1. Introduction
Membrane crystallization (MCr) is emerging as a promising and environmentally sustainable technology for recovering high-value compounds. This innovative approach combines membrane technology with the crystallization process [
1]. Specifically, MCr follows the membrane distillation step, which is used to recover fresh water and concentrate the feed solution until it reaches saturation [
1]. In a membrane crystallizer, a solution containing a non-volatile solute, such as brines, lithium salt-enriched solutions, or mother liquors resulting from separation processes, is brought into contact with the distillate side through a hydrophobic microporous membrane [
2]. The membrane prevents mass transfer in the liquid phase, allowing only vapor-phase transfer driven by a vapor pressure gradient [
3]. As a result, the continuous evaporation of the solvent leads to the supersaturation of the solute, facilitating crystallization with the aid of the membrane [
4,
5,
6]. The driving force for this process is the transmembrane vapor pressure gradient, which can be induced through the application of a thermal gradient in thermal membrane crystallizers or by a concentration gradient in osmotic membrane crystallizers [
7,
8]
This method is attracting increasing interest, since numerous studies have shown that it offers well-controlled nucleation and growth kinetics, high crystallization rates, and reduced induction times. Additionally, it possesses potential advantages in terms of crystal quality and energy efficiency, as it can operate at relatively low temperatures and utilize alternative heat sources [
1,
9,
10,
11].
A supersaturated solution that undergoes crystallization can be viewed as a dynamic, multi-component system where many solute molecules move within a lattice of solvent molecules. Collisions between solute molecules may initiate the formation of self-assembly clusters and pre-nucleation aggregates. However, transitioning to stable cluster formation is thermodynamically constrained by an energetic barrier that must be overcome. In this context, the membrane surpasses its role as a mere physical barrier separating two distinct phases; it intervenes by reducing the Gibbs free energy required for the formation of stable critical nuclei, thereby increasing the probability of nucleation in its vicinity compared to other areas (heterogeneous nucleation) [
7].
In recent years, MCr has been explored as a promising method for lithium recovery from brines, owing to the growing interest in lithium for energy storage applications. Lithium is an essential element in the production of lithium-ion batteries (LiBs), which are crucial for a wide range of electronic devices and electric vehicles due to their high energy density, power output, and long cycle life. The lithium-ion battery market is rapidly expanding, driven by the increasing commitment to promote sustainable mobility through the electrification of transportation. Projections suggest that, by 2030, lithium supply may fall short of global demand.
Thus, the need for lithium recovery arises from a combination of increasing technological relevance, limited availability of primary resources, and significant environmental and economic advantages. Specifically, recycling spent batteries presents a valuable commercial opportunity, as producing cathode materials (including lithium, nickel, cobalt, and manganese) constitutes a significant portion of the total cost of manufacturing an LiB cell [
12].
The growing demand for lithium, along with climate change and environmental concerns, has emphasized the need for more sustainable processes, such as MCr, in lithium-ion battery recycling. This poses stringent requirements on membrane performance. Specifically, membranes must operate under high salinity, temperature, and pH without undergoing fouling, wetting, or morphological degradation.
The performance of MCr is critically dependent on selected operating conditions, including temperature, pressure, the concentration and nature of the precipitating species, and most importantly, the physical and morphological characteristics of the membrane barrier [
7,
13]. For example, temperature significantly impacts both the vapor pressure gradient across the membrane, which is the driving force for the process, and the solubility of the components in the solution. Feed flow rates affect the system’s hydrodynamics and mixing efficiency, while the concentration level (or degree of saturation) directly influences crystal yield. Higher levels of supersaturation can enhance nucleation rates and contribute to improved crystal purity [
14].
Recently, Jiang et al. [
15] investigated the engineering and effective control of the MCr process, conceptualizing membranes used in this process as microscale interfacial technology. The study demonstrated how various morphological attributes—such as porosity, pore size distribution, the orientation of polymer chains, surface contact angle, roughness, and additional parameters—can significantly influence the overall performance of the separation process. By modulating these properties, it is possible to ensure a uniform degree of micromixing within the interfacial liquid layer, adjust the contact area, enhance nucleation activity, improve selectivity, accelerate crystal growth kinetics, and influence the permeability of the liquid phase in the membrane structure.
