
Fertility Protection in Female Cancer Patients: From Molecular Mechanisms of Gonadotoxic Therapies to Pharmacotherapeutic Possibilities

Abstract
1. Introduction
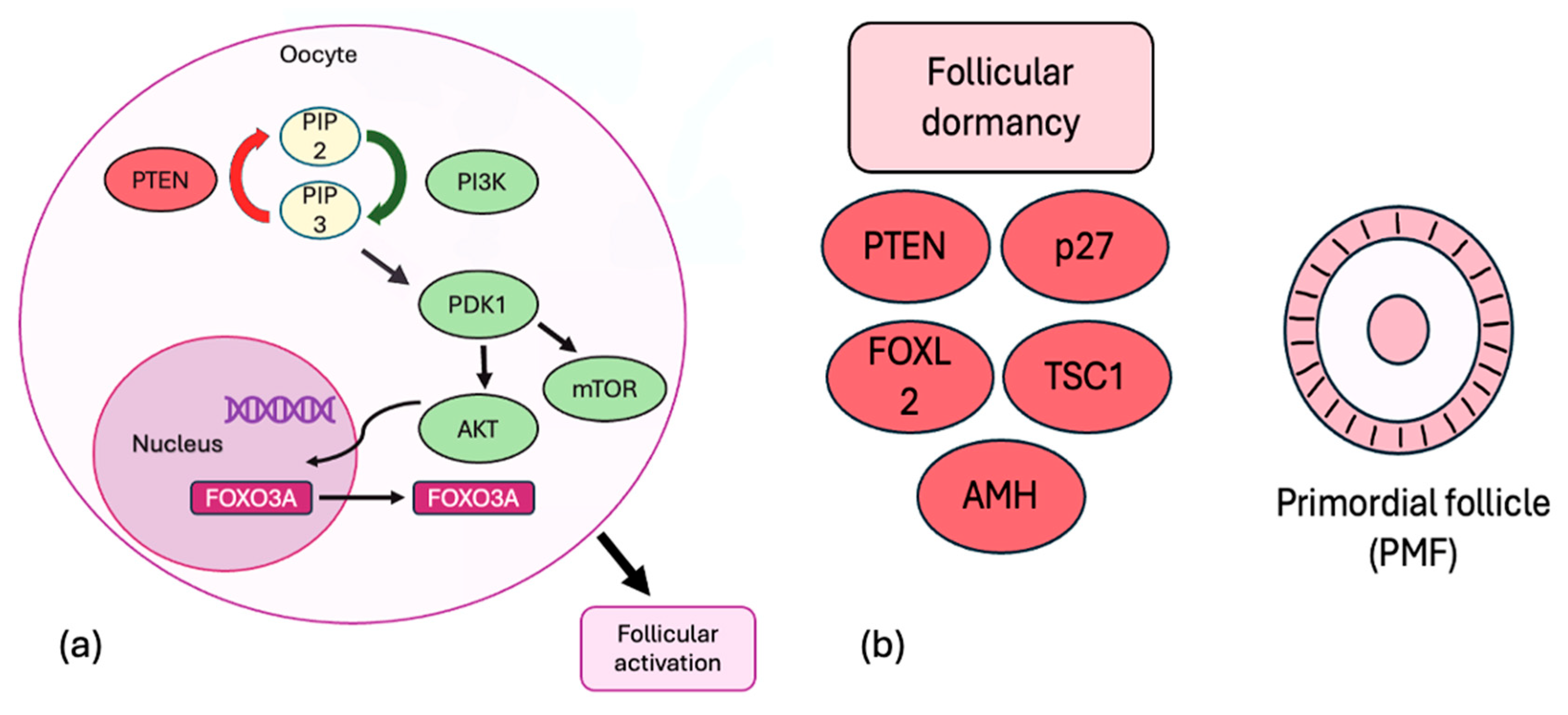
2. Regulation of Ovarian Reserve
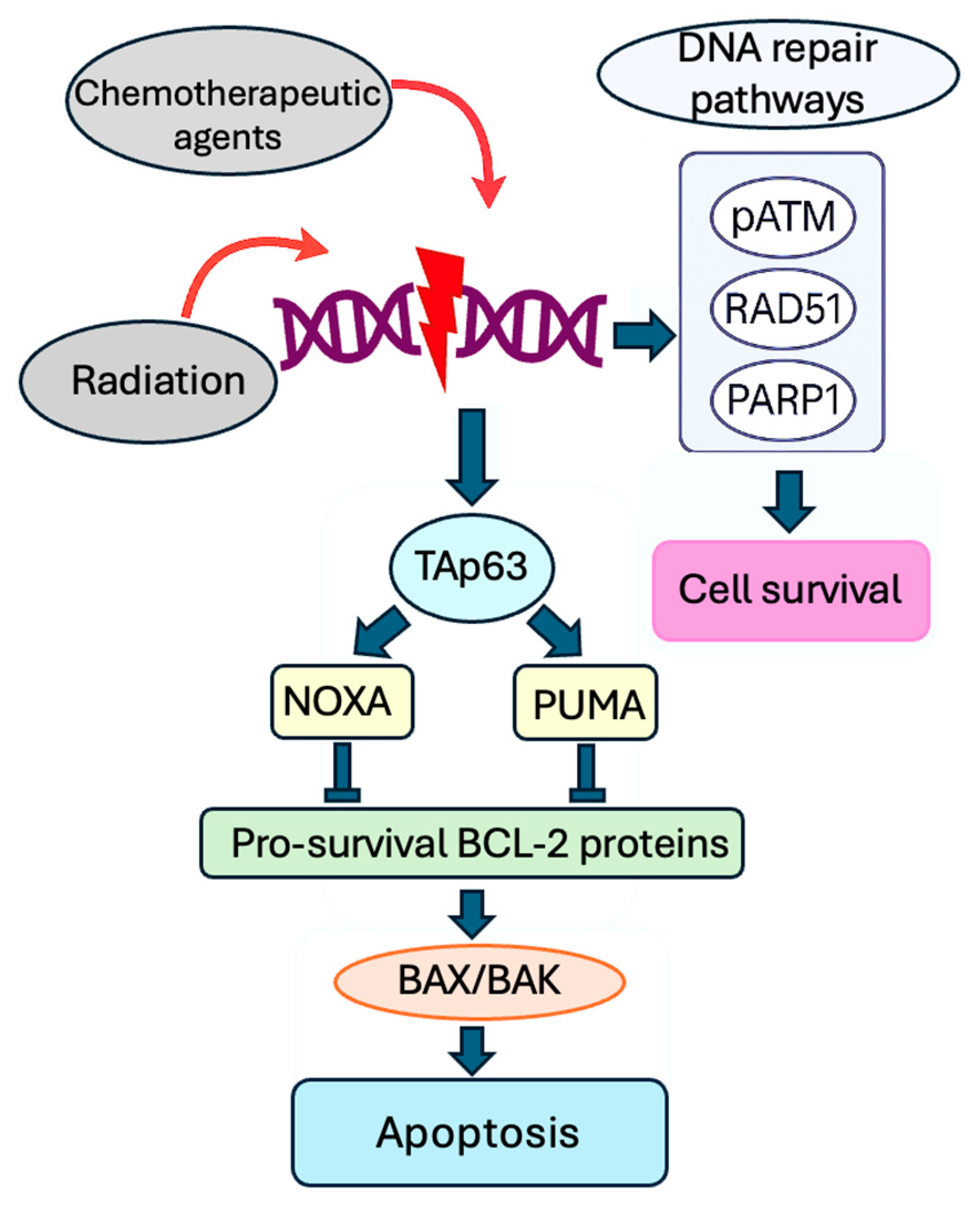
