocrine 2025, 1–6. [Google Scholar] [CrossRef]
- Beiswenger, K.K.; Calcutt, N.A.; Mizisin, A.P. Dissociation of thermal hypoalgesia and epidermal denervation in streptozotocin-diabetic mice. Neurosci. Lett. 2008, 442, 267–272. [Google Scholar] [CrossRef]
- Christianson, J.A.; Ryals, J.M.; Johnson, M.S.; Dobrowsky, R.T.; Wright, D.E. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience 2007, 145, 303–313. [Google Scholar] [CrossRef]
- Mizisin, A.P.; Nelson, R.W.; Sturges, B.K.; Vernau, K.M.; Lecouteur, R.A.; Williams, D.C.; Burgers, M.L.; Shelton, G.D. Comparable myelinated nerve pathology in feline and human diabetes mellitus. Acta Neuropathol. 2007, 113, 431–442. [Google Scholar] [CrossRef]
- Mizisin, A.P.; Shelton, G.D.; Wagner, S.; Rusbridge, C.; Powell, H.C. Myelin splitting, Schwann cell injury and demyelination in feline diabetic neuropathy. Acta Neuropathol. 1998, 95, 171–174. [Google Scholar] [CrossRef]
- Yorek, M.S.; Coppey, L.J.; Shevalye, H.; Obrosov, A.; Kardon, R.H.; Yorek, M.A. Effect of Treatment with Salsalate, Menhaden Oil, Combination of Salsalate and Menhaden Oil, or Resolvin D1 of C57Bl/6J Type 1 Diabetic Mouse on Neuropathic Endpoints. J. Nutr. Metab. 2016, 2016, 5905891. [Google Scholar] [CrossRef] [PubMed]
- Elafros, M.A.; Andersen, H.; Bennett, D.L.; Savelieff, M.G.; Viswanathan, V.; Callaghan, B.C.; Feldman, E.L. Towards prevention of diabetic peripheral neuropathy: Clinical presentation, pathogenesis, and new treatments. Lancet Neurol. 2022, 21, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.A.; Jolivalt, C.G.; Goor, J.; Mizisin, A.P.; Calcutt, N.A. Hypertension-induced peripheral neuropathy and the combined effects of hypertension and diabetes on nerve structure and function in rats. Acta Neuropathol. 2012, 124, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gholipourmalekabadi, M.; Shafikhani, S.H. Animal models for type 1 and type 2 diabetes: Advantages and limitations. Front. Endocrinol. 2024, 15, 1359685. [Google Scholar] [CrossRef]
- Chen, X.; Graham, J.; Dabbah, M.A.; Petropoulos, I.N.; Ponirakis, G.; Asghar, O.; Alam, U.; Marshall, A.; Fadavi, H.; Ferdousi, M.; et al. Small nerve fiber quantification in the diagnosis of diabetic sensorimotor polyneuropathy: Comparing corneal confocal microscopy with intraepidermal nerve fiber density. Diabetes Care 2015, 38, 1138–1144. [Google Scholar] [CrossRef]
- Dhage, S.; Ferdousi, M.; Adam, S.; Ho, J.H.; Kalteniece, A.; Azmi, S.; Alam, U.; Ponirakis, G.; Petropoulos, I.; Atkinson, A.J.; et al. Corneal confocal microscopy identifies small fibre damage and progression of diabetic neuropathy. Sci. Rep. 2021, 11, 1859. [Google Scholar] [CrossRef]
- Galiero, R.; Caturano, A.; Vetrano, E.; Beccia, D.; Brin, C.; Alfano, M.; Di Salvo, J.; Epifani, R.; Piacevole, A.; Tagliaferri, G.; et al. Peripheral Neuropathy in Diabetes Mellitus: Pathogenetic Mechanisms and Diagnostic Options. Int. J. Mol. Sci. 2023, 24, 3554. [Google Scholar] [CrossRef]
- Rawat, A.; Morrison, B.M. Metabolic Transporters in the Peripheral Nerve-What, Where, and Why? Neurotherapeutics 2021, 18, 2185–2199. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef]
- Peng, W.; Tan, C.; Mo, L.; Jiang, J.; Zhou, W.; Du, J.; Zhou, X.; Liu, X.; Chen, L. Glucose transporter 3 in neuronal glucose metabolism: Health and diseases. Metabolism 2021, 123, 154869. [Google Scholar] [CrossRef]
- Mizukami, H.; Osonoi, S. Pathogenesis and Molecular Treatment Strategies of Diabetic Neuropathy Collateral Glucose-Utilizing Pathways in Diabetic Polyneuropathy. Int. J. Mol. Sci. 2020, 22, 94. [Google Scholar] [CrossRef]
- Mengstie, M.A.; Chekol Abebe, E.; Behaile Teklemariam, A.; Tilahun Mulu, A.; Agidew, M.M.; Teshome Azezew, M.; Zewde, E.A.; Agegnehu Teshome, A. Endogenous advanced glycation end products in the pathogenesis of chronic diabetic complications. Front. Mol. Biosci. 2022, 9, 1002710. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, Z.; Luo, Y.; Liu, Y.; Luo, W.; Du, X.; Luo, Z.; Hu, J.; Peng, S. Diabetic peripheral neuropathy: Pathogenetic mechanisms and treatment. Front. Endocrinol. 2023, 14, 1265372. [Google Scholar] [CrossRef]
