

XYLT1 Deficiency of Human Mesenchymal Stem Cells: Impact on Osteogenic, Chondrogenic, and Adipogenic Differentiation
 ,
,  ,
,
Abstract
1. Introduction
2. Results
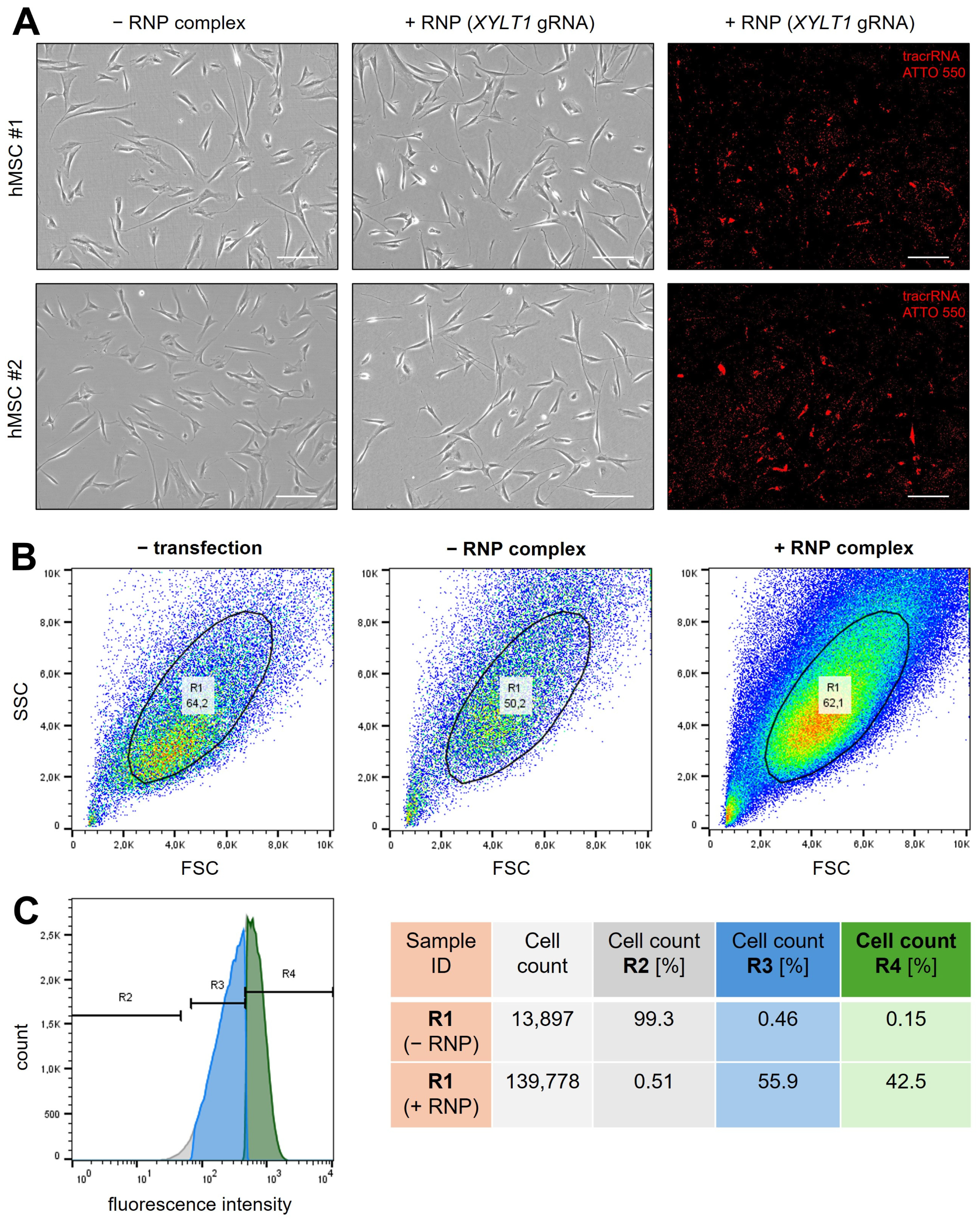
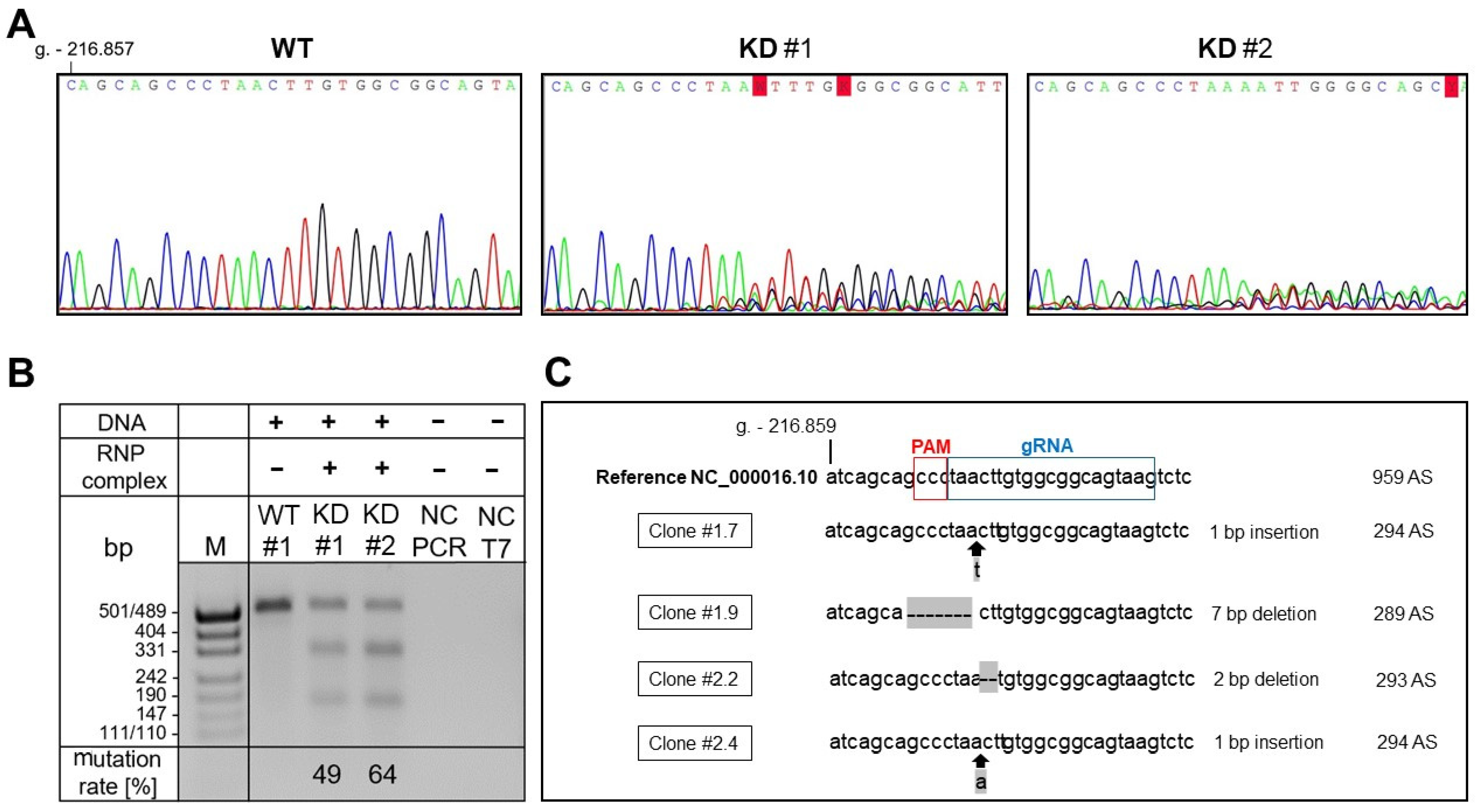
2.1. Successful Generation of XYLT1-Deficient hMSCs by CRISPR-Cas9 Genome Editing
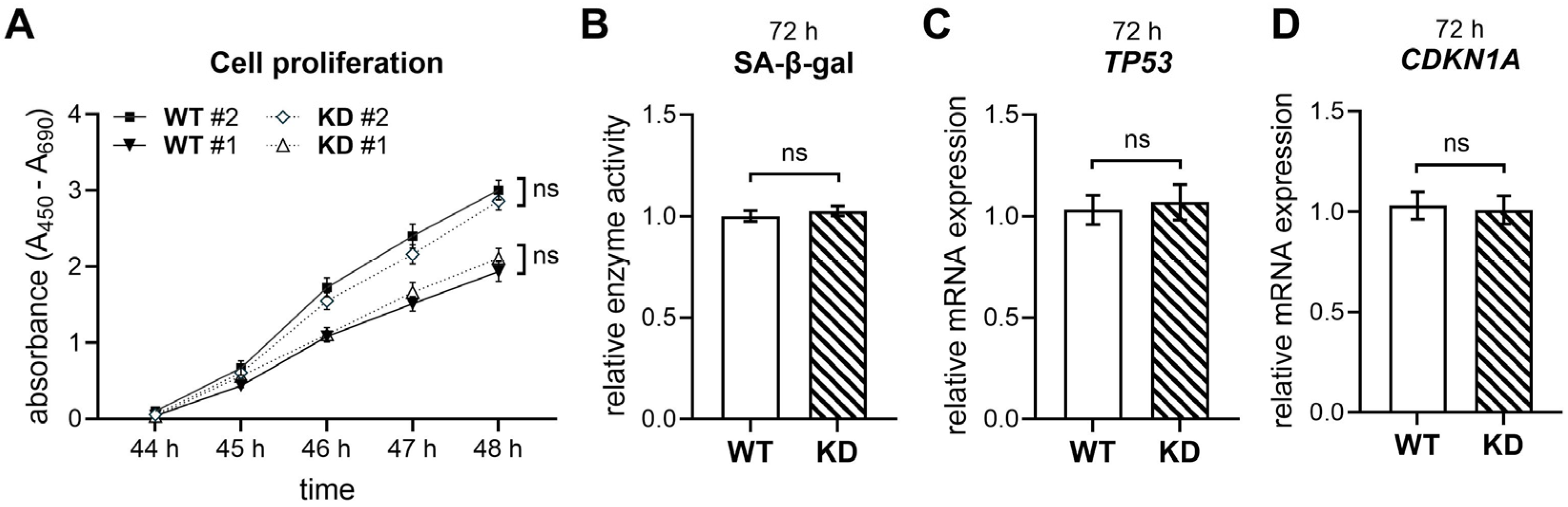
2.2. XYLT1 Deficiency Does Not Impact the Cellular Proliferation and Senescence of hMSCs
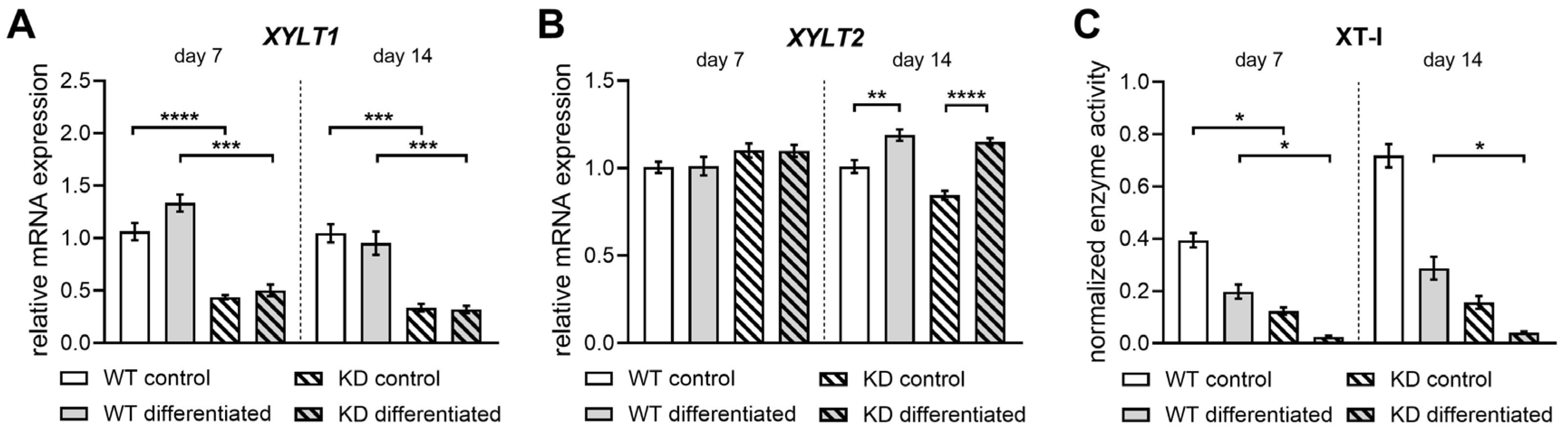
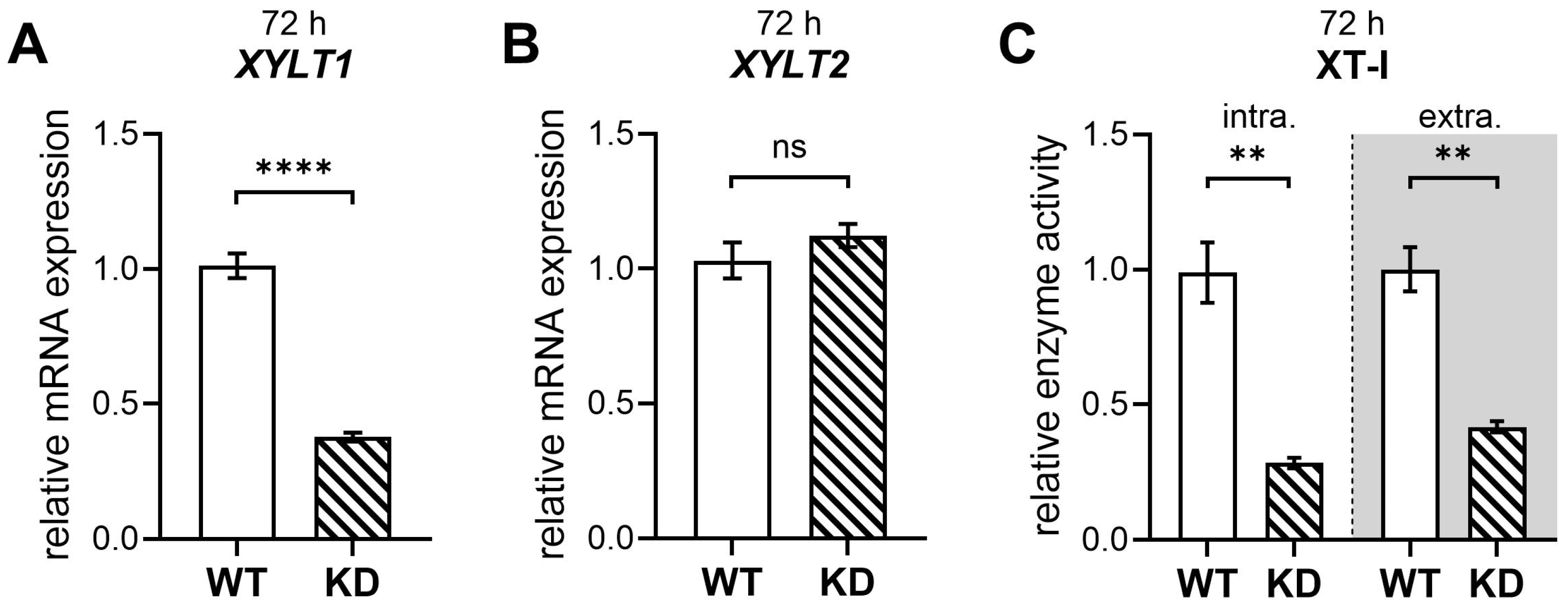
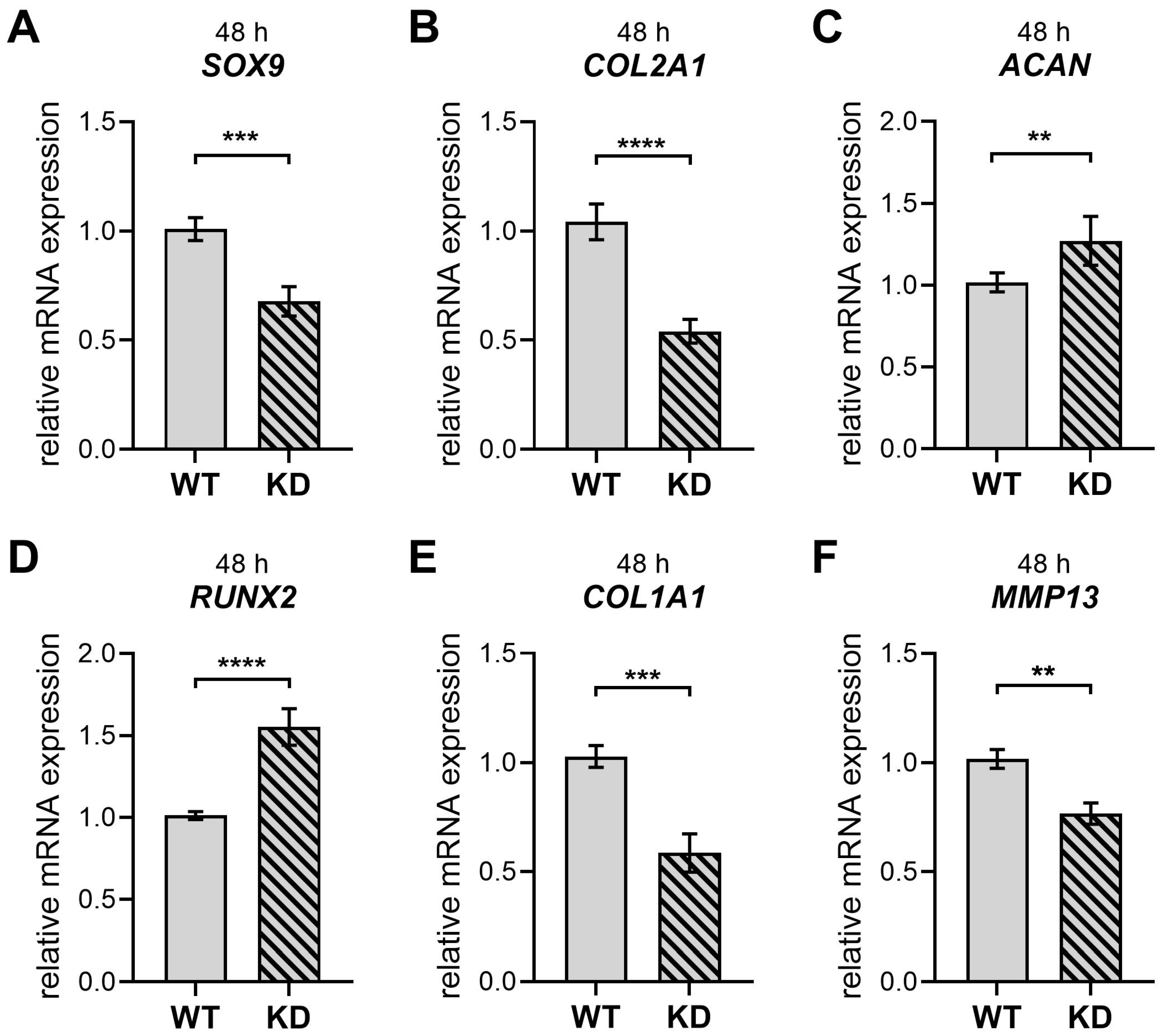
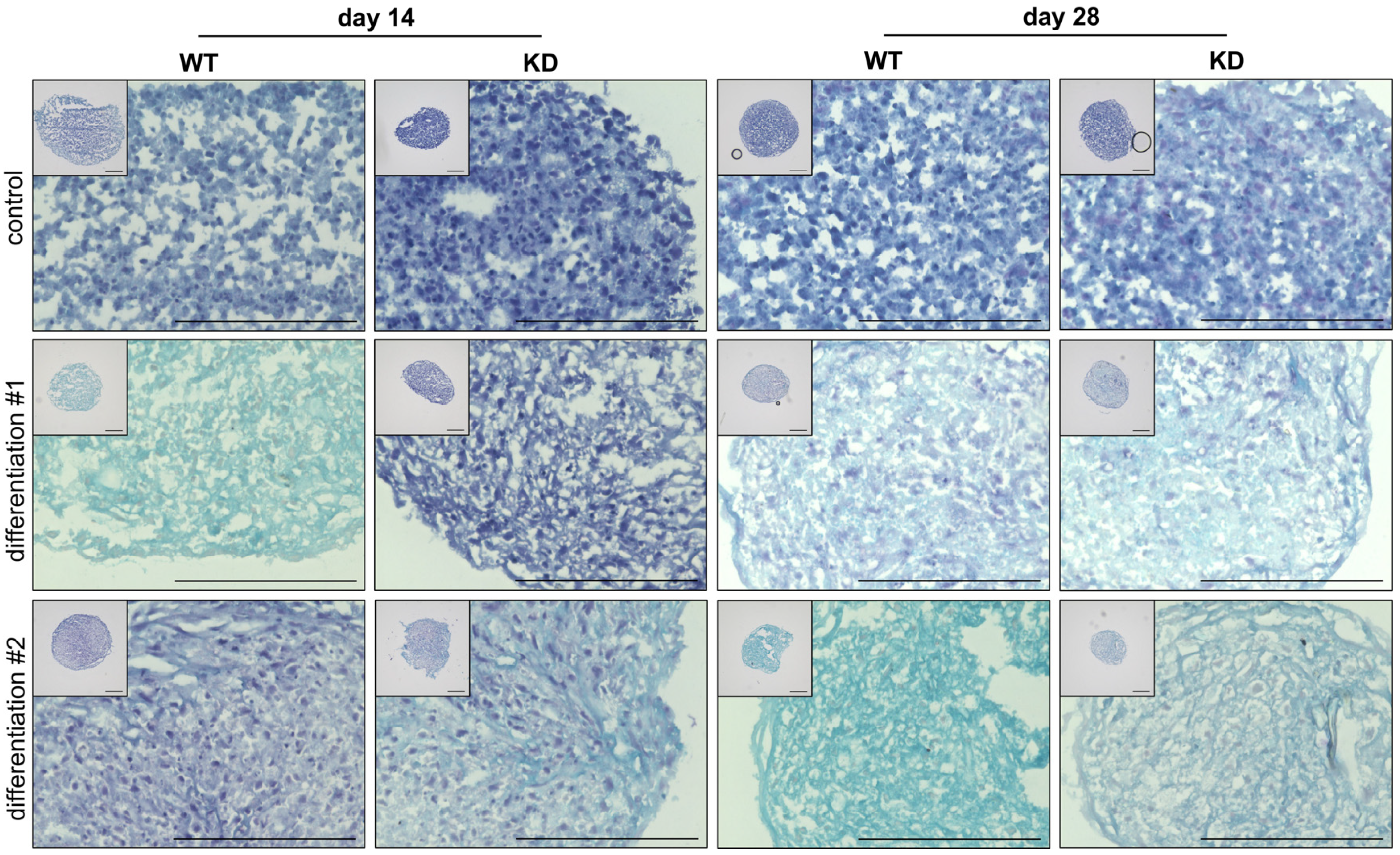
2.3. Diminished XYLT1 mRNA Expression of hMSCs Affects Their Chondrogenic Differentiation Potential
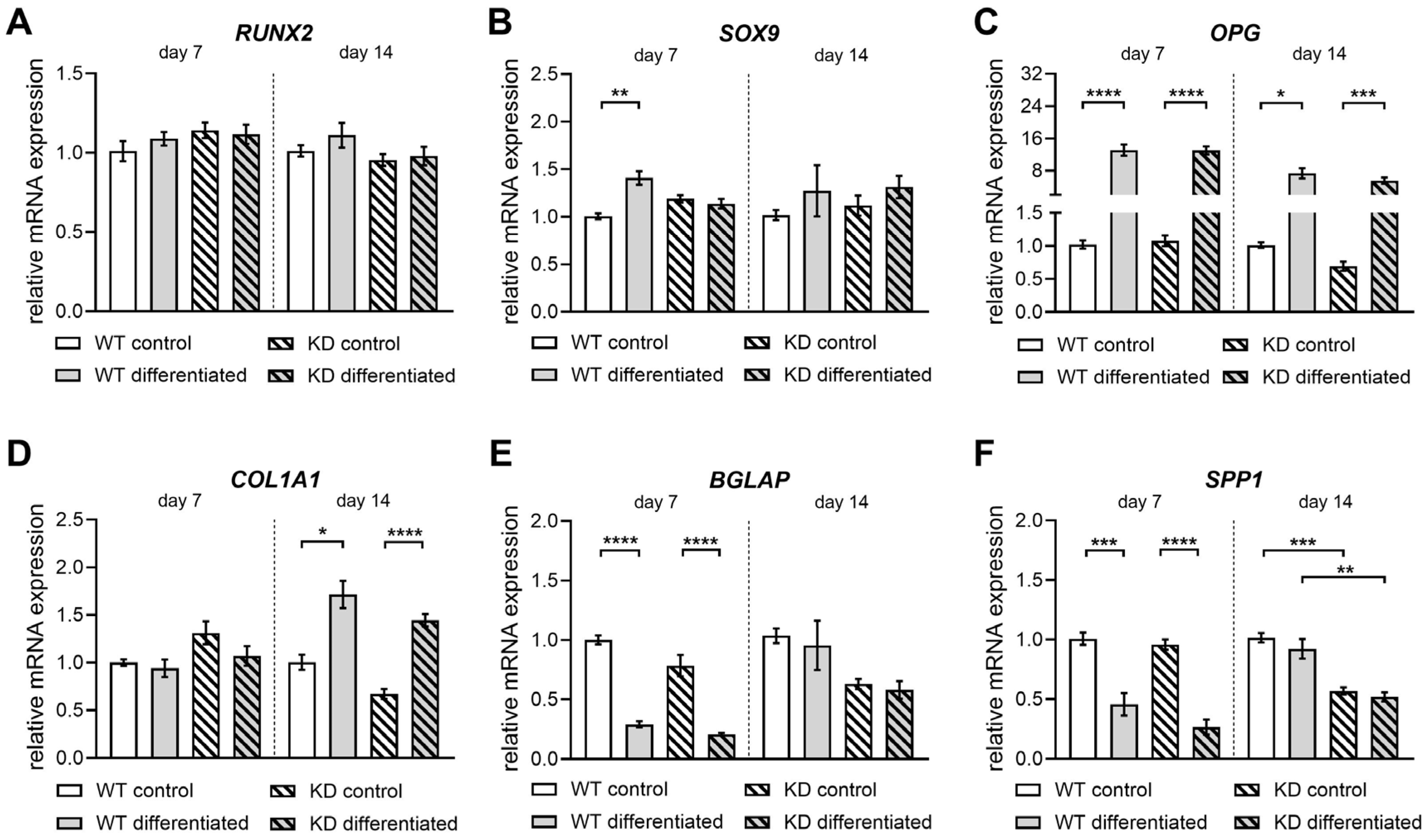
2.4. Diminished XYLT1 Expression of hMSCs Does Not Critically Impair Their Differentiation into Osteoblasts
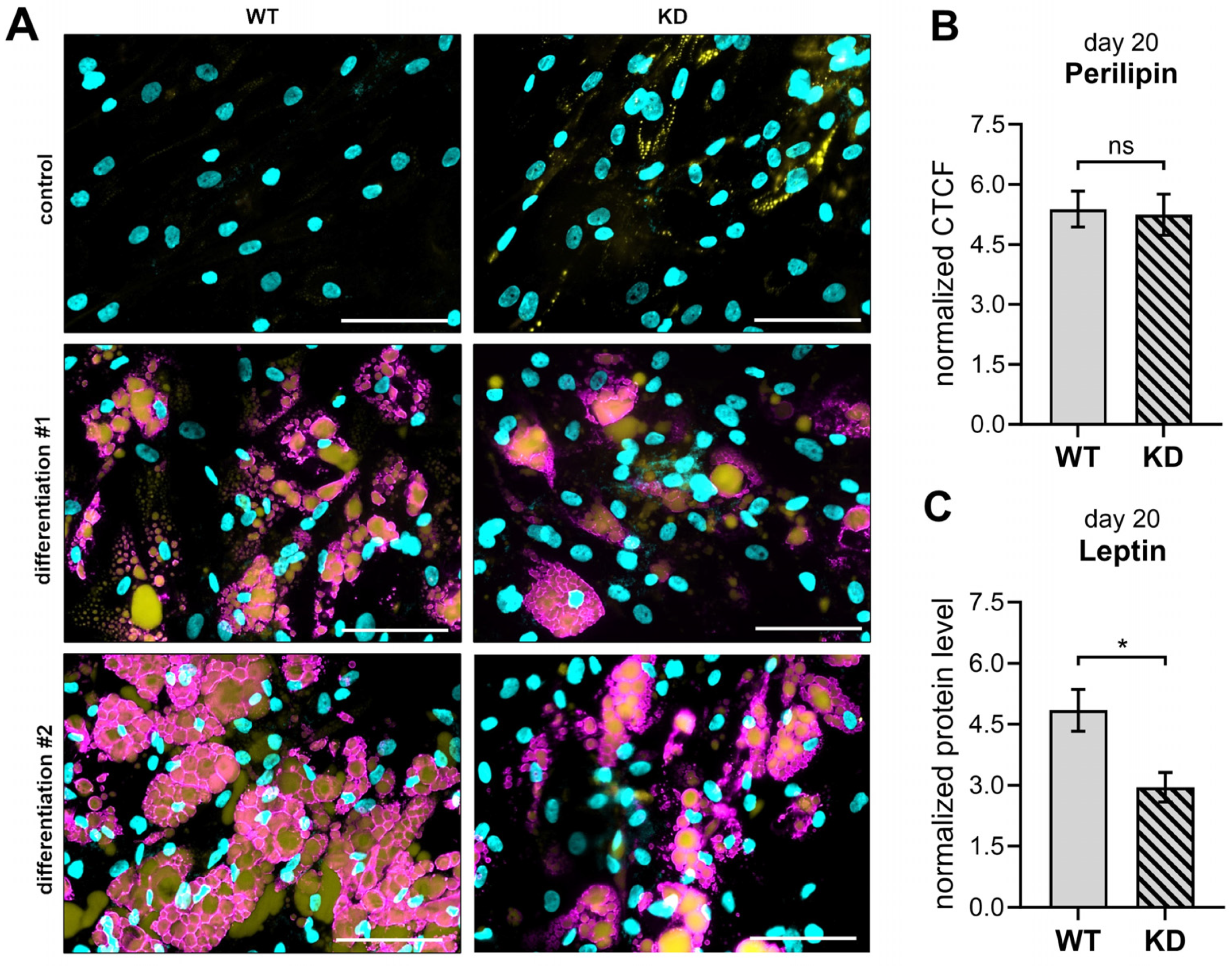
2.5. XYLT1-Deficient Adipogenically Differentiated hMSCs Possess a Diminished Leptin Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Ribonucleoprotein-Based CRISPR-Cas9-Mediated XYLT1 KD in hMSCs
4.3. Fluorescence-Activated Cell Sorting
4.4. Genomic DNA Amplification
4.5. Determining Genome Targeting Efficiency Using T7 Endonuclease I
4.6. TA Cloning for Single Allele Sequencing
4.7. Sanger Sequencing
4.8. Nucleic Acid Extraction, Quantification, and Synthesis of Complementary DNA
4.9. Quantitative Real-Time PCR Analysis
4.10. Bicinchoninic Acid Assay (BCA)
4.11. Mass Spectrometric XT-I Selective Activity Assay
4.12. Cell Proliferation Assay
4.13. Quantitative Determination of the Senescence-Associated β-Galactosidase (SA-β-gal) Activity
4.14. Cryosection Procedure
4.15. Alcian Blue Staining
4.16. Picro-Sirius Red Staining
4.17. Alkaline Phosphatase Activity Assay in Cell Lysates
4.18. Quantification of Inorganic Phosphate from Cell Culture Supernatants
4.19. Enzyme-Linked Immunosorbent Assay
4.20. Alizarin Red Staining
4.21. Immunostaining and BODIPY Staining for Fluorescence Microscopy
4.22. Experimental Setup and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Handa, A.; Grigelioniene, G.; Nishimura, G. Skeletal dysplasia families: A stepwise approach to diagnosis. Radiographics 2023, 43, e220067. [Google Scholar] [CrossRef]
- Neto, G.d.P.C.; Alves, C.D.A.D. Demystifying skeletal dysplasias: A practical approach for the pediatric endocrinologist. Horm. Res. Paediatr. 2024, 98, 214–225. [Google Scholar] [CrossRef]