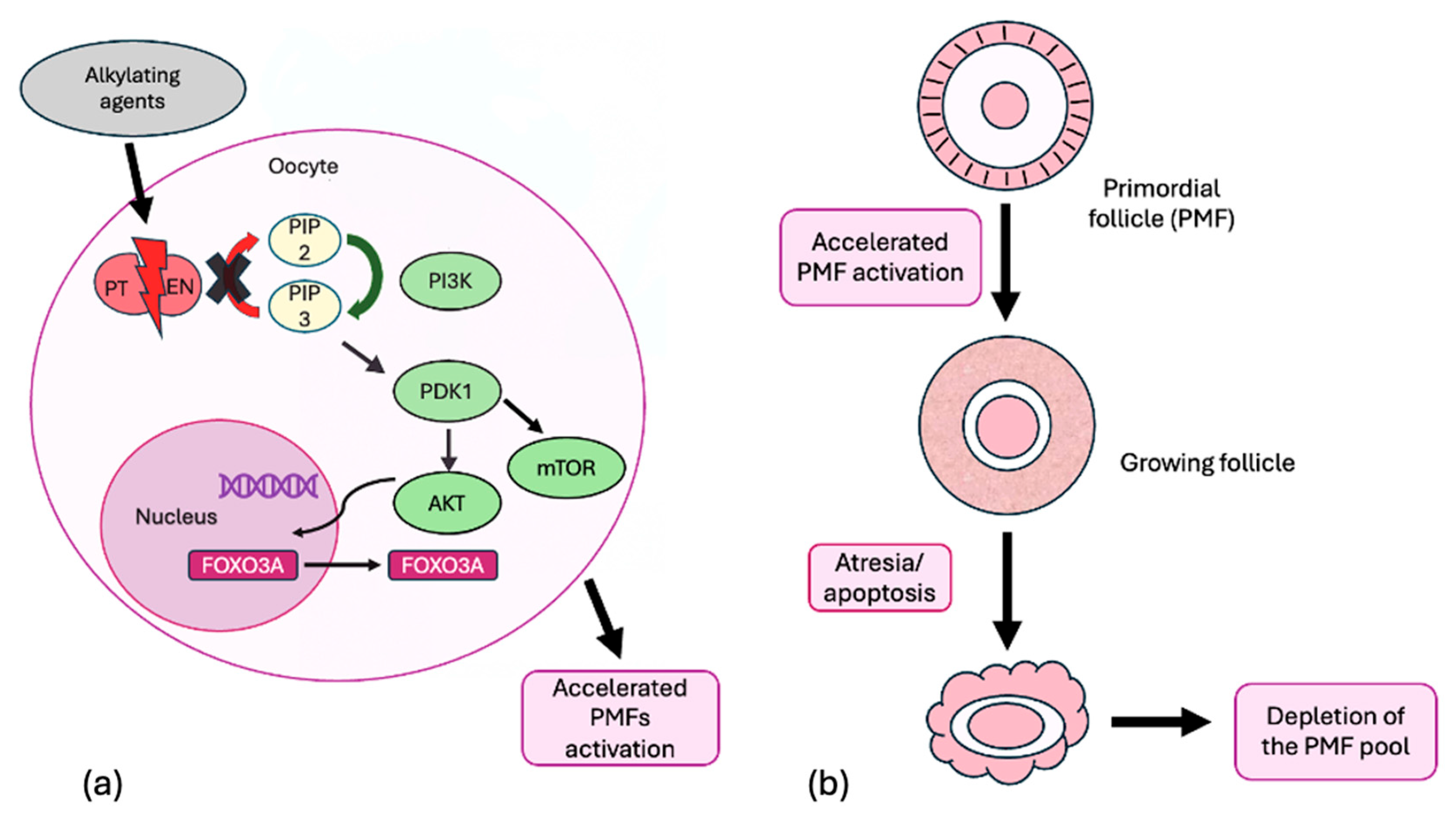
3. Molecular Mechanism of Gonadotoxicity of Radiation and Chemotherapy
4. Fertoprotective Agents
- Gonadotropin-releasing hormone (GnRH)
- Anti-Müllerian hormone (AMH)
- Melatonin
- Imatinib
- Granulocyte colony-stimulating factor (G-CSF)
- Sphingosine-1-phosphate (S1P) and ceramide-1-phosphate (C1P)
- mTOR Inhibitors
- AS101
- Luteinizing hormone (LH)
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2018. Available online: https://seer.cancer.gov/csr/1975_2016 (accessed on 9 April 2020).