To enhance durability and performance, bioinspired and surface-engineered membranes have been developed. For instance, Jiang et al. [
16], developed bioinspired composite membranes with a hybrid micro- and nanostructure to be used in membrane distillation of high-salinity water. They demonstrated the feasibility of modulating the morphological properties of polymeric membranes to prevent salt crystal deposition during membrane distillation processes. Specifically, commercial polypropylene (PP) membranes underwent various treatments, including plasma treatment, to increase pore size and introduce active graft-specific groups, loading with SiO
2 nanoparticles, and fluorination to enhance their hydrophobicity. The combination of these treatments and morphological modifications resulted in a membrane with a complex surface morphology and exceptional hydrophobicity (with a water contact angle of 160.59°), effectively inhibiting the wetting of the porous surface, counteracting salt crystal deposition, and significantly reducing fouling. In another approach, Al-Shaeli et al. [
17] focused on inorganic layered polymer membranes and surface engineering strategies. One notable modification involved the incorporation of zinc oxide (ZnO) nanorods along with the application of a perfluorodecyltriethoxysilane (PDTS) coating, which produced membranes with outstanding anti-fouling and anti-wetting properties. Likewise, membranes modified with titanium dioxide (TiO
2) nanorods achieved salt rejection rates exceeding 99.9% in membrane distillation applications for treating high-salinity solutions. In contrast, untreated pristine membranes displayed a gradual decline in separation performance due to pore wetting and foreshortening during operation. Another modification strategy involved incorporating halloysite nanotubes (m-HNTs) [
17], which can be deposited on porous polyacrylonitrile (PAN) membranes due to their hollow tubular structure formed by rolled aluminosilicate sheets. The presence of a dense, well-aligned, and oriented layer increased the surface area and improved the mechanical stability of the membrane. Furthermore, the hollow structure of these nanotubes, with negative surface charges, provided a high level of selectivity for molecular transport.
The study conducted by Li et al. [
1] showed that the hydrophobicity of a membrane, a crucial feature in both membrane distillation and MCr, is primarily influenced by its surface energy and roughness. A reduction in surface energy leads to lower affinity between the membrane and the liquid phase, resulting in increased hydrophobicity. Similarly, as described by the Cassie–Baxter equation, an increase in the surface roughness of the material expands the gas–liquid contact area and enhances the interfacial repulsion effect towards the liquid phase.
One surface modification strategy involves grafting sulfobetaine methacrylate (SBMA) onto a hydrophobic polyvinylidene fluoride (PVDF) membrane. This modification imparts anti-fouling and anti-wetting properties to the membrane, yielding a slightly lower contact angle while achieving a higher transmembrane flux than the unmodified membrane. Other examples of surface grafting utilize materials such as perfluoroalkylsilane (PFAS), fluoroalkylsilane (FAS), polydimethylsiloxane (PDMS), polyethylene glycol (PEG), and ethyl acrylate (EA). These examples highlight the versatility of surface modification strategies in adjusting membrane properties.
Additionally, the study explores blending fluorinated polymers with inorganic nanoparticles, including titanium dioxide (TiO
2), silicon dioxide (SiO
2), and calcium carbonate (CaCO
3). This approach involves incorporating modifying materials into the casting solutions, resulting in membranes with enhanced hydrophobicity, improved mechanical strength, and better anti-fouling capabilities. Such modifications directly and significantly influence membrane performance in membrane distillation and MCr processes [
1].
Perrotta et al. [
18] also demonstrated that different crystalline phases of PVDF—amorphous, α, and β forms—exhibit distinct morphological characteristics that significantly affect the nucleation and growth behavior of sodium chloride (NaCl) crystals during membrane crystallization. Their study shows that amorphous PVDF (density 1.7 ± 0.015 g/cm
3) promotes a higher nucleation rate, leading to shorter induction times (0 ns) and the formation of a greater number of smaller yet more regular clusters compared to the α and β forms (densities of 1.925 g/cm
3 and 1.931 g/cm
3, respectively). This behavior is attributed to the polymer chains’ morphological characteristics: amorphous PVDF has a free volume fraction of approximately 16.54% between the chains (compared to 2.3 Å for the β phase and 2.2 Å for the α phase), which allows water molecules to move more freely between them. This mobility facilitates the adsorption of water onto the surface and promotes more rapid supersaturation of the feed solution. The water uptake for the amorphous phase reaches significantly higher values (2.55 ± 0.55%) compared to the α and β phases, which have more compact and tightly interconnected crystalline structures that hinder water molecule mobility.