- Alford, A.I.; Kozloff, K.M.; Hankenson, K.D. Extracellular matrix networks in bone remodeling. Int. J. Biochem. Cell Biol. 2015, 65, 20–31. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.; Huber, C.; Tuysuz, B.; Alanay, Y.; Bole-Feysot, C.; Leroy, J.G.; Mortier, G.; Nitschke, P.; Munnich, A.; Cormier-Daire, V. XYLT1 Mutations in Desbuquois dysplasia type 2. Am. J. Hum. Genet. 2014, 94, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Prante, C.; Gastens, M.; Kuhn, J.; Kleesiek, K.; Götting, C. Increased levels of xylosyltransferase I correlate with the mineralization of the extracellular matrix during osteogenic differentiation of mesenchymal stem cells. Matrix Biol. 2008, 27, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Patil, S.; Gao, Y.-G.; Qian, A. The bone extracellular matrix in bone formation and regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef] [PubMed]
- Chouery, E.; Karam, R.; Mrad, Y.N.; Mehawej, C.; El Jalbout, N.D.; Bleik, J.; Mahfoud, D.; Megarbane, A. Spondyloocular syndrome: A report of an additional family and phenotypic spectrum delineation. Genes 2023, 14, 497. [Google Scholar] [CrossRef]
- Paganini, C.; Costantini, R.; Superti-Furga, A.; Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: A panoramic view. FEBS J. 2019, 286, 3008–3032. [Google Scholar] [CrossRef]
- Lamoureux, F.; Baud’huin, M.; Duplomb, L.; Heymann, D.; Rédini, F. Proteoglycans: Key partners in bone cell biology. BioEssays 2007, 29, 758–771. [Google Scholar] [CrossRef]
- Michelacci, Y.M.; Baccarin, R.Y.A.; Rodrigues, N.N.P. Chondrocyte homeostasis and differentiation: Transcriptional control and signaling in healthy and osteoarthritic conditions. Life 2023, 13, 1460. [Google Scholar] [CrossRef]
- Akune, T.; Ohba, S.; Kamekura, S.; Yamaguchi, M.; Chung, U.-I.; Kubota, N.; Terauchi, Y.; Harada, Y.; Azuma, Y.; Nakamura, K.; et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J. Clin. Investig. 2004, 113, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, A.A.; Al Mamun, A. Current advances in regulation of bone homeostasis. FASEB BioAdvances 2020, 2, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Baud’Huin, M.; Charrier, C.; Bougras, G.; Brion, R.; Lezot, F.; Padrines, M.; Heymann, D. Proteoglycans and Osteolysis; Rédini, F., Ed.; Proteoglycans, Humana Press: Totowa, NJ, USA, 2012; pp. 323–337. [Google Scholar] [CrossRef]
- Kodama, J.; Wilkinson, K.J.; Iwamoto, M.; Otsuru, S.; Enomoto-Iwamoto, M. The role of hypertrophic chondrocytes in regulation of the cartilage-to-bone transition in fracture healing. Bone Rep. 2022, 17, 101616. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.; Wang, X.; Qiu, X.; Wu, Z.; Gao, B.; Liu, L.; Liang, G.; Zhou, H.; Yang, X.; Peng, Y.; et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1−SMAD1 interaction. Bone Res. 2019, 7, 8. [Google Scholar] [CrossRef]
- Maes, C.; Kronenberg, H.M. Bone Development and Remodeling. In Endocrinology: Adult and Pediatric; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1038–1062.e8. [Google Scholar] [CrossRef]
- Tschaffon, M.E.A.; Reber, S.O.; Schoppa, A.; Nandi, S.; Cirstea, I.C.; Aszodi, A.; Ignatius, A.; Haffner-Luntzer, M. A novel in vitro assay to study chondrocyte-to-osteoblast transdifferentiation. Endocrine 2022, 75, 266–275. [Google Scholar] [CrossRef]
