
Junctional Epidermolysis Bullosa Caused by a Hemiallelic Nonsense Mutation in LAMA3 Revealed by 18q11.2 Microdeletion

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
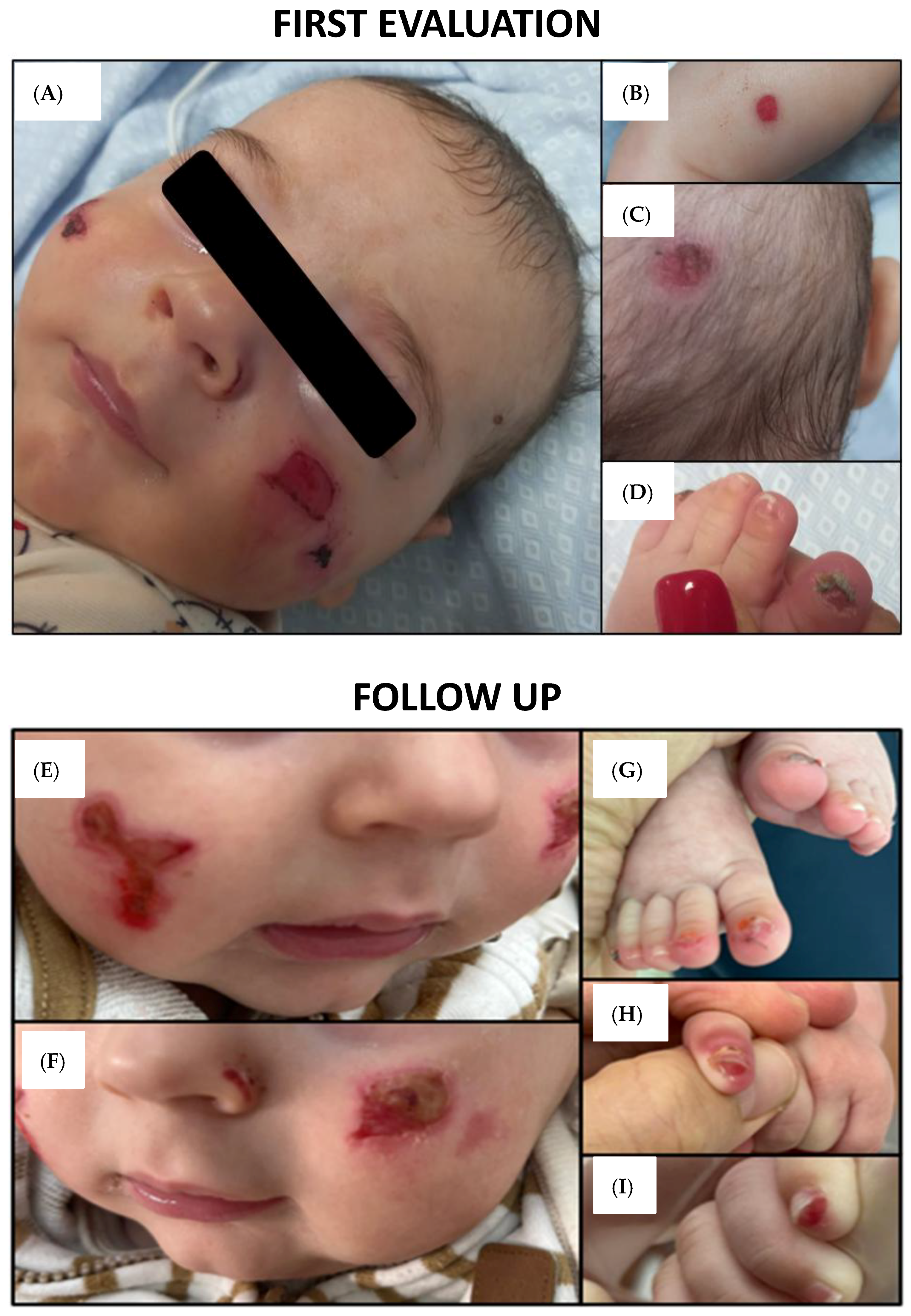
2.1. Clinical Features and Findings
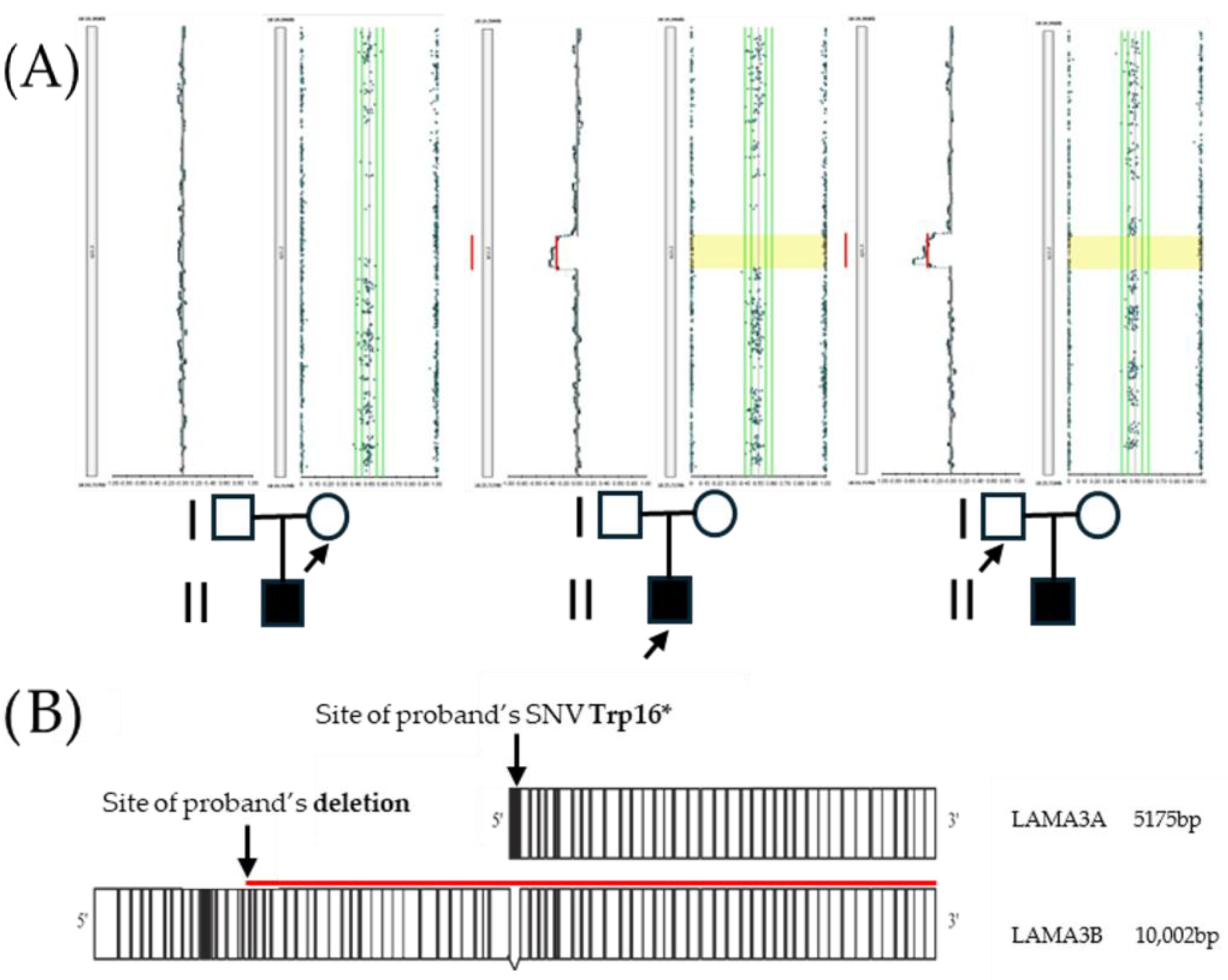
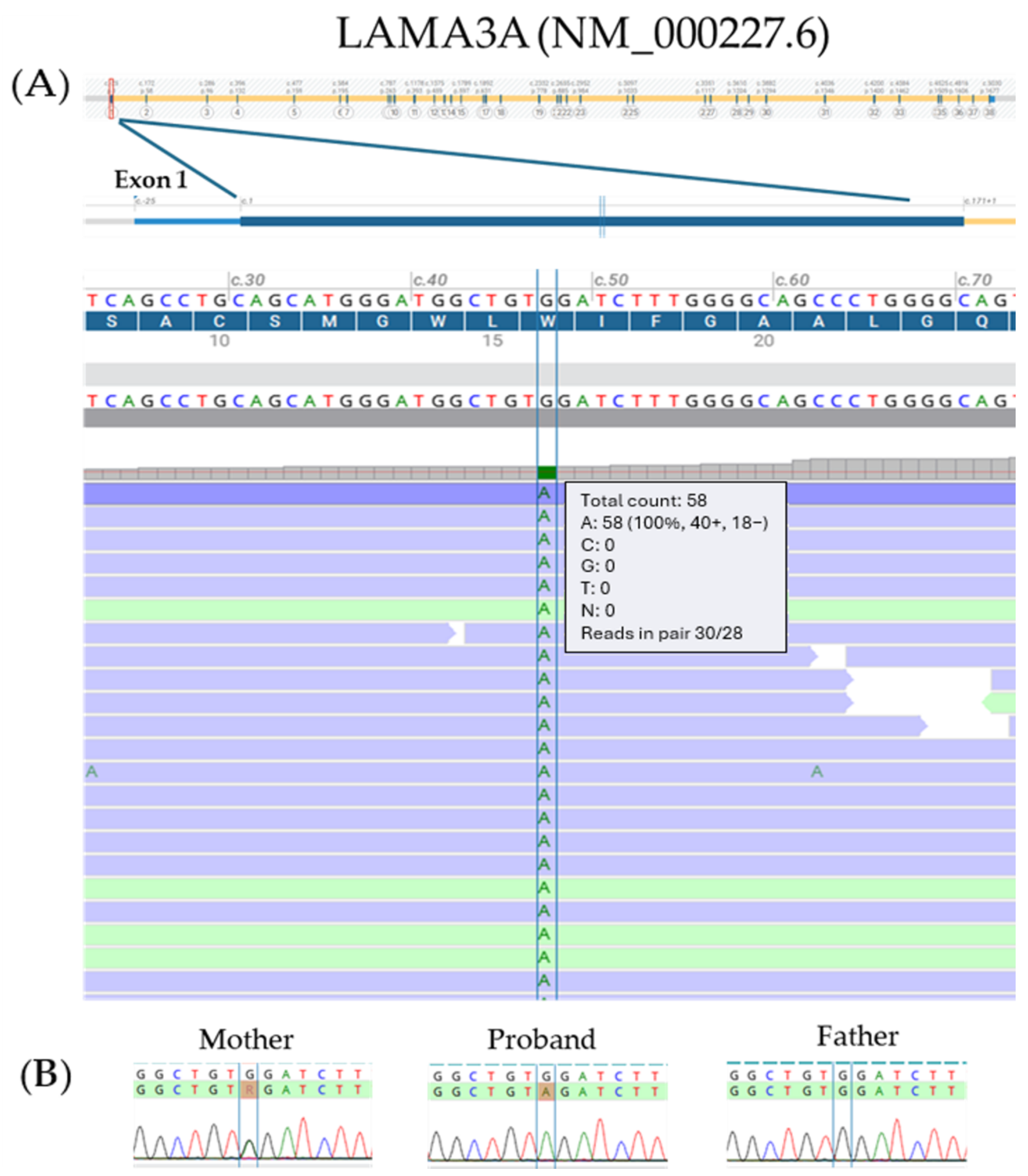
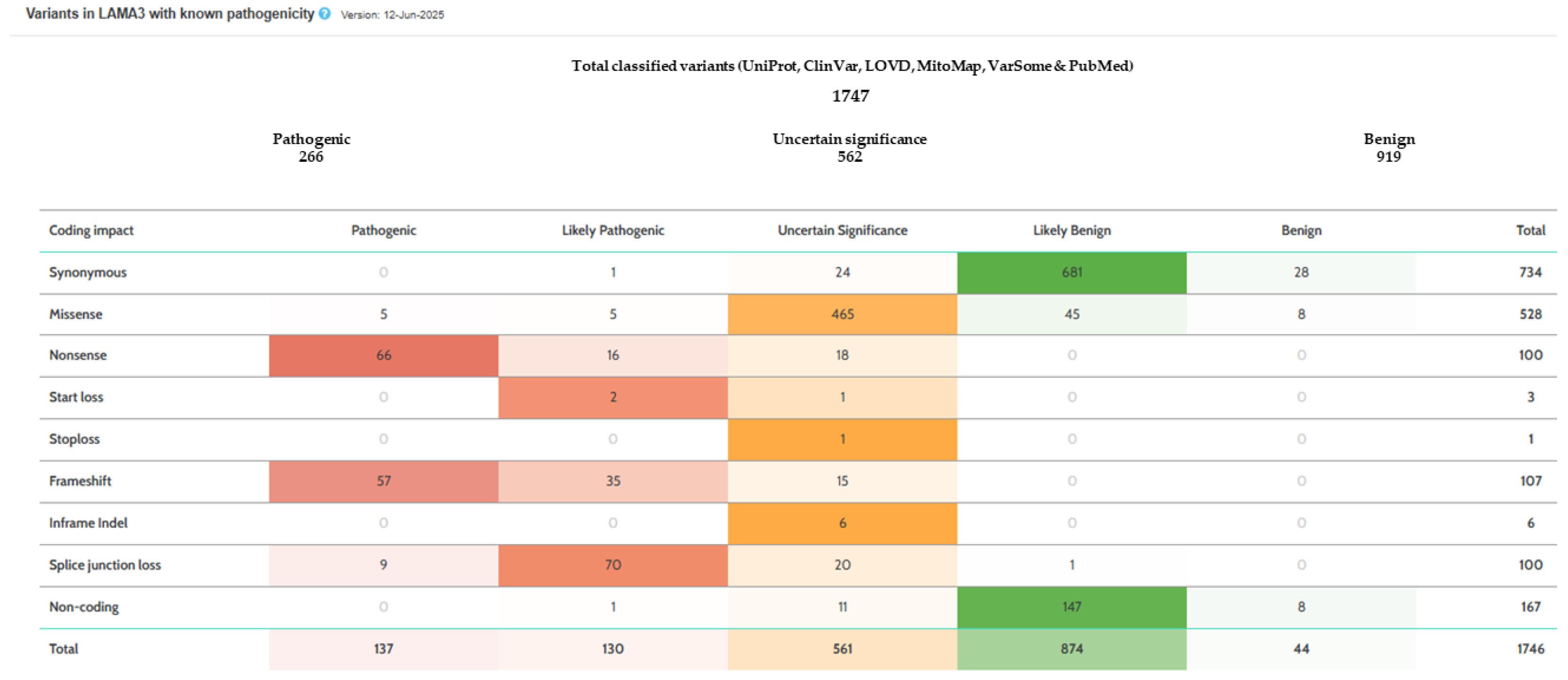
2.2. Molecular Analysis
3. Discussion
4. Materials and Methods
4.1. Patient and Family Members
4.2. Genomic DNA (gDNA) Sample Extraction
4.3. Clinical Exome Sequencing (CES)
4.4. Sanger Sequencing Analysis
4.5. SNP Array Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonamonte, D.; Filoni, A.; De Marco, A.; Lospalluti, L.; Nacchiero, E.; Ronghi, V.; Colagrande, A.; Giudice, G.; Cazzato, G. Squamous Cell Carcinoma in Patients with Inherited Epidermolysis Bullosa: Review of Current Literature. Cells 2022, 11, 1365. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, A.; Bruckner-Tuderman, L.; Chapple, I.L.C.; Fine, J.D.; Harper, N.; Has, C.; Magin, T.M.; Marinkovich, M.P.; Marshall, J.F.; McGrath, J.A.; et al. Epidermolysis Bullosa. Nat. Rev. Dis. Primers 2020, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Filoni, A.; Cicco, G.; Lospalluti, L.; Maglietta, A.; Foti, C.; Annichiarico, G.; Resta, L.; Bonamonte, D. Morphological