- Mu, X.; Yang, M.; Ling, P.; Wu, A.; Zhou, H.; Jiang, J. Acylcarnitines: Can They Be Biomarkers of Diabetic Nephropathy? Diabetes Metab. Syndr. Obes. 2022, 15, 247–256. [Google Scholar] [CrossRef]
- Rumora, A.E.; LoGrasso, G.; Hayes, J.M.; Mendelson, F.E.; Tabbey, M.A.; Haidar, J.A.; Lentz, S.I.; Feldman, E.L. The Divergent Roles of Dietary Saturated and Monounsaturated Fatty Acids on Nerve Function in Murine Models of Obesity. J. Neurosci. 2019, 39, 3770–3781. [Google Scholar] [CrossRef] [PubMed]
- Sajic, M.; Rumora, A.E.; Kanhai, A.A.; Dentoni, G.; Varatharajah, S.; Casey, C.; Brown, R.D.R.; Peters, F.; Hinder, L.M.; Savelieff, M.G.; et al. High Dietary Fat Consumption Impairs Axonal Mitochondrial Function In Vivo. J. Neurosci. 2021, 41, 4321–4334. [Google Scholar] [CrossRef] [PubMed]
- Handzlik, M.K.; Gengatharan, J.M.; Frizzi, K.E.; McGregor, G.H.; Martino, C.; Rahman, G.; Gonzalez, A.; Moreno, A.M.; Green, C.R.; Guernsey, L.S.; et al. Insulin-regulated serine and lipid metabolism drive peripheral neuropathy. Nature 2023, 614, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Hakim, S.; Jain, A.; Adamson, S.S.; Petrova, V.; Indajang, J.; Kim, H.W.; Kawaguchi, R.; Wang, Q.; Duran, E.S.; Nelson, D.; et al. Macrophages protect against sensory axon loss in peripheral neuropathy. Nature 2025, 640, 212–220. [Google Scholar] [CrossRef]
- Yao, X.; Wang, X.; Zhang, R.; Kong, L.; Fan, C.; Qian, Y. Dysregulated mast cell activation induced by diabetic milieu exacerbates the progression of diabetic peripheral neuropathy in mice. Nat. Commun. 2025, 16, 4170. [Google Scholar] [CrossRef]
- Cashman, C.R.; Höke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2015, 596, 33–50. [Google Scholar] [CrossRef]
- Fernyhough, P.; McGavock, J. Mechanisms of disease: Mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb. Clin. Neurol. 2014, 126, 353–377. [Google Scholar] [CrossRef]
- Wang, Y.; Dai, X.; Li, H.; Jiang, H.; Zhou, J.; Zhang, S.; Guo, J.; Shen, L.; Yang, H.; Lin, J.; et al. The role of mitochondrial dynamics in disease. MedComm (2020) 2023, 4, e462. [Google Scholar] [CrossRef] [PubMed]
- Varughese, J.T.; Buchanan, S.K.; Pitt, A.S. The Role of Voltage-Dependent Anion Channel in Mitochondrial Dysfunction and Human Disease. Cells 2021, 10, 1737. [Google Scholar] [CrossRef] [PubMed]
- Mannella, C.A. Consequences of Folding the Mitochondrial Inner Membrane. Front. Physiol. 2020, 11, 536. [Google Scholar] [CrossRef] [PubMed]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca(2+) Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Kalichman, M.W.; Powell, H.C.; Mizisin, A.P. Reactive, degenerative, and proliferative Schwann cell responses in experimental galactose and human diabetic neuropathy. Acta Neuropathol. 1998, 95, 47–56. [Google Scholar] [CrossRef]
- Kamiya, H.; Zhang, W.; Sima, A.A. Degeneration of the Golgi and neuronal loss in dorsal root ganglia in diabetic BioBreeding/Worcester rats. Diabetologia 2006, 49, 2763–2774. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Green, K.G.; Snipes, L.L.; Feng, D. Neuritic dystrophy and neuronopathy in Akita (Ins2(Akita)) diabetic mouse sympathetic ganglia. Exp. Neurol. 2009, 216, 207–218. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Parvin, C.A.; Green, K.G. Synaptic ultrastructural alterations anticipate the development of neuroaxonal dystrophy in sympathetic ganglia of aged and diabetic mice. J. Neuropathol. Exp. Neurol. 2008, 67, 1166–1186. [Google Scholar] [CrossRef]
- Schroer, J.A.; Plurad, S.B.; Schmidt, R.E. Fine structure of presynaptic axonal terminals in sympathetic autonomic ganglia of aging and diabetic human subjects. Synapse 1992, 12, 1–13. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal 2020, 4, Ns20200008. [Google Scholar] [CrossRef]
- Galloway, C.A.; Yoon, Y. Mitochondrial morphology in metabolic diseases. Antioxid. Redox Signal. 2013, 19, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.K.; Zherebitskaya, E.; Smith, D.R.; Akude, E.; Chattopadhyay, S.; Jolivalt, C.G.; Calcutt, N.A.; Fernyhough, P. Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 2010, 59, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Roy Chowdhury, S.K.; Smith, D.R.; Saleh, A.; Schapansky, J.; Marquez, A.; Gomes, S.; Akude, E.; Morrow, D.; Calcutt, N.A.; Fernyhough, P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 2012, 135, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.J.; Pan, P.; Farmer, K.L.; Zhao, H.; Blagg, B.S.; Dobrowsky, R.T. Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp. Neurol. 2012, 235, 388–396. [Google Scholar] [CrossRef]