- McCoy, S.Y.; Falgowski, K.A.; Srinivasan, P.P.; Thompson, W.R.; Selva, E.M.; Kirn-Safran, C.B. Serum xylosyltransferase 1 level increases during early posttraumatic osteoarthritis in mice with high bone forming potential. Bone 2012, 51, 224–231. [Google Scholar] [CrossRef]
- Eames, B.F.; Yan, Y.-L.; Swartz, M.E.; Levic, D.S.; Knapik, E.W.; Postlethwait, J.H.; Kimmel, C.B. Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet. 2011, 7, e1002246. [Google Scholar] [CrossRef]
- Mis, E.K.; Liem, K.F.; Kong, Y.; Schwartz, N.B.; Domowicz, M.; Weatherbee, S.D. Forward genetics defines Xylt1 as a key, conserved regulator of early chondrocyte maturation and skeletal length. Dev. Biol. 2014, 385, 67–82. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells-current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Al-Azab, M.; Safi, M.; Idiiatullina, E.; Al-Shaebi, F.; Zaky, M.Y. Aging of mesenchymal stem cell: Machinery, markers, and strategies of fighting. Cell. Mol. Biol. Lett. 2022, 27, 69. [Google Scholar] [CrossRef]
- Kleine, A.; Kühle, M.; Ly, T.-D.; Schmidt, V.; Faust-Hinse, I.; Knabbe, C.; Fischer, B. Xylosyltransferase-deficiency in human dermal fibroblasts induces compensatory myofibroblast differentiation and long-term ECM reduction. Biomedicines 2024, 12, 572. [Google Scholar] [CrossRef]
- Turinetto, V.; Vitale, E.; Giachino, C. Senescence in human mesenchymal stem cells: Functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci. 2016, 17, 1164. [Google Scholar] [CrossRef]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.-X. Senescence; aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Levinson, C.; Hertl, M.; Broguiere, N.; Brück, O.; Mustjoki, S.; Gerstenberg, A.; Weber, D.; Salzmann, G.; Steinwachs, M.; et al. Characterization of polydactyly chondrocytes and their use in cartilage engineering. Sci. Rep. 2019, 9, 4275. [Google Scholar] [CrossRef] [PubMed]
- Hamidouche, Z.; Fromigue, O.; Ringe, J.; Srouji, S.; Livne, E.; Marie, P.J. Priming integrin alpha5 promotes human mesenchymal stromal cell osteoblast differentiation and osteogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 18587–18591. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Pérez, A.; Guillán-Fresco, M.; Franco-Trepat, E.; Jorge-Mora, A.; López-Fagúndez, M.; Pazos-Pérez, A.; Crespo-Golmar, A.; Caeiro-Rey, J.R.; Gómez, R. Improved protocol to study osteoblast and adipocyte differentiation balance. Biomedicines 2022, 11, 31. [Google Scholar] [CrossRef]
- Reid, I.R.; Baldock, P.A.; Cornish, J. Effects of leptin on the skeleton. Endocr. Rev. 2018, 39, 938–959. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Garty, N.B.; Blanchette-Mackie, E.J.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar] [CrossRef]
- Upadhyay, J.; Farr, O.M.; Mantzoros, C.S. The role of leptin in regulating bone metabolism. Metabolism 2015, 64, 105–113. [Google Scholar] [CrossRef]