Further investigation into the nucleation and growth of NaCl crystals via MCr was conducted using PVDF membranes functionalized with graphene (at 0.5%, 5%, and 10% weight percentages of graphene nanoparticles) [
19]. The incorporation of graphene induces morphological changes in the membrane structure, promoting the formation of cavities between PVDF chains and graphene flakes. These void regions allow for the infiltration of water molecules. The study demonstrates that graphene functionalization enhances the hydrophobicity of the membrane, reduces crystal nucleation time by up to 190%, increases water removal from the feed solution, and shortens the distance between ionic species within the resulting crystalline aggregates [
19].
Regarding the influence of membrane morphology on nucleation and crystal growth, another promising approach involves functionalizing membranes with plasmonic or photothermal materials. This increasingly common procedure in membrane technologies leads to morphological changes that can significantly alter membrane performance in relevant applications. The primary goal of this type of functionalization is to elevate the temperature at the membrane–feed interface through light irradiation, thereby reducing or eliminating thermal polarization (TP) and enhancing the driving force for mass transfer. However, integration processes, such as the homogeneous dispersion of nanoparticles, incorporation of nanofibers, and surface coatings, can induce morphological modifications that affect key properties like wettability, porosity, surface roughness, and flow resistance. These factors are critical in membrane distillation and MCr operations [
20]. Utilizing intrinsically hydrophilic materials like polydopamine (PDA) while incorporating nanoparticles to achieve greater surface roughness can enhance membrane hydrophilicity. However, introducing nanomaterials such as silver nanoparticles (Ag NPs), MXenes, titanium nitride (TiN), antimony tin oxide (ATO), and magnetite (Fe
3O
4) may introduce additional resistance to mass flow, with reductions of up to 13%, despite the photothermal benefits [
20,
21]. A comprehensive analysis of these properties is essential in understanding how the morphological characteristics of the employed membranes influence key phenomena such as supersaturation control, nucleation kinetics, and crystal growth dynamics [
22].
In a previous study, Prenesti et al. [
22] selected polypropylene (PP) membranes for MCr operations involving lithium salts. This choice was primarily due to their highly hydrophobic nature, which promotes crystal nucleation and growth within the membrane pores, thereby enhancing process efficiency. Recent studies on the MCr of NaCl salts have shown that the morphology of PP membranes, characterized by a sponge-like internal structure and cavity-like features on the outer surface, favors the formation of anchoring sites where crystals can grow and adhere [
10,
23].
Quist-Jensen et al. [
24] utilized commercially available PP membrane modules to conduct a comparative analysis of various MCr and MD configurations aimed at recovering lithium chloride (LiCl) from highly concentrated aqueous solutions. Their findings indicated that using PP membranes in vapor membrane distillation (VMD) allows for the treatment of solutions up to saturation levels, thereby facilitating the potential for crystallization.
Nevertheless, the influence of concentrated ionic environments on the structural response of polymeric membranes remains poorly understood at the molecular scale. While experimental studies provide valuable performance indicators, they cannot fully elucidate how polymer chains rearrange upon exposure to hypersaline solution, especially during the early stages of contact. This gap is particularly relevant when analyzing local morphological descriptors such as anisotropy, tortuosity, and surface charge distribution, which may respond to ionic strength and temperature variations prior to the onset of macroscopic degradation.
This study addresses this knowledge gap by combining atomistic molecular dynamics (MD) simulations with computational fluid dynamics (CFD) to investigate the short-term morphological response of isotactic polypropylene (PP) membranes in contact with LiF solutions. The work focuses on the nanoscopic evolution of key structural descriptors (tortuosity, connectivity, void fraction, anisotropy, and surface charge) under hypersaline conditions (5.8 M and 8.9 M) and across a temperature range (300–353 K).
In particular, the effective diffusivity tensor and the corresponding diffusional tortuosity tensor of the polymer matrix are computed using a methodology recently developed by Caravella et al. [
25], based on a CFD approach implemented within the OpenFOAM
® (
https://www.openfoam.com/) software suite. The membrane morphologies subjected to CFD analysis were extracted from MD trajectories at selected simulation times.
The MD simulations were carried out under a set of clearly defined assumptions [
22]:
1. The membrane is modeled as a dense, non-porous, and non-reactive polymeric matrix, with no flux, porosity, or degradation pathways considered. This idealized setup allows for isolating local physical rearrangements of polymer chains in response to ionic strength and thermal stimuli.