- Huang, T.J.; Price, S.A.; Chilton, L.; Calcutt, N.A.; Tomlinson, D.R.; Verkhratsky, A.; Fernyhough, P. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes 2003, 52, 2129–2136. [Google Scholar] [CrossRef]
- Srinivasan, S.; Stevens, M.; Wiley, J.W. Diabetic peripheral neuropathy: Evidence for apoptosis and associated mitochondrial dysfunction. Diabetes 2000, 49, 1932–1938. [Google Scholar] [CrossRef]
- Akude, E.; Zherebitskaya, E.; Chowdhury, S.K.; Smith, D.R.; Dobrowsky, R.T.; Fernyhough, P. Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 2011, 60, 288–297. [Google Scholar] [CrossRef]
- Ma, J.; Farmer, K.L.; Pan, P.; Urban, M.J.; Zhao, H.; Blagg, B.S.; Dobrowsky, R.T. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J. Pharmacol. Exp. Ther. 2014, 348, 281–292. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Smith, D.R.; Fernyhough, P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 2013, 51, 56–65. [Google Scholar] [CrossRef]
- Vincent, A.M.; Olzmann, J.A.; Brownlee, M.; Sivitz, W.I.; Russell, J.W. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes 2004, 53, 726–734. [Google Scholar] [CrossRef]
- Casanova-Molla, J.; Morales, M.; Garrabou, G.; Solà-Valls, N.; Soriano, A.; Calvo, M.; Grau, J.M.; Valls-Solé, J. Mitochondrial loss indicates early axonal damage in small fiber neuropathies. J. Peripher. Nerv. Syst. 2012, 17, 147–157. [Google Scholar] [CrossRef]
- Hamid, H.S.; Mervak, C.M.; Münch, A.E.; Robell, N.J.; Hayes, J.M.; Porzio, M.T.; Singleton, J.R.; Smith, A.G.; Feldman, E.L.; Lentz, S.I. Hyperglycemia- and neuropathy-induced changes in mitochondria within sensory nerves. Ann. Clin. Transl. Neurol. 2014, 1, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Fernyhough, P.; Calcutt, N.A. Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium 2010, 47, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Klocke, B.; Krone, K.; Tornes, J.; Moore, C.; Ott, H.; Pitychoutis, P.M. Insights into the role of intracellular calcium signaling in the neurobiology of neurodevelopmental disorders. Front. Neurosci. 2023, 17, 1093099. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The Interplay between Ca(2+) Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef]
- Shkryl, V.M. Endoplasmic Reticulum Calcium Signaling in Hippocampal Neurons. Biomolecules 2024, 14, 1617. [Google Scholar] [CrossRef]
- Zherebitskaya, E.; Schapansky, J.; Akude, E.; Smith, D.R.; Van der Ploeg, R.; Solovyova, N.; Verkhratsky, A.; Fernyhough, P. Sensory neurons derived from diabetic rats have diminished internal Ca2+ stores linked to impaired re-uptake by the endoplasmic reticulum. ASN Neuro 2012, 4, AN20110038. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef]
- George, D.S.; Hackelberg, S.; Jayaraj, N.D.; Ren, D.; Edassery, S.L.; Rathwell, C.A.; Miller, R.E.; Malfait, A.M.; Savas, J.N.; Miller, R.J.; et al. Mitochondrial calcium uniporter deletion prevents painful diabetic neuropathy by restoring mitochondrial morphology and dynamics. Pain 2022, 163, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Emery, S.M.; Dobrowsky, R.T. Promoting Neuronal Tolerance of Diabetic Stress: Modulating Molecular Chaperones. Int. Rev. Neurobiol. 2016, 127, 181–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, H.; Blagg, B.S.; Dobrowsky, R.T. C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J. Proteome Res. 2012, 11, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Dia, M.; Gomez, L.; Thibault, H.; Tessier, N.; Leon, C.; Chouabe, C.; Ducreux, S.; Gallo-Bona, N.; Tubbs, E.; Bendridi, N.; et al. Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 2020, 115, 74. [Google Scholar] [CrossRef]
- Nilius, B.; Talavera, K.; Verkhratsky, A. T-type calcium channels: The never ending story. Cell Calcium 2006, 40, 81–88. [Google Scholar] [CrossRef]