- Geenen, M.M.; Cardous-Ubbink, M.C.; Kremer, L.C.; van den Bos, C.; van der Pal, H.J.; Heinen, R.C.; Jaspers, M.W.; Koning, C.C.; Oldenburger, F.; Langeveld, N.E.; et al. Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA 2007, 297, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Sklar, C.A.; Mertens, A.C.; Mitby, P.; Whitton, J.; Stovall, M.; Kasper, C.; Mulder, J.; Green, D.; Nicholson, H.S.; Yasui, Y.; et al. Premature menopause in survivors of childhood cancer: A report from the childhood cancer survivor study. J. Natl. Cancer Inst. 2006, 98, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Ogilvy-Stuart, A.L.; Shalet, S.M. Effect of radiation on the human reproductive system. Environ. Health Perspect. 1993, 101 (Suppl. S2), 109–116. [Google Scholar] [PubMed]
- Norwitz, E.R.; Stern, H.M.; Grier, H.; Lee-Parritz, A. Placenta percreta and uterine rupture associated with prior whole body radiation therapy. Obstet. Gynecol. 2001, 98, 929–931. [Google Scholar]
- Müller, J. Impact of Cancer therapy on the reproductive Axis. Horm. Res. Paediatr. 2003, 59 (Suppl. S1), 12–20. [Google Scholar] [CrossRef]
- Rose, S.R.; Schreiber, R.E.; Kearney, N.S.; Lustig, R.H.; Danish, R.K.; Burghen, G.A.; Hudson, M.M. Hypothalamic dysfunction after chemotherapy. J. Pediatr. Endocrinol. Metab. JPEM 2004, 17, 55–66. [Google Scholar] [CrossRef]
- Wallace, W.H.; Kelsey, T.W. Human ovarian reserve from conception to the menopause. PLoS ONE 2010, 5, e8772. [Google Scholar] [CrossRef]
- Meirow, D.; Biederman, H.; Anderson, R.A.; Wallace, W.H.B. Toxicity of chemotherapy and radiation on female reproduction. Clin. Obstet. Gynecol. 2010, 53, 727–739. [Google Scholar] [CrossRef]
- Bedoschi, G.; Navarro, P.A.; Oktay, K. Chemotherapy induced damage to ovary: Mechanisms and clinical impact. Future Oncol. Lond. Engl. 2016, 12, 2333–2344. [Google Scholar] [CrossRef]
- Jacobson, M.H.; Mertens, A.C.; Spencer, J.B.; Manatunga, A.K.; Howards, P.P. Menses resumption after cancer treatment-induced amenorrhea occurs early or not at all. Fertil. Steril. 2016, 105, 765–772. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Webber, L.; Davies, M.; Anderson, R.; Bartlett, J.; Braat, D.; Cartwright, B.; Cifkova, R.; de Muinck Keizer-Schrama, S.; Hogervorst, E.; Janse, F.; et al. ESHRE Guideline: Management of women with premature ovarian insufficiency. Hum. Reprod. 2016, 31, 926–937. [Google Scholar] [CrossRef] [PubMed]
- ESHRE Guideline Group on Female Fertility Preservation; Anderson, R.A.; Amant, F.; Braat, D.; D’Angelo, A.; Chuva de Sousa Lopes, S.M.; Demeestere, I.; Dwek, S.; Frith, L.; Lambertini, M.; et al. ESHRE guideline: Female fertility preservation. Hum. Reprod. Open 2020, 2020, hoaa052. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yeh, J. Onco-fertility and personalized testing for potential for loss of ovarian reserve in patients undergoing chemotherapy: Proposed next steps for development of genetic testing to predict changes in ovarian reserve. Fertil. Res. Pract. 2021, 7, 13. [Google Scholar] [CrossRef]
- Schover, L.R.; Rybicki, L.A.; Martin, B.A.; Bringelsen, K.A. Having children after cancer. Cancer 1999, 86, 697–709. [Google Scholar] [CrossRef]