and Morphometric Analysis of Cutaneous Squamous Cell Carcinoma in Patients with Recessive Dystrophic Epidermolysis Bullosa: A Retrospective Study. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1707–1714. [Google Scholar] [CrossRef]
- El Hachem, M.; Diociaiuti, A.; Bonamonte, D.; Brena, M.; Lospalluti, L.; Magnoni, C.; Neri, I.; Peris, K.; Tadini, G.; Zambruno, G.; et al. Taking Care of Patients with Recessive Dystrophic Epidermolysis Bullosa from Birth to Adulthood: A Multidisciplinary Italian Delphi Consensus. Orphanet J. Rare Dis. 2025, 20, 128. [Google Scholar] [CrossRef]
- Wen, D.; Hunjan, M.; Bardhan, A.; Harper, N.; Ogboli, M.; Ozoemena, L.; Liu, L.; Fine, J.D.; Chapple, I.; Balacco, D.L.; et al. Genotype–Phenotype Correlation in Junctional Epidermolysis Bullosa: Signposts to Severity. J. Investig. Dermatol. 2024, 144, 1334–1343.e14. [Google Scholar] [CrossRef]
- Prodinger, C.; Chottianchaiwat, S.; Mellerio, J.E.; McGrath, J.A.; Ozoemena, L.; Liu, L.; Moore, W.; Laimer, M.; Petrof, G.; Martinez, A.E. The Natural History of Laryngo-Onycho-Cutaneous Syndrome: A Case Series of Six Pediatric Patients and Literature Review. Pediatr. Dermatol. 2021, 38, 1094–1101. [Google Scholar] [CrossRef]