- Huang, T.J.; Sayers, N.M.; Fernyhough, P.; Verkhratsky, A. Diabetes-induced alterations in calcium homeostasis in sensory neurones of streptozotocin-diabetic rats are restricted to lumbar ganglia and are prevented by neurotrophin-3. Diabetologia 2002, 45, 560–570. [Google Scholar] [CrossRef]
- Kostyuk, E.; Pronchuk, N.; Shmigol, A. Calcium signal prolongation in sensory neurones of mice with experimental diabetes. Neuroreport 1995, 6, 1010–1012. [Google Scholar] [CrossRef]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef]
- Smiles, W.J.; Ovens, A.J.; Oakhill, J.S.; Kofler, B. The metabolic sensor AMPK: Twelve enzymes in one. Mol. Metab. 2024, 90, 102042. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.; Jaiswal, M.; Martin, C.; Pop-Busui, R. Glucose control and diabetic neuropathy: Lessons from recent large clinical trials. Curr. Diabetes Rep. 2014, 14, 528. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Scott, J.W.; Kemp, B.E. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol. Metab. 2012, 23, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Li, Y. Pleiotropic Regulation of PGC-1α in Tumor Initiation and Progression. Antioxid. Redox Signal. 2024, 41, 557–572. [Google Scholar] [CrossRef]
- Feige, J.N.; Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007, 17, 292–301. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Aghanoori, M.R.; Smith, D.R.; Shariati-Ievari, S.; Ajisebutu, A.; Nguyen, A.; Desmond, F.; Jesus, C.H.A.; Zhou, X.; Calcutt, N.A.; Aliani, M.; et al. Insulin-like growth factor-1 activates AMPK to augment mitochondrial function and correct neuronal metabolism in sensory neurons in type 1 diabetes. Mol. Metab. 2019, 20, 149–165. [Google Scholar] [CrossRef]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, S.S. NF-kappaB inhibitory action of resveratrol: A probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochem. Biophys. Res. Commun. 2010, 394, 360–365. [Google Scholar] [CrossRef]
- Yadav, J.P.; Verma, A.; Pathak, P.; Dwivedi, A.R.; Singh, A.K.; Kumar, P.; Khalilullah, H.; Jaremko, M.; Emwas, A.H.; Patel, D.K. Phytoconstituents as modulators of NF-κB signalling: Investigating therapeutic potential for diabetic wound healing. Biomed. Pharmacother. 2024, 177, 117058. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Choi, J.; Salimian, M.; Hedayat, A.F.; Russell, J.W. Administration of AICAR, an AMPK Activator, Prevents and Reverses Diabetic Polyneuropathy (DPN) by Regulating Mitophagy. Int. J. Mol. Sci. 2024, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Isop, L.M.; Neculau, A.E.; Necula, R.D.; Kakucs, C.; Moga, M.A.; Dima, L. Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals 2023, 16, 1714. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef]
- McAloon, L.M.; Muller, A.G.; Nay, K.; Lu, E.L.; Smeuninx, B.; Means, A.R.; Febbraio, M.A.; Scott, J.W. CaMKK2: Bridging the gap between Ca2+ signaling and energy-sensing. Essays Biochem. 2024, 68, 309–320. [Google Scholar] [CrossRef]
- Saleh, A.; Sabbir, M.G.; Aghanoori, M.R.; Smith, D.R.; Roy Chowdhury, S.K.; Tessler, L.; Brown, J.; Gedarevich, E.; Kassahun, M.Z.; Frizzi, K.; et al. Muscarinic Toxin 7 Signals Via Ca(2+)/Calmodulin-Dependent Protein Kinase Kinase β to Augment Mitochondrial Function and Prevent Neurodegeneration. Mol. Neurobiol. 2020, 57, 2521–2538. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): Advances in structures, mechanisms, and drug discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, A.J.; Munguba, H.; Levitz, J. Emerging modes of regulation of neuromodulatory G protein-coupled receptors. Trends Neurosci. 2024, 47, 635–650. [Google Scholar] [CrossRef]
- Chen, S.R.; Wess, J.; Pan, H.L. Functional activity of the M2 and M4 receptor subtypes in the spinal cord studied with muscarinic acetylcholine receptor knockout mice. J. Pharmacol. Exp. Ther. 2005, 313, 765–770. [Google Scholar] [CrossRef]