- Hess, R.; Pino, A.M.; Ríos, S.; Fernández, M.; Rodríguez, J.P. High affinity leptin receptors are present in human mesenchymal stem cells (MSCs) derived from control and osteoporotic donors. J. Cell. Biochem. 2005, 94, 50–57. [Google Scholar] [CrossRef]
- Picó, C.; Palou, M.; Pomar, C.A.; Rodríguez, A.M.; Palou, A. Leptin as a key regulator of the adipose organ. Rev. Endocr. Metab. Disord. 2022, 23, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; Pereira, J.A.d.S.; de Almeida, C.J.; Moraes-Vieira, P.M.M.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin induces proadipogenic and proinflammatory signaling in adipocytes. Front. Endocrinol. 2019, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Nakada, S.; Yoshihara, T.; Nara, T.; Furuya, N.; Miida, T.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan, a heparan sulfate proteoglycan, regulates systemic metabolism with dynamic changes in adipose tissue and skeletal muscle. Sci. Rep. 2018, 8, 7766. [Google Scholar] [CrossRef]
- Guo, L.; Elcioglu, N.H.; Iida, A.; Demirkol, Y.K.; Aras, S.; Matsumoto, N.; Nishimura, G.; Miyake, N.; Ikegawa, S. Novel and recurrent XYLT1 mutations in two Turkish families with Desbuquois dysplasia, type 2. J. Hum. Genet. 2017, 62, 447–451. [Google Scholar] [CrossRef]
- Rajabi, F.; Bereshneh, A.H.; Ramezanzadeh, M.; Garshasbi, M. Novel compound heterozygous variants in XYLT1 gene caused Desbuquois dysplasia type 2 in an aborted fetus: A case report. BMC Pediatr. 2022, 22, 63. [Google Scholar] [CrossRef]
- Schreml, J.; Durmaz, B.; Cogulu, O.; Keupp, K.; Beleggia, F.; Pohl, E.; Milz, E.; Coker, M.; Ucar, S.K.; Nürnberg, G.; et al. The missing “link”: An autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum. Genet. 2014, 133, 29–39. [Google Scholar] [CrossRef]
- Van Koningsbruggen, S.; Knoester, H.; Bakx, R.; Mook, O.; Knegt, L.; Cobben, J.M. Complete and partial XYLT1 deletion in a patient with neonatal short limb skeletal dysplasia. Am. J. Med. Genet. Part A 2016, 170, 510–514. [Google Scholar] [CrossRef]
- Baratela, W.A.R.; Bober, M.B.; Tiller, G.E.; Okenfuss, E.; Ditro, C.; Duker, A.; Krakow, D.; Stabley, D.L.; Sol-Church, K.; Mackenzie, W.; et al. A newly recognized syndrome with characteristic facial features, skeletal dysplasia, and developmental delay. Am. J. Med. Genet. Part A 2012, 158A, 1815–1822. [Google Scholar] [CrossRef]
- LaCroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. GGC Repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef]
- Mizumoto, S.; Yamada, S. An overview of in vivo functions of chondroitin sulfate and dermatan sulfate revealed by their deficient mice. Front. Cell Dev. Biol. 2021, 9, 764781. [Google Scholar] [CrossRef] [PubMed]