- Has, C.; Nyström, A. Epidermal Basement Membrane in Health and Disease. Curr. Top. Membr. 2015, 76, 117–170. [Google Scholar] [CrossRef]
- Wang, R.; Sun, L.; Habulieti, X.; Liu, J.; Guo, K.; Yang, X.; Ma, D.; Zhang, X. Novel Variants in LAMA3 and COL7A1 and Recurrent Variant in KRT5 Underlying Epidermolysis Bullosa in Five Chinese Families. Front. Med. 2022, 16, 808–814. [Google Scholar] [CrossRef]
- McLean, W.H.I.; Irvine, A.D.; Hamill, K.J.; Whittock, N.V.; Coleman-Campbell, C.M.; Mellerio, J.E.; Ashton, G.S.; Dopping-Hepenstal, P.J.H.; Eady, R.A.J.; Jamil, T.; et al. An Unusual N-Terminal Deletion of the Laminin A3a Isoform Leads to the Chronic Granulation Tissue Disorder Laryngo-Onycho-Cutaneous Syndrome. Hum. Mol. Genet 2003, 12, 2395–2409. [Google Scholar] [CrossRef]
- Mosallaei, D.; Hao, M.; Antaya, R.J.; Levian, B.; Kwong, A.; Cogan, J.; Hamilton, C.; Schwieger-Briel, A.; Tan, C.; Tang, X.; et al. Molecular and Clinical Outcomes After Intravenous Gentamicin Treatment for Patients With Junctional Epidermolysis Bullosa Caused by Nonsense Variants. JAMA Dermatol. 2022, 158, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin Induces Functional Type VII Collagen in Recessive Dystrophic Epidermolysis Bullosa Patients. J. Clin. Investig. 