- Zhao, L.X.; Ge, Y.H.; Xiong, C.H.; Tang, L.; Yan, Y.H.; Law, P.Y.; Qiu, Y.; Chen, H.Z. M1 muscarinic receptor facilitates cognitive function by interplay with AMPA receptor GluA1 subunit. FASEB J. 2018, 32, 4247–4257. [Google Scholar] [CrossRef]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic acetylcholine receptors: Mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.W.; Dencker, D.; Grannan, M.; Bubser, M.; Zhan, X.; Wess, J.; Xiang, Z.; Locuson, C.; Lindsley, C.W.; Conn, P.J.; et al. Role for the M1 Muscarinic Acetylcholine Receptor in Top-Down Cognitive Processing Using a Touchscreen Visual Discrimination Task in Mice. ACS Chem. Neurosci. 2015, 6, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Islam, M.R.; Mim, S.A.; Sultana, N.; Chellappan, D.K.; Dua, K.; Kamal, M.A.; Sharma, R.; Emran, T.B. Insights into the Promising Prospect of G Protein and GPCR-Mediated Signaling in Neuropathophysiology and Its Therapeutic Regulation. Oxid. Med. Cell Longev. 2022, 2022, 8425640. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.; Shiferaw, Y. A structure-based computational model of IP(3)R1 incorporating Ca and IP3 regulation. Biophys. J. 2024, 123, 1274–1288. [Google Scholar] [CrossRef]
- Callender, J.A.; Newton, A.C. Conventional protein kinase C in the brain: 40 years later. Neuronal Signal 2017, 1, Ns20160005. [Google Scholar] [CrossRef]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Rüdiger, T.; Bolz, J. Acetylcholine influences growth cone motility and morphology of developing thalamic axons. Cell Adhes. Migr. 2008, 2, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Erskine, L.; McCaig, C.D. Growth cone neurotransmitter receptor activation modulates electric field-guided nerve growth. Dev. Biol. 1995, 171, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci. 2003, 23, 1–6. [Google Scholar] [CrossRef]
- Sabbir, M.G.; Calcutt, N.A.; Fernyhough, P. Muscarinic Acetylcholine Type 1 Receptor Activity Constrains Neurite Outgrowth by Inhibiting Microtubule Polymerization and Mitochondrial Trafficking in Adult Sensory Neurons. Front. Neurosci. 2018, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Young, S.H.; Poo, M.M. Spontaneous release of transmitter from growth cones of embryonic neurones. Nature 1983, 305, 634–637. [Google Scholar] [CrossRef]
- Hagg, T. Collateral sprouting as a target for improved function after spinal cord injury. J. Neurotrauma 2006, 23, 281–294. [Google Scholar] [CrossRef]
- Calcutt, N.A.; Smith, D.R.; Frizzi, K.; Sabbir, M.G.; Chowdhury, S.K.; Mixcoatl-Zecuatl, T.; Saleh, A.; Muttalib, N.; Van der Ploeg, R.; Ochoa, J.; et al. Selective antagonism of muscarinic receptors is neuroprotective in peripheral neuropathy. J. Clin. Investig. 2017, 127, 608–622. [Google Scholar] [CrossRef]
- Bellier, J.P.; Kimura, H. Acetylcholine synthesis by choline acetyltransferase of a peripheral type as demonstrated in adult rat dorsal root ganglion. J. Neurochem. 2007, 101, 1607–1618. [Google Scholar] [CrossRef]
- Hanada, K.; Kishimoto, S.; Bellier, J.P.; Kimura, H. Peripheral choline acetyltransferase in rat skin demonstrated by immunohistochemistry. Cell Tissue Res. 2013, 351, 497–510. [Google Scholar] [CrossRef]
- Bernardini, N.; Tomassy, G.S.; Tata, A.M.; Augusti-Tocco, G.; Biagioni, S. Detection of basal and potassium-evoked acetylcholine release from embryonic DRG explants. J. Neurochem. 2004, 88, 1533–1539. [Google Scholar] [CrossRef]
- Smith, D.S.; Skene, J.H. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 1997, 17, 646–658. [Google Scholar] [CrossRef]
- Scarr, E. Muscarinic M1 receptor agonists: Can they improve cognitive performance? Int. J. Neuropsychopharmacol. 2013, 16, 717–720. [Google Scholar] [CrossRef]
- Jolivalt, C.G.; Frizzi, K.E.; Han, M.M.; Mota, A.J.; Guernsey, L.S.; Kotra, L.P.; Fernyhough, P.; Calcutt, N.A. Topical Delivery of Muscarinic Receptor Antagonists Prevents and Reverses Peripheral Neuropathy in Female Diabetic Mice. J. Pharmacol. Exp. Ther. 2020, 374, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, A.; Peräkylä, M.; Akerman, K.E.; Näsman, J. Muscarinic toxin 7 selectivity is dictated by extracellular receptor loops. J. Biol. Chem. 2004, 279, 50923–50929. [Google Scholar] [CrossRef] [PubMed]