- Plümers, R.; Osterhage, M.R.; Lindenkamp, C.; Knabbe, C.; Hendig, D. Targeting ABCC6 in mesenchymal stem cells: Impairment of mature adipocyte lipid homeostasis. Int. J. Mol. Sci. 2022, 23, 9218. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, J.M.; Lee, S.; Cho, H.B.; Park, J.-I.; Kim, J.-H.; Park, J.S.; Park, K.-H. Efficient CRISPR-Cas9-based knockdown of RUNX2 to induce chondrogenic differentiation of stem cells. Biomater. Sci. 2022, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.K.; Ogando, C.R.; See, C.W.; Chang, T.-Y.; Barabino, G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef]
- Fischer, B.; Ly, T.-D.; Hendig, D.; Kuhn, J.; Pécheur, E.-I.; Reungoat, E.; Knabbe, C.; Faust, I. First description of a compensatory xylosyltransferase I induction observed after an antifibrotic UDP-treatment of normal human dermal fibroblasts. Biochem. Biophys. Res. Commun. 2019, 512, 7–13. [Google Scholar] [CrossRef]
- Denu, R.A.; Nemcek, S.; Bloom, D.D.; Goodrich, A.D.; Kim, J.; Mosher, D.F.; Hematti, P. Fibroblasts and mesenchymal stromal/stem cells are phenotypically indistinguishable. Acta Haematol. 2016, 136, 85–97. [Google Scholar] [CrossRef]
- Ichim, T.E.; O’Heeron, P.; Kesari, S. Fibroblasts as a practical alternative to mesenchymal stem cells. J. Transl. Med. 2018, 16, 212. [Google Scholar] [CrossRef]
- Frippiat, C.; Dewelle, J.; Remacle, J.; Toussaint, O. Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic. Biol. Med. 2002, 33, 1334–1346. [Google Scholar] [CrossRef]
- Ugurlu, B.; Karaoz, E. Comparison of similar cells: Mesenchymal stromal cells and fibroblasts. Acta Histochem. 2020, 122, 151634. [Google Scholar] [CrossRef]
- Ly, T.-D.; Wolny, M.; Lindenkamp, C.; Birschmann, I.; Hendig, D.; Knabbe, C.; Faust-Hinse, I. The human myofibroblast marker xylosyltransferase-I: A new indicator for macrophage polarization. Biomedicines 2022, 10, 2869. [Google Scholar] [CrossRef] [PubMed]
- Prante, C.; Bieback, K.; Funke, C.; Schön, S.; Kern, S.; Kuhn, J.; Gastens, M.; Kleesiek, K.; Götting, C. The formation of extracellular matrix during chondrogenic differentiation of mesenchymal stem cells correlates with increased levels of xylosyltransferase I. Stem Cells 2006, 24, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, R.W.H.; Lam, Y.; Chan, W.C.W.; Chan, D. Hypertrophic chondrocytes at the junction of musculoskeletal structures. Bone Rep. 2023, 19, 101698. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Park, K.-H. Regulation and function of SOX9 during cartilage development and regeneration. Semin. Cancer Biol. 2020, 67, 12–23. [Google Scholar] [CrossRef]
- Taieb, M.; Ghannoum, D.; Barré, L.; Ouzzine, M. Xylosyltransferase I mediates the synthesis of proteoglycans with long glycosaminoglycan chains and controls chondrocyte hypertrophy and collagen fibers organization of in the growth plate. Cell Death Dis. 2023, 14, 355. [Google Scholar] [CrossRef]