2017, 127, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, V.; Cogan, J.; Hou, Y.; Hirsch, M.; Hao, M.; Alexeev, V.; De Luca, M.; De Rosa, L.; Bauer, J.W.; Woodley, D.T.; et al. Gentamicin Induces LAMB3 Nonsense Mutation Readthrough and Restores Functional Laminin 332 in Junctional Epidermolysis Bullosa. Proc. Natl. Acad. Sci. USA 2018, 115, E6536–E6545. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Jiang, Q.; Prisco, M.; Gruber, C.; Piñón Hofbauer, J.; Chen, M.; Has, C.; Bruckner-Tuderman, L.; McGrath, J.A.; Uitto, J.; et al. Amlexanox Enhances Premature Termination Codon Read-Through in COL7A1 and Expression of Full Length Type VII Collagen: Potential Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2017, 137, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.; Cogan, J.; Hou, Y.; Antaya, R.; Hao, M.; Kim, G.; Lincoln, V.; Chen, Q.; Woodley, D.T.; Chen, M. Gentamicin Induces Laminin 332 and Improves Wound Healing in Junctional Epidermolysis Bullosa Patients with Nonsense Mutations. Mol. Ther. 2020, 28, 1327–1338. [Google Scholar] [CrossRef]
- Turczynski, S.; Titeux, M.; Pironon, N.; Hovnanian, A. Antisense-Mediated Exon Skipping to Reframe Transcripts. Methods Mol. Biol. 2012, 867, 221–238. [Google Scholar] [CrossRef]