- el-Fakahany, E.E.; Cioffi, C.L.; Abdellatif, M.M.; Miller, M.M. Competitive interaction of pirenzepine with rat brain muscarinic acetylcholine receptors. Eur. J. Pharmacol. 1986, 131, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Toth, C.; Brussee, V.; Cheng, C.; Zochodne, D.W. Diabetes mellitus and the sensory neuron. J. Neuropathol. Exp. Neurol. 2004, 63, 561–573. [Google Scholar] [CrossRef]
- Casselini, C.M.; Parson, H.K.; Frizzi, K.E.; Marquez, A.; Smith, D.R.; Guernsey, L.; Nemmani, R.; Tayarani, A.; Jolivalt, C.G.; Weaver, J.; et al. A muscarinic receptor antagonist reverses multiple indices of diabetic peripheral neuropathy: Preclinical and clinical studies using oxybutynin. Acta Neuropathol. 2024, 147, 60. [Google Scholar] [CrossRef]
- Gavazzi, I.; Kumar, R.D.; McMahon, S.B.; Cohen, J. Growth responses of different subpopulations of adult sensory neurons to neurotrophic factors in vitro. Eur. J. Neurosci. 1999, 11, 3405–3414. [Google Scholar] [CrossRef]
- Jolivalt, C.G.; Han, M.M.; Nguyen, A.; Desmond, F.; Alves Jesus, C.H.; Vasconselos, D.C.; Pedneault, A.; Sandlin, N.; Dunne-Cerami, S.; Frizzi, K.E.; et al. Using Corneal Confocal Microscopy to Identify Therapeutic Agents for Diabetic Neuropathy. J. Clin. Med. 2022, 11, 2307. [Google Scholar] [CrossRef]
- Sivadasan, A.; Fernyhough, P.; Calcutt, N.A.; Frizzi, K.E.; Gardner, K.; Hansen, A.; Breiner, A.; Zochodne, D.W.; McInnes, N.; Punthakee, Z.; et al. Topical Application of the Antimuscarinic Pirenzepine, Increased Lower Limb Nerve Fiber Density in a Phase 2a Study in Type 2 Diabetic Patients with Peripheral Neuropathy. Ebiomedicine 2025. accepted subject to minor revision. [Google Scholar]
- Ziegler, D.; Papanas, N.; Zhivov, A.; Allgeier, S.; Winter, K.; Ziegler, I.; Brüggemann, J.; Strom, A.; Peschel, S.; Köhler, B.; et al. Early detection of nerve fiber loss by corneal confocal microscopy and skin biopsy in recently diagnosed type 2 diabetes. Diabetes 2014, 63, 2454–2463. [Google Scholar] [CrossRef]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial Dysfunction in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef]
- Lee, K.T.; Bulls, H.W.; Hoogland, A.I.; James, B.W.; Colon-Echevarria, C.B.; Jim, H.S.L. Chemotherapy-Induced Peripheral Neuropathy (CIPN): A Narrative Review and Proposed Theoretical Model. Cancers 2024, 16, 2571. [Google Scholar] [CrossRef]
- Cerles, O.; Gonçalves, T.C.; Chouzenoux, S.; Benoit, E.; Schmitt, A.; Bennett Saidu, N.E.; Kavian, N.; Chéreau, C.; Gobeaux, C.; Weill, B.; et al. Preventive action of benztropine on platinum-induced peripheral neuropathies and tumor growth. Acta Neuropathol. Commun. 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Han, M.M.; Frizzi, K.E.; Ellis, R.J.; Calcutt, N.A.; Fields, J.A. Prevention of HIV-1 TAT Protein-Induced Peripheral Neuropathy and Mitochondrial Disruption by the Antimuscarinic Pirenzepine. Front. Neurol. 2021, 12, 663373. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef] [PubMed]
- Cao, E. Structural mechanisms of transient receptor potential ion channels. J. Gen. Physiol. 2020, 152, e201811998. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tu, S.; Zhang, J.; Shao, A. Roles of TRP Channels in Neurological Diseases. Oxid. Med. Cell Longev. 2020, 2020, 7289194. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Fleig, A.; Penner, R. The TRPM ion channel subfamily: Molecular, biophysical and functional features. Trends Pharmacol. Sci. 2004, 25, 633–639. [Google Scholar] [CrossRef]
- Himmel, N.J.; Cox, D.N. Transient receptor potential channels: Current perspectives on evolution, structure, function and nomenclature. Proc. Biol. Sci. 2020, 287, 20201309. [Google Scholar] [CrossRef]
- Chubanov, V.; Köttgen, M.; Touyz, R.M.; Gudermann, T. TRPM channels in health and disease. Nat. Rev. Nephrol. 2024, 20, 175–187. [Google Scholar] [CrossRef]
- Uchida, K. TRPM3, TRPM4, and TRPM5 as thermo-sensitive channels. J. Physiol. Sci. 2024, 74, 43. [Google Scholar] [CrossRef]
- Behrendt, M. Implications of TRPM3 and TRPM8 for sensory neuron sensitisation. Biol. Chem. 2024, 405, 583–599. [Google Scholar] [CrossRef]
- Aloi, V.D.; Pinto, S.; Van Bree, R.; Luyten, K.; Voets, T.; Vriens, J. TRPM3 as a novel target to alleviate acute oxaliplatin-induced peripheral neuropathic pain. Pain 2023, 164, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Vandewauw, I.; De Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; Van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018, 555, 662–666. [Google Scholar] [CrossRef]