2. Micropores and the distillation step are deliberately excluded from the model. The simulations focus exclusively on crystallization dynamics in a static, supersaturated solution, where salt concentration remains constant throughout. All morphological descriptors refer to spacing and topological reorganization among polymer chains, and do not account for actual porosity or long-term degradation phenomena. Moreover, no transport through pores or reactive aging processes is considered.
3. Simulations start from a pre-defined supersaturated state, enabling the direct observation of crystallization behavior modulated by the presence of the polymeric membrane.
This approach enables us to selectively explore how local interactions between the dense polymer matrix and the surrounding ionic solution drive morphological changes. By excluding mass flux and chemical degradation, the analysis focuses solely on short-term, interaction-driven phenomena. It is important to emphasize that the conclusions drawn in this study are strictly limited to the modeled conditions and should not be extrapolated to long-term performance scenarios.
Overall, the aim of this work is to establish a foundational understanding of how polymeric membranes initially respond—at the nanoscopic scale—to elevated salinity and temperature. These insights are intended to support the rational design of membranes and modeling frameworks for next-generation membrane-assisted crystallization processes.
4. Conclusions
This study addressed the short-term morphological evolution of a dense isotactic polypropylene (PP) membrane model upon contact with highly concentrated LiF aqueous solutions under controlled thermodynamic conditions. The investigation was motivated by the increasing interest in understanding how solid–liquid interfacial interactions may influence membrane structure and performance during crystallization processes involving highly supersaturated systems. While experimental studies have highlighted the susceptibility of polymeric membranes to structural changes under such conditions, the molecular-level mechanisms involved remain largely unexplored.
To bridge this gap, we employed a multiscale computational strategy that integrates atomistic molecular dynamics (MD) simulations with an innovative computational fluid dynamic procedure (CFD). Developed within our research group, it was applied to different structural architectures extracted at successive times (0 ns, 150 ns, and 300 ns).
The adopted model intentionally neglects membrane porosity, ion transport, fluid permeation, or chemical degradation mechanisms. Instead, it focuses exclusively on probing local morphological descriptors in a dense, static membrane system over short simulation times (up to 300 ns). This approach allows for isolating and characterizing subtle physical rearrangements induced by temperature and solute concentration at the polymer–solution interface.
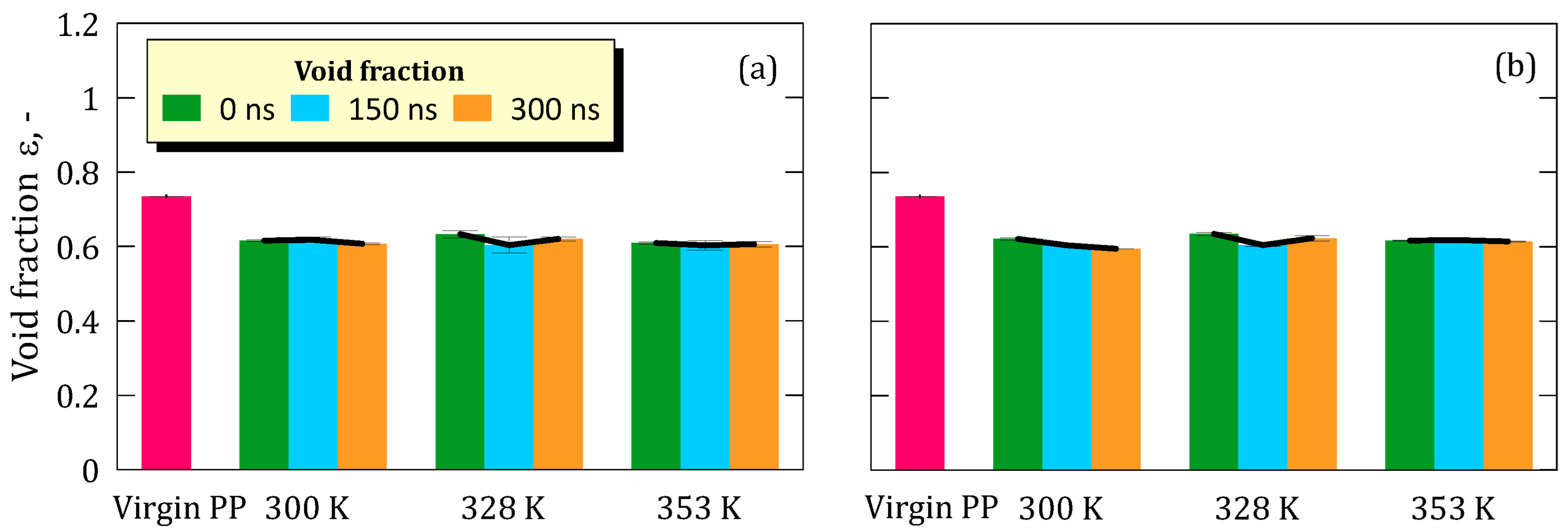
Our results demonstrate that global morphological descriptors such as tortuosity, connectivity, and void fraction remain essentially unchanged across all simulated conditions: temperature range (300–353 K) and LiF concentrations (5.8 M and 8.9 M). These observations suggest that the bulk structure of the PP matrix remains topologically stable in the early stages of exposure to hypersaline environments. Notably, no progressive trends were observed across the 300 ns time window, further supporting the conclusion that short-term interfacial interactions do not significantly disrupt the membrane’s internal architecture.