- Donnez, J.; Dolmans, M.M.; Pellicer, A.; Diaz-Garcia, C.; Sanchez Serrano, M.; Schmidt, K.T.; Ernst, E.; Luyckx, V.; Andersen, C.Y. Restoration of ovarian activity and pregnancy after transplantation of cryopreserved ovarian tissue: A review of 60 cases of reimplantation. Fertil. Steril. 2013, 99, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Khattak, H.; Malhas, R.; Craciunas, L.; Afifi, Y.; Amorim, C.A.; Fishel, S.; Silber, S.; Gook, D.; Demeestere, I.; Bystrova, O.; et al. Fresh and cryopreserved ovarian tissue transplantation for preserving reproductive and endocrine function: A systematic review and individual patient data meta-analysis. Hum. Reprod. Update 2022, 28, 400–416. [Google Scholar] [CrossRef]
- Woodruff, T.K. A win-win for women’s reproductive health: A nonsteroidal contraceptive and fertoprotective neoadjuvant. Proc. Natl. Acad. Sci. USA 2017, 114, 2101–2102. [Google Scholar] [CrossRef]
- Faddy, M.J.; Gosden, R.G.; Gougeon, A.; Richardson, S.J.; Nelson, J.F. Accelerated disappearance of ovarian follicles in mid-life: Implications for forecasting menopause. Hum. Reprod. 1992, 7, 1342–1346. [Google Scholar] [CrossRef]
- Reddy, P.; Zheng, W.; Liu, K. Mechanisms maintaining the dormancy and survival of mammalian primordial follicles. Trends. Endocrinol. Metab. 2010, 21, 96–103. [Google Scholar] [CrossRef]
- Grosbois, J.; Devos, M.; Demeestere, I. Implications of Nonphysiological Ovarian Primordial Follicle Activation for Fertility Preservation. Endocr. Rev. 2020, 41, bnaa020. [Google Scholar] [CrossRef]
- Bhardwaj, J.K.; Paliwal, A.; Saraf, P.; Sachdeva, S.N. Role of autophagy in follicular development and maintenance of primordial follicular pool in the ovary. J. Cell. Physiol. 2022, 237, 1157–1170. [Google Scholar] [CrossRef]
- Song, Z.H.; Yu, H.Y.; Wang, P.; Mao, G.K.; Liu, W.X.; Li, M.N.; Wang, H.N.; Shang, Y.L.; Liu, C.; Xu, Z.L.; et al. Germ cell-specific Atg7 knockout results in primary ovarian insufficiency in female mice. Cell Death Dis. 2015, 6, e1589. [Google Scholar] [CrossRef]
- Gawriluk, T.R.; Hale, A.N.; Flaws, J.A.; Dillon, C.P.; Green, D.R.; Rucker, E.B., 3rd. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 2011, 141, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Dólleman, M.; Verschuren, W.M.; Eijkemans, M.J.; Dollé, M.E.; Jansen, E.H.; Broekmans, F.J.; van der Schouw, Y.T. Reproductive and lifestyle determinants of anti-Müllerian hormone in a large population-based study. J. Clin. Endocrinol. Metab. 2013, 98, 2106–2115. [Google Scholar] [CrossRef]
- Kelsey, T.W.; Hua, C.H.; Wyatt, A.; Indelicato, D.; Wallace, W.H. A predictive model of the effect of therapeutic radiation on the human ovary. PLoS ONE 2022, 17, e0277052. [Google Scholar] [CrossRef]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA damage induced by alkylating agents and repair pathways. J. Nucleic. Acids. 2010, 2010, 543531. [Google Scholar] [CrossRef]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response: Convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta. 2014, 1845, 84–89. [Google Scholar] [CrossRef]