- Jamsheer, A.; Olech, E.M.; Kozłowski, K.; Niedziela, M.; Sowińska-Seidler, A.; Obara-Moszyńska, M.; Latos-Bieleńska, A.; Karczewski, M.; Zemojtel, T. Exome sequencing reveals two novel compound heterozygous XYLT1 mutations in a Polish patient with Desbuquois dysplasia type 2 and growth hormone deficiency. J. Hum. Genet. 2016, 61, 577–583. [Google Scholar] [CrossRef]
- Özdemir, M.; Kavak, R.P. Radiographic findings of Desbuquois dysplasia. BJR Case Rep. 2021, 7, 20200137. [Google Scholar] [CrossRef]
- Piwar, H.; Ordak, M.; Bujalska-Zadrozny, M. Clinical and genetic insights into Desbuquois dysplasia: Review of 111 case reports. Int. J. Mol. Sci. 2024, 25, 9700. [Google Scholar] [CrossRef]
- Chawla, S.; Mainardi, A.; Majumder, N.; Dönges, L.; Kumar, B.; Occhetta, P.; Martin, I.; Egloff, C.; Ghosh, S.; Bandyopadhyay, A.; et al. Chondrocyte hypertrophy in osteoarthritis: Mechanistic studies and models for the identification of new therapeutic strategies. Cells 2022, 11, 4034. [Google Scholar] [CrossRef]
- Komori, T. Molecular mechanism of Runx2-dependent bone development. Mol. Cells 2020, 43, 168–175. [Google Scholar] [CrossRef]
- Venkatesan, N.; Barré, L.; Bourhim, M.; Magdalou, J.; Mainard, D.; Netter, P.; Fournel-Gigleux, S.; Ouzzine, M. Xylosyltransferase-I regulates glycosaminoglycan synthesis during the pathogenic process of human osteoarthritis. PLoS ONE 2012, 7, e34020. [Google Scholar] [CrossRef]
- Holm, E.; Gleberzon, J.S.; Liao, Y.; Sørensen, E.S.; Beier, F.; Hunter, G.K.; Goldberg, H.A. Osteopontin mediates mineralization and not osteogenic cell development in vitro. Biochem. J. 2014, 464, 355–364. [Google Scholar] [CrossRef]
- Rendina-Ruedy, E.; Guntur, A.R.; Rosen, C.J. Intracellular lipid droplets support osteoblast function. Adipocyte 2017, 6, 250–258. [Google Scholar] [CrossRef]
- Chen, Q.; Shou, P.; Zhang, L.; Xu, C.; Zheng, C.; Han, Y.; Li, W.; Huang, Y.; Zhang, X.; Shao, C.; et al. An osteopontin-integrin interaction plays a critical role in directing adipogenesis and osteogenesis by mesenchymal stem cells. Stem Cells 2014, 32, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Sivasami, P.; Poudel, N.; Munteanu, M.C.; Hudson, J.; Lovern, P.; Liu, L.; Griffin, T.; Hinsdale, M.E. Adipose tissue loss and lipodystrophy in xylosyltransferase II deficient mice. Int. J. Obes. 2019, 43, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Wilsie, L.C.; Chanchani, S.; Navaratna, D.; Orlando, R.A. Cell surface heparan sulfate proteoglycans contribute to intracellular lipid accumulation in adipocytes. Lipids Health Dis. 2005, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Kalita, M.; Villanueva-Meyer, J.; Ohkawa, Y.; Kalyanaraman, C.; Chen, K.; Mohamed, E.; Parker, M.F.L.; Jacobson, M.P.; Phillips, J.J.; Evans, M.J.; et al. Synthesis and screening of α-xylosides in human glioblastoma cells. Mol. Pharm. 2021, 18, 451–460. [Google Scholar] [CrossRef]