- Krügel, U.; Straub, I.; Beckmann, H.; Schaefer, M. Primidone inhibits TRPM3 and attenuates thermal nociception in vivo. Pain 2017, 158, 856–867. [Google Scholar] [CrossRef]
- Thiel, G.; Müller, I.; Rössler, O.G. Signal transduction via TRPM3 channels in pancreatic β-cells. J. Mol. Endocrinol. 2013, 50, R75–R83. [Google Scholar] [CrossRef]
- Mian, M.U.; Afzal, M.; Butt, A.A.; Ijaz, M.; Khalil, K.; Abbasi, M.; Fatima, M.; Asif, M.; Nadeem, S.; Jha, S.; et al. Neuropharmacology of Neuropathic Pain: A Systematic Review. Cureus 2024, 16, e69028. [Google Scholar] [CrossRef]
- Burglen, L.; Van Hoeymissen, E.; Qebibo, L.; Barth, M.; Belnap, N.; Boschann, F.; Depienne, C.; De Clercq, K.; Douglas, A.G.L.; Fitzgerald, M.P.; et al. Gain-of-function variants in the ion channel gene TRPM3 underlie a spectrum of neurodevelopmental disorders. eLife 2023, 12, e81032. [Google Scholar] [CrossRef]
- Zhao, S.; Yudin, Y.; Rohacs, T. Disease-associated mutations in the human TRPM3 render the channel overactive via two distinct mechanisms. eLife 2020, 9, e55634. [Google Scholar] [CrossRef]
- Held, K.; Kichko, T.; De Clercq, K.; Klaassen, H.; Van Bree, R.; Vanherck, J.C.; Marchand, A.; Reeh, P.W.; Chaltin, P.; Voets, T.; et al. Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc. Natl. Acad. Sci. USA 2015, 112, E1363–E1372. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Gruss, F.; Aloi, V.D.; Janssens, A.; Ulens, C.; Voets, T.; Vriens, J. Mutations in the voltage-sensing domain affect the alternative ion permeation pathway in the TRPM3 channel. J. Physiol. 2018, 596, 2413–2432. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Kraft, R.; Sauerbruch, S.; Schultz, G.; Harteneck, C. Molecular and functional characterization of the melastatin-related cation channel TRPM3. J. Biol. Chem. 2003, 278, 21493–21501. [Google Scholar] [CrossRef]
- Wagner, T.F.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Düfer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat. Cell Biol. 2008, 10, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Held, K.; Janssens, A.; Tóth, B.I.; Kerselaers, S.; Nilius, B.; Vennekens, R.; Voets, T. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat. Chem. Biol. 2014, 10, 188–195. [Google Scholar] [CrossRef]
- Straub, I.; Krügel, U.; Mohr, F.; Teichert, J.; Rizun, O.; Konrad, M.; Oberwinkler, J.; Schaefer, M. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol. Pharmacol. 2013, 84, 736–750. [Google Scholar] [CrossRef]
- Badheka, D.; Yudin, Y.; Borbiro, I.; Hartle, C.M.; Yazici, A.; Mirshahi, T.; Rohacs, T. Inhibition of Transient Receptor Potential Melastatin 3 ion channels by G-protein βγ subunits. eLife 2017, 6, e26147. [Google Scholar] [CrossRef]
- Quallo, T.; Alkhatib, O.; Gentry, C.; Andersson, D.A.; Bevan, S. G protein βγ subunits inhibit TRPM3 ion channels in sensory neurons. eLife 2017, 6, e26138. [Google Scholar] [CrossRef]
- Badheka, D.; Borbiro, I.; Rohacs, T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J. Gen. Physiol. 2015, 146, 65–77. [Google Scholar] [CrossRef]
- Tóth, B.I.; Konrad, M.; Ghosh, D.; Mohr, F.; Halaszovich, C.R.; Leitner, M.G.; Vriens, J.; Oberwinkler, J.; Voets, T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J. Gen. Physiol. 2015, 146, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Smith, D.R.; Shariati-Ievari, S.; Srivastava, A.; Dhingra, S.; Aliani, M.; Fernyhough, P. Muscarinic acetylcholine type 1 receptor antagonism activates TRPM3 to augment mitochondrial function and drive axonal repair in adult sensory neurons. Mol. Metab. 2025, 92, 102083. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Calmodulin Regulates Transient Receptor Potential TRPM3 and TRPM8-Induced Gene Transcription. Int. J. Mol. Sci. 2023, 24, 7902. [Google Scholar] [CrossRef] [PubMed]
- Holakovska, B.; Grycova, L.; Jirku, M.; Sulc, M.; Bumba, L.; Teisinger, J. Calmodulin and S100A1 protein interact with N terminus of TRPM3 channel. J. Biol. Chem. 2012, 287, 16645–16655. [Google Scholar] [CrossRef]