- Winship, A.L.; Stringer, J.M.; Liew, S.H.; Hutt, K.J. The importance of DNA repair for maintaining oocyte quality in response to anti-cancer treatments, environmental toxins and maternal ageing. Hum. Reprod. Update 2018, 24, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, S.W.; Han, S.J.; Lee, S.; Park, H.T.; Song, J.Y.; Kim, T. Molecular mechanism and prevention strategy of chemotherapy- and radiotherapy-induced ovarian damage. Int. J. Mol. Sci. 2021, 22, 7484. [Google Scholar] [CrossRef]
- Kerr, J.B.; Hutt, K.J.; Michalak, E.M.; Cook, M.; Vandenberg, C.J.; Liew, S.H.; Bouillet, P.; Mills, A.; Scott, C.L.; Findlay, J.K.; et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol. Cell 2012, 48, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Bellusci, G.; Mattiello, L.; Iannizzotto, V.; Ciccone, S.; Maiani, E.; Villani, V.; Diederich, M.; Gonfloni, S. Kinase-independent inhibition of cyclophosphamide-induced pathways protects the ovarian reserve and prolongs fertility. Cell Death Dis. 2019, 10, 726. [Google Scholar] [CrossRef]
- Bolcun-Filas, E.; Rinaldi, V.D.; White, M.E.; Schimenti, J.C. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 2014, 343, 533–536. [Google Scholar] [CrossRef]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hötte, K.; Hoffmeister, M.; Schäfer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat. Struct. Mol. Biol. 2018, 25, 261–269. [Google Scholar] [CrossRef]
- Nguyen, Q.N.; Zerafa, N.; Liew, S.H.; Morgan, F.H.; Strasser, A.; Scott, C.L.; Findlay, J.K.; Hickey, M.; Hutt, K.J. Loss of PUMA protects the ovarian reserve during DNA-damaging chemotherapy and preserves fertility. Cell Death Dis. 2018, 9, 618. [Google Scholar] [CrossRef]
- Gebel, J.; Tuppi, M.; Sänger, N.; Schumacher, B.; Dötsch, V. DNA Damaged Induced Cell Death in Oocytes. Molecules 2020, 25, 5714. [Google Scholar] [CrossRef]
- Blum, R.H.; Carter, S.K. Adriamycin. A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1974, 80, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Xia, H.X.; Guan, H.Y.; Li, B.; Zhang, W. Follicle Loss and Apoptosis in Cyclophosphamide-Treated Mice: What’s the Matter? Int. J. Mol. Sci. 2016, 17, 836. [Google Scholar] [CrossRef]
- Lande, Y.; Fisch, B.; Tsur, A.; Farhi, J.; Prag-Rosenberg, R.; Ben-Haroush, A.; Kessler-Icekson, G.; Zahalka, M.A.; Ludeman, S.M.; Abir, R. Short-term exposure of human ovarian follicles to cyclophosphamide metabolites seems to promote follicular activation in vitro. Reprod. Biomed. Online 2017, 34, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, K.J.; Tan, X.; Oktay, K. Unraveling the mechanisms of chemotherapy-induced damage to human primordial follicle reserve: Road to developing therapeutics for fertility preservation and reversing ovarian aging. Mol. Hum. Reprod. 2020, 26, 553–566. [Google Scholar] [CrossRef]
- Meirow, D.; Epstein, M.; Lewis, H.; Nugent, D.; Gosden, R.G. Administration of cyclophosphamide at different stages of follicular maturation in mice: Effects on reproductive performance and fetal malformations. Hum. Reprod. 2001, 16, 632–637. [Google Scholar] [CrossRef]