- Maier, K.; He, Y.; Esser, P.R.; Thriene, K.; Sarca, D.; Kohlhase, J.; Dengjel, J.; Martin, L.; Has, C. Single Amino Acid Deletion in Kindlin-1 Results in Partial Protein Degradation Which Can Be Rescued by Chaperone Treatment. J. Investig. Dermatol. 2016, 136, 920–929. [Google Scholar] [CrossRef]
- Condrat, I.; He, Y.; Cosgarea, R.; Has, C. Junctional Epidermolysis Bullosa: Allelic Heterogeneity and Mutation Stratification for Precision Medicine. Front. Med. 2019, 6, 363. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly, Ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020; ISBN 9781491975190. [Google Scholar]
- Gargano, M.A.; Matentzoglu, N.; Coleman, B.; Addo-Lartey, E.B.; Anagnostopoulos, A.V.; Anderton, J.; Avillach, P.; Bagley, A.M.; Bakštein, E.; Balhoff, J.P.; et al. The Human Phenotype Ontology in 2024: Phenotypes around the World. Nucleic Acids Res. 2024, 52, D1333–D1346. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020) Reprint of “Cytogenetic and Genome Research 2020, Vol. 160, No. 7-8”; S. Karger AG: Basel, Switzerland, 2020; ISBN 978-3-318-06706-4. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacoviello, M.; Piglionica, M.; Tabaku, O.; Garganese, A.; De Marco, A.; Cardinale, F.; Bonamonte, D.; Resta, N. Junctional Epidermolysis Bullosa Caused by a Hemiallelic Nonsense Mutation in LAMA3 Revealed by 18q11.2 Microdeletion. Int. J. Mol. Sci. 2025, 26, 7343. https://doi.org/10.3390/ijms26157343
Iacoviello M, Piglionica M, Tabaku O, Garganese A, De Marco A, Cardinale F, Bonamonte D, Resta N. Junctional Epidermolysis Bullosa Caused by a Hemiallelic Nonsense Mutation in LAMA3 Revealed by 18q11.2 Microdeletion. International Journal of Molecular Sciences. 2025; 26(15):7343. https://doi.org/10.3390/ijms26157343
Chicago/Turabian StyleIacoviello, Matteo, Marilidia Piglionica, Ornella Tabaku, Antonella Garganese, Aurora De Marco, Fabio Cardinale, Domenico Bonamonte, and Nicoletta Resta. 2025. "Junctional Epidermolysis Bullosa Caused by a Hemiallelic Nonsense Mutation in LAMA3 Revealed by 18q11.2 Microdeletion" International Journal of Molecular Sciences 26, no. 15: 7343. https://doi.org/10.3390/ijms26157343
APA StyleIacoviello, M., Piglionica, M., Tabaku, O., Garganese, A., De Marco, A., Cardinale, F., Bonamonte, D., & Resta, N. (2025). Junctional Epidermolysis Bullosa Caused by a Hemiallelic Nonsense Mutation in LAMA3 Revealed by 18q11.2 Microdeletion. International Journal of Molecular Sciences, 26(15), 7343. https://doi.org/10.3390/ijms26157343