- Wang, D.; Gao, Q.; Schaefer, I.; Moerz, H.; Hoheisel, U.; Rohr, K.; Greffrath, W.; Treede, R.D. TRPM3-mediated dynamic mitochondrial activity in nerve growth factor-induced latent sensitization of chronic low back pain. Pain 2022, 163, e1115–e1128. [Google Scholar] [CrossRef]
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J.; et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 2014, 26, 738–753. [Google Scholar] [CrossRef]
- Hatsuda, A.; Kurisu, J.; Fujishima, K.; Kawaguchi, A.; Ohno, N.; Kengaku, M. Calcium signals tune AMPK activity and mitochondrial homeostasis in dendrites of developing neurons. Development 2023, 150, dev201930. [Google Scholar] [CrossRef]
- Liu, L.; Yuan, H.; Denton, K.; Li, X.J.; McCullough, L.; Li, J. Calcium/calmodulin-dependent protein kinase kinase β is neuroprotective in stroke in aged mice. Eur. J. Neurosci. 2016, 44, 2139–2146. [Google Scholar] [CrossRef]
- Wang, D.; Treede, R.D.; Köhr, G. Electrophysiological evidence that TRPM3 is a candidate in latent spinal sensitization of chronic low back pain. Neurosci. Lett. 2023, 816, 137509. [Google Scholar] [CrossRef]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Naznin, F.; Waise, T.M.Z.; Fernyhough, P. Antagonism of the Muscarinic Acetylcholine Type 1 Receptor Enhances Mitochondrial Membrane Potential and Expression of Respiratory Chain Components via AMPK in Human Neuroblastoma SH-SY5Y Cells and Primary Neurons. Mol. Neurobiol. 2022, 59, 6754–6770. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.M.; Lorrain, K.I.; Stebbins, K.J.; Edu, G.C.; Broadhead, A.R.; Lorenzana, A.J.; Roppe, J.R.; Baccei, J.M.; Baccei, C.S.; Chen, A.C.; et al. Targeting the muscarinic M1 receptor with a selective, brain-penetrant antagonist to promote remyelination in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2024, 121, e2407974121. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Aigrot, M.S.; Lamari, F.; Bachelin, C.; Lubetzki, C.; Nait Oumesmar, B.; Zalc, B.; Stankoff, B. Teriflunomide Promotes Oligodendroglial 8,9-Unsaturated Sterol Accumulation and CNS Remyelination. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1091. [Google Scholar] [CrossRef]
- Attoh-Mensah, E.; Loggia, G.; Schumann-Bard, P.; Morello, R.; Descatoire, P.; Marcelli, C.; Chavoix, C. Adverse Effects of Anticholinergic Drugs on Cognition and Mobility: Cutoff for Impairment in a Cross-Sectional Study in Young-Old and Old-Old Adults. Drugs Aging 2020, 37, 301–310. [Google Scholar] [CrossRef]
- Becker, A.; Mannebach, S.; Mathar, I.; Weissgerber, P.; Freichel, M.; Loodin, A.P.; Fecher-Trost, C.; Belkacemi, A.; Beck, A.; Philipp, S.E. Control of Insulin Release by Transient Receptor Potential Melastatin 3 (TRPM3) Ion Channels. Cell Physiol. Biochem. 2020, 54, 1115–1131. [Google Scholar] [CrossRef]
- Held, K.; Voets, T.; Vriens, J. TRPM3 in temperature sensing and beyond. Temperature 2015, 2, 201–213. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, S.; Calcutt, N.A.; Fernyhough, P. Muscarinic Receptor Antagonism and TRPM3 Activation as Stimulators of Mitochondrial Function and Axonal Repair in Diabetic Sensorimotor Polyneuropathy. Int. J. Mol. Sci. 2025, 26, 7393. https://doi.org/10.3390/ijms26157393
Chauhan S, Calcutt NA, Fernyhough P. Muscarinic Receptor Antagonism and TRPM3 Activation as Stimulators of Mitochondrial Function and Axonal Repair in Diabetic Sensorimotor Polyneuropathy. International Journal of Molecular Sciences. 2025; 26(15):7393. https://doi.org/10.3390/ijms26157393
Chicago/Turabian StyleChauhan, Sanjana, Nigel A. Calcutt, and Paul Fernyhough. 2025. "Muscarinic Receptor Antagonism and TRPM3 Activation as Stimulators of Mitochondrial Function and Axonal Repair in Diabetic Sensorimotor Polyneuropathy" International Journal of Molecular Sciences 26, no. 15: 7393. https://doi.org/10.3390/ijms26157393
APA StyleChauhan, S., Calcutt, N. A., & Fernyhough, P. (2025). Muscarinic Receptor Antagonism and TRPM3 Activation as Stimulators of Mitochondrial Function and Axonal Repair in Diabetic Sensorimotor Polyneuropathy. International Journal of Molecular Sciences, 26(15), 7393. https://doi.org/10.3390/ijms26157393