- Goldman, K.N.; Chenette, D.; Arju, R.; Duncan, F.E.; Keefe, D.L.; Grifo, J.A.; Schneider, R.J. mTORC1/2 inhibition preserves ovarian function and fertility during genotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2017, 114, 3186–3191. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, H.; Gorre, N.; Risal, S.; Shen, Y.; Liu, K. Two classes of ovarian primordial follicles exhibit distinct developmental dynamics and physiological functions. Hum. Mol. Genet. 2014, 23, 920–928. [Google Scholar] [CrossRef]
- Devos, M.; Diaz Vidal, P.; Bouziotis, J.; Anckaert, E.; Dolmans, M.M.; Demeestere, I. Impact of first chemotherapy exposure on follicle activation and survival in human cryopreserved ovarian tissue. Hum. Reprod. 2023, 38, 408–420. [Google Scholar] [CrossRef]
- Titus, S.; Szymanska, K.J.; Musul, B.; Turan, V.; Taylan, E.; Garcia-Milian, R.; Mehta, S.; Oktay, K. Individual-oocyte transcriptomic analysis shows that genotoxic chemotherapy depletes human primordial follicle reserve in vivo by triggering proapoptotic pathways without growth activation. Sci. Rep. 2021, 11, 407. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Erden, M.; Oktay, K.H. Does gonadotoxic chemotherapy deplete the ovarian reserve through activation of primordial follicles? Hum. Reprod. 2025, 40, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, T.; Moravec, R.; Niles, A.; O’Brien, M.; Riss, T.; Corporation, P. Timing your apoptosis assays. Protoc. Appl. Guid. 2006, 16, 18–21. [Google Scholar]
- Oktem, O.; Oktay, K. A novel ovarian xenografting model to characterize the impact of chemotherapy agents on human primordial follicle reserve. Cancer Res. 2007, 67, 10159–10162. [Google Scholar] [CrossRef] [PubMed]
- Roness, H.; Spector, I.; Leichtmann-Bardoogo, Y.; Savino, A.M.; Dereh-Haim, S.; Meirow, D. Pharmacological administration of recombinant human AMH rescues ovarian reserve and preserves fertility in a mouse model of chemotherapy, without interfering with anti-tumoural effects. J. Assist. Reprod. Genet. 2019, 36, 1793–1803. [Google Scholar] [CrossRef]
- Durlinger, A.L.; Kramer, P.; Karels, B.; de Jong, F.H.; Uilenbroek, J.T.; Grootegoed, J.A.; Themmen, A.P. Control of primordial follicle recruitment by anti-Müllerian hormone in the mouse ovary. Endocrinology 1999, 140, 5789–5796. [Google Scholar] [CrossRef]
- Durlinger, A.L.; Gruijters, M.J.; Kramer, P.; Karels, B.; Ingraham, H.A.; Nachtigal, M.W.; Uilenbroek, J.T.; Grootegoed, J.A.; Themmen, A.P. Anti-Müllerian hormone inhibits initiation of primordial follicle growth in the mouse ovary. Endocrinology 2002, 143, 1076–1084. [Google Scholar] [CrossRef]
- Hayes, E.; Kushnir, V.; Ma, X.; Biswas, A.; Prizant, H.; Gleicher, N.; Sen, A. Intra-cellular mechanism of antiMüllerian hormone (AMH) in regulation of follicular development. Mol. Cell. Endocrinol. 2016, 433, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Sosulski, A.E.; Zhang, L.; Saatcioglu, H.D.; Wang, D.; Nagykery, N.; Sabatini, M.E.; Gao, G.; Donahoe, P.K.; Pépin, D. AMH/MIS as a contraceptive that protects the ovarian reserve during chemotherapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1688–E1697. [Google Scholar] [CrossRef] [PubMed]