- Jernås, M.; Palming, J.; Sjöholm, K.; Jennische, E.; Svensson, P.-A.; Gabrielsson, B.G.; Levin, M.; Sjögren, A.; Rudemo, M.; Lystig, T.C.; et al. Separation of human adipocytes by size: Hypertrophic fat cells display distinct gene expression. FASEB J. 2006, 20, 1540–1542. [Google Scholar] [CrossRef]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef]
- Nagai, N.; Shioiri, T.; Hatano, S.; Sugiura, N.; Watanabe, H. Regulatory role of heparan sulfate in leptin signaling. Cell. Signal. 2024, 124, 111456. [Google Scholar] [CrossRef]
- Zhang, Y.; Dallner, O.S.; Nakadai, T.; Fayzikhodjaeva, G.; Lu, Y.-H.; Lazar, M.A.; Roeder, R.G.; Friedman, J.M. A noncanonical PPARγ/RXRα-binding sequence regulates leptin expression in response to changes in adipose tissue mass. Proc. Natl. Acad. Sci. USA 2018, 115, E6039–E6047. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shao, Y.Y.; Ballock, R.T. Leptin antagonizes peroxisome proliferator-activated receptor-γ signaling in growth plate chondrocytes. PPAR Res. 2012, 2012, 756198. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, E.; Buetti-Dinh, A.; Licandro, G.; Ahmad, P.; Basoli, V.; Alini, M.; Stoddart, M.J. Dexamethasone induces changes in osteogenic differentiation of human mesenchymal stromal cells via SOX9 and PPARG, but not RUNX2. Int. J. Mol. Sci. 2021, 22, 4785. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xu, Y.; Wang, Y.; Jiang, Y. Differential expression profiles of long noncoding RNAs and mRNAs in human bone marrow mesenchymal stem cells after exposure to a high dosage of dexamethasone. Stem Cell Res. Ther. 2021, 12, 9. [Google Scholar] [CrossRef]
- Banas, K.; Rivera-Torres, N.; Bialk, P.; Yoo, B.-C.; Kmiec, E.B. Kinetics of nuclear uptake and site-specific DNA cleavage during CRISPR-directed gene editing in solid tumor cells. Mol. Cancer Res. 2020, 18, 891–902. [Google Scholar] [CrossRef]
- Ly, T.-D.; Kleine, A.; Fischer, B.; Schmidt, V.; Hendig, D.; Kuhn, J.; Knabbe, C.; Faust, I. Identification of putative non-substrate-based XT-I inhibitors by natural product library screening. Biomolecules 2020, 10, 1467. [Google Scholar] [CrossRef]
- Ly, T.-D.; Lindenkamp, C.; Kara, E.; Schmidt, V.; Kleine, A.; Fischer, B.; Hendig, D.; Knabbe, C.; Faust-Hinse, I. The impact of inflammatory stimuli on xylosyltransferase-I regulation in primary human dermal fibroblasts. Biomedicines 2022, 10, 1451. [Google Scholar] [CrossRef]
- Gary, R.K.; Kindell, S.M. Quantitative assay of senescence-associated β-galactosidase activity in mammalian cell extracts. Anal. Biochem. 2005, 343, 329–334. [Google Scholar] [CrossRef]
- Schmidt, V.; Ohmes, J.; Ly, T.-D.; Fischer, B.; Kleine, A.; Knabbe, C.; Faust-Hinse, I. Human Xylosyltransferase I—An important linker between acute senescence and fibrogenesis. Biomedicines 2023, 11, 460. [Google Scholar] [CrossRef]
- Scott, J.E.; Dorling, J. Differential staining of acid glycosaminoglycans (mucopolysaccharides) by Alcian blue in salt solutions. Histochemie 1965, 5, 221–233. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lot Number | Donor Age | Sex | Designation | Race | Tissue/Localization | Surface Markers * |