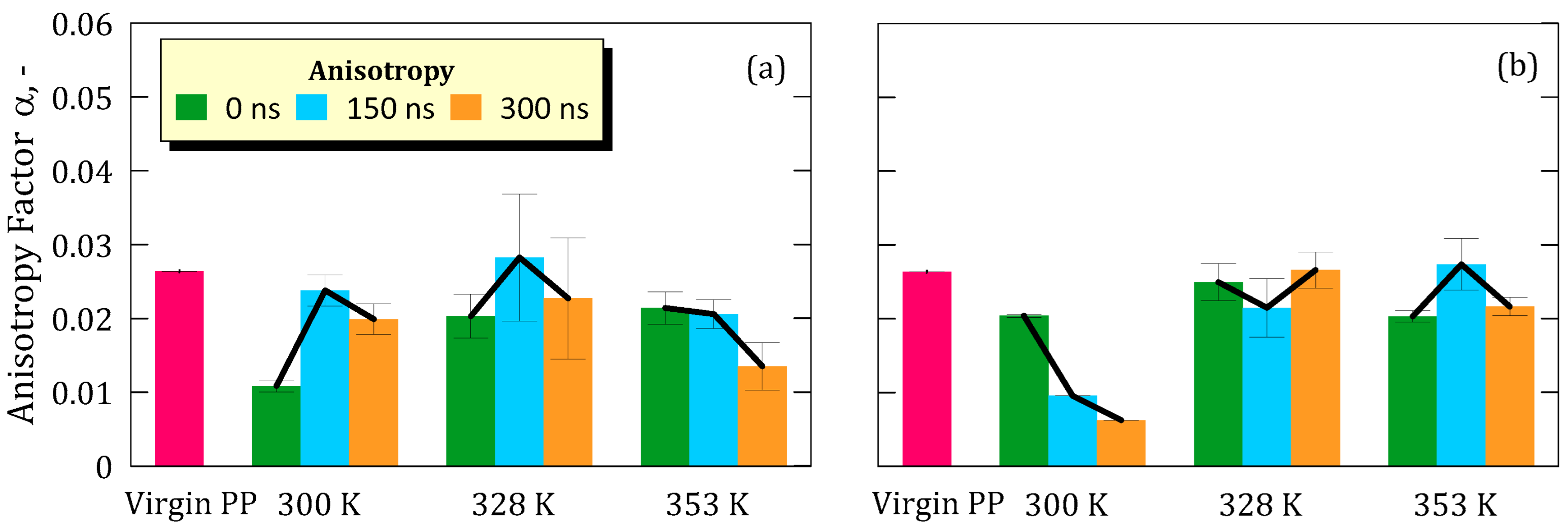
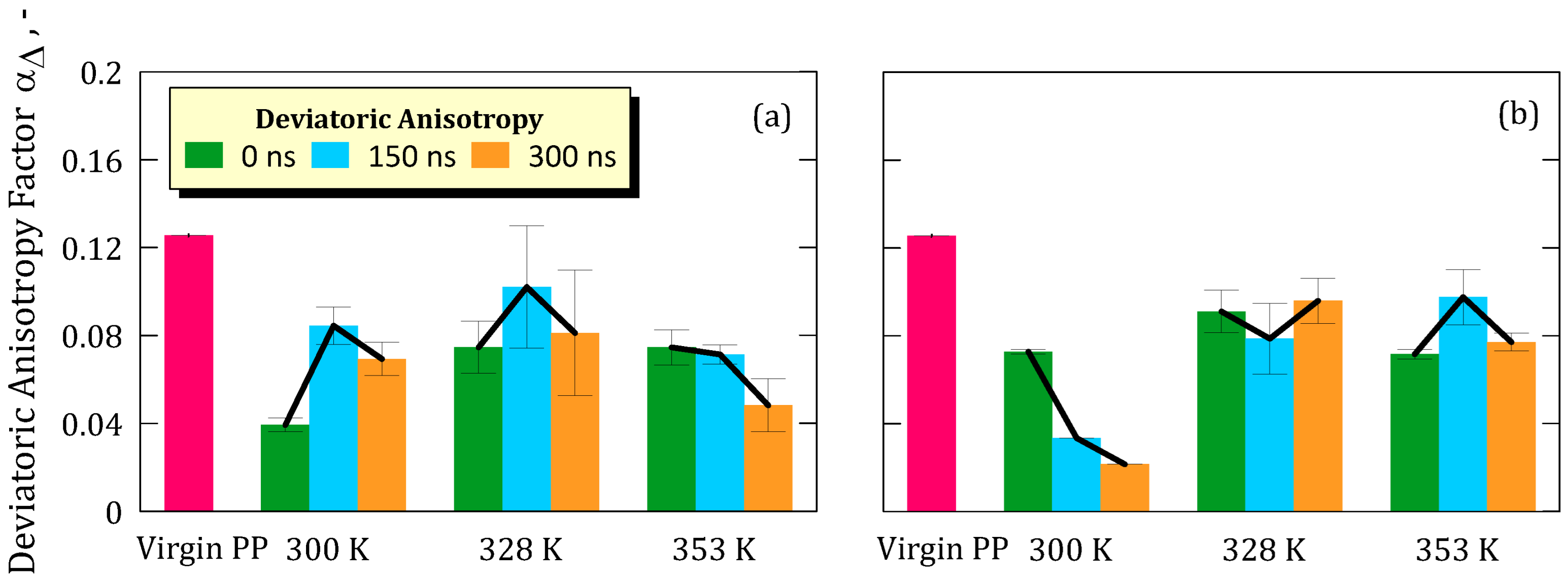
However, local structural rearrangements were observed through variations in anisotropy (α) and deviatoric anisotropy (αΔ), indicating that the polymer chains undergo non-negligible conformational adjustments when immersed in the ionic solution. These changes were more pronounced at lower temperatures and lower LiF concentrations, suggesting that thermal energy and electrostatic interactions modulate the degree of chain mobility. Although anisotropy values partially recovered toward the end of the simulation, they did not return to the levels of the pristine membrane, implying that the observed rearrangements are not fully reversible on the simulated timescale and result in a modest but detectable compaction and alignment of polymer chains.
The analysis of electrostatic surface charge distributions revealed additional effects of ion–polymer interaction. At a 5.8 M concentration, we observed a redistribution of partial atomic charges on the polymer surface, consistent with chain reorganization and exposure of terminal groups. This effect was attenuated at higher temperatures, likely due to the increased kinetic energy of the system. In contrast, simulations at 8.9 M displayed minimal variation in surface charge, pointing to the dominant role of strong Coulombic interactions in restricting chain mobility and preserving surface structure. Across all conditions, the PP surface retained a net negative character, consistent with experimental zeta potential values and relevant for understanding possible influences on heterogeneous nucleation.
In conclusion, this work presents a computational investigation focused on the initial physical interactions between a dense PP membrane and a hypersaline solution of LiF. Within the boundaries of the applied model, which excludes fluid transport and degradation phenomena, this study confirms that polypropylene preserves its overall morphological framework in the early stages of exposure. At the same time, it highlights measurable local reorganizations of chain orientation and electrostatic properties that may influence subsequent interfacial processes. These findings do not imply long-term structural stability but rather contribute to clarifying the short-timescale morphological response of the material, offering a computational framework that may support more complex future simulations and experimental comparisons aimed at optimizing membrane design for crystallization and separation technologies.



 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}