- Sonigo, C.; Beau, I.; Grynberg, M.; Binart, N. AMH prevents primordial ovarian follicle loss and fertility alteration in cyclophosphamide-treated mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 33, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Lecot-Connan, T.; Boumerdassi, Y.; Magnin, F.; Binart, N.; Kamenický, P.; Sonigo, C.; Beau, I. Anti-Müllerian hormone induces autophagy to preserve the primordial follicle pool in mice. FASEB J. 2024, 38, e23506. [Google Scholar] [CrossRef]
- Roness, H.; Kashi, O.; Meirow, D. Prevention of chemotherapy-induced ovarian damage. Fertil. Steril. 2016, 105, 20–29. [Google Scholar] [CrossRef]
- Man, L.; Lustgarten, N.; Kallinos, E.; Zangi, L.; Arazi, L.; Rosenwaks, Z.; James, D. Administration of modified rna encoding anti-mullerian hormone (modrna-amh) before cyclophosphamide bridges protein loss until resumption of secretion by a critical mass of growing follicles. Fertil. Steril. 2023, 120, e119. [Google Scholar] [CrossRef]
- El-Missiry, M.A.; Othman, A.I.; Alabdan, M.A. Melatonin for protection against ionizing radiation. In Current Topics in Ionizing Radiation Research; Nenoi, M., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef]
- Amer, M.E.; Othman, A.I.; Abozaid, H.M.; El-Missiry, M.A. Utility of melatonin in mitigating ionizing radiation-induced testis injury through synergistic interdependence of its biological properties. Biol. Res. 2022, 55, 33. [Google Scholar] [CrossRef]
- Mills, E.; Wu, P.; Seely, D.; Guyatt, G. Melatonin in the treatment of cancer: A systematic review of randomized controlled trials and meta-analysis. J. Pineal Res. 2005, 39, 360–366. [Google Scholar] [CrossRef]
- Jang, H.; Lee, O.H.; Lee, Y.; Yoon, H.; Chang, E.M.; Park, M.; Lee, J.W.; Hong, K.; Kim, J.O.; Kim, N.K.; et al. Melatonin prevents cisplatin-induced primordial follicle loss via suppression of PTEN/AKT/FOXO3a pathway activation in the mouse ovary. J. Pineal Res. 2016, 60, 336–347. [Google Scholar] [CrossRef]
- Jang, H.; Na, Y.; Hong, K.; Lee, S.; Moon, S.; Cho, M.; Park, M.; Lee, O.H.; Chang, E.M.; Lee, D.R.; et al. Synergistic effect of melatonin and ghrelin in preventing cisplatin-induced ovarian damage via regulation of FOXO3a phosphorylation and binding to the p27Kip1promoter in primordial follicles. J. Pineal Res. 2017, 63, e12432. [Google Scholar] [CrossRef] [PubMed]
- Shedid, S.M.; Abdel-Aziz, N.; Algeda, F.R.; Saada, H.N. The Mitigating Effect of Melatonin Against Radiation-Induced Inflammation and Disturbance of Reproductive Hormones in Female Albino Rats. Dose-Response 2025, 23, 15593258251323796. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kimura, F.; Zheng, L.; Kaku, S.; Takebayashi, A.; Kasahara, K.; Tsuji, S.; Murakami, T. Protective effect of a mechanistic target of rapamycin inhibitor on an in vivo model ofcisplatin-induced ovarian gonadotoxicity. Exp. Anim. 2018, 67, 493–500. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tanaka, Y.; Amano, T.; Nakamura, A.; Yoshino, F.; Takebayashi, A.; Takahashi, A.; Yamanaka, H.; Inatomi, A.; Hanada, T.; Yoneoka, Y.; et al. Rapamycin prevents cyclophosphamide-induced ovarian follicular loss and potentially inhibits tumour proliferation in a breast cancer xenograft mouse model. Hum. Reprod. 2024, 39, 1519–1532. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kashi, O.; Roness, H.; Spector, I.; Derech-Haim