|---|---|---|---|---|---|---|
| 465Z016 | 63 | male | #1 | Caucasian | Human bone marrow/femoral head | CD73+, CD90+, CD105+, CD14−, CD34−, CD45−, CD19−, HLA-DR− |
| 467Z023.5 | 68 | |||||
| 466Z020 | 44 | female | #2 | |||
| 451Z012.3 | 66 |
| Gene | 5′ to 3′ Sequence | TA [°C] | Efficiency |
|---|---|---|---|
| ACAN | CACCCCATGCAATTTGAG GCCACTGTGCCCTTTTTA | 63 | 2.00 |
| B2M | TGTGCTCGCGCTACTCTCTCTT CGGATGGATGAAACCCAGACA | 63 | 1.87 |
| BGLAP | CGCCTGGGTCTCTTCACTAC CTCACACTCCTCGCCCTATT | 66 | 1.84 |
| CDKN1A | GCTTCATGCCAGCTAACTTCC CCCTTCAAAGTGCCATCTGT | 66 | 2.00 |
| COL1A1 | GATGTGCCACTCTGACT GGGTTCTTGCTGATG | 63 | 1.74 |
| COL2A1 | GGGCTCCCGCAAGAA GCAGGCGTAGGAAGGTCA | 63 | 1.84 |
| GAPDH | AGGTCGGAGTCAACGGAT TCCTGGAAGATGGTGATG | 59 | 1.83 |
| HMBS | CTGCCAGAGAAGAGTGTG AGCTGTTGCCAGGATGAT | 63 | 1.92 |
| MMP13 | AGCTGGACTCATTGTCGGGC AGGTAGCGCTCTGCAAACTGG | 63 | 1.75 |
| OPG | CGGGAAAGAAAGTGGGAGCAG CTTCAAGGTGTCTTGGTCGCCAT | 63 | 1.99 |
| RPL13A | CGGAAGGTGGTGGTCGTA CTCGGGAAGGGTTGGTGT | 63 | 1.87 |
| RUNX2 | AGAAGGCACAGACAGAAGCTTGA AGGAATGCGCCCTAAATCACT | 61 | 1.82 |
| SOX9 | TACCCGCACTTGCACAAC TCTCGCTCTCGTTCAGAAGTC | 63 | 2.00 |
| SPP1 | TGATGACCATGTGGACAG ACCATTCAACTCCTCGCT | 61 | 1.90 |
| TP53 | AGATAGCGATGGTCTGGC TTGGGCAGTGCTCGCTTAGT | 63 | 2.00 |
| XYLT1 | GAAGCCGTGGTGAATCAG CGGTCAGCAAGGAAGTAG | 63 | 2.00 |
| XYLT2 | ACACAGATGACCCGCTTGTGG TTGGTGACCCGCAGGTTGTTG | 63 | 1.95 |
| YWHAZ | CTCCCGTTTCCGAGCCATAA AAGATGACCTACGGGCTCCT | 63 | 2.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ly, T.-D.; Schmidt, V.; Kühle, M.; Böker, K.O.; Fischer, B.; Knabbe, C.; Faust-Hinse, I. XYLT1 Deficiency of Human Mesenchymal Stem Cells: Impact on Osteogenic, Chondrogenic, and Adipogenic Differentiation. Int. J. Mol. Sci. 2025, 26, 7363. https://doi.org/10.3390/ijms26157363
Ly T-D, Schmidt V, Kühle M, Böker KO, Fischer B, Knabbe C, Faust-Hinse I. XYLT1 Deficiency of Human Mesenchymal Stem Cells: Impact on Osteogenic, Chondrogenic, and Adipogenic Differentiation. International Journal of Molecular Sciences. 2025; 26(15):7363. https://doi.org/10.3390/ijms26157363
Chicago/Turabian StyleLy, Thanh-Diep, Vanessa Schmidt, Matthias Kühle, Kai Oliver Böker, Bastian Fischer, Cornelius Knabbe, and Isabel Faust-Hinse. 2025. "XYLT1 Deficiency of Human Mesenchymal Stem Cells: Impact on Osteogenic, Chondrogenic, and Adipogenic Differentiation" International Journal of Molecular Sciences 26, no. 15: 7363. https://doi.org/10.3390/ijms26157363
APA StyleLy, T.-D., Schmidt, V., Kühle, M., Böker, K. O., Fischer, B., Knabbe, C., & Faust-Hinse, I. (2025). XYLT1 Deficiency of Human Mesenchymal Stem Cells: Impact on Osteogenic, Chondrogenic, and Adipogenic Differentiation. International Journal of Molecular Sciences, 26(15), 7363. https://doi.org/10.3390/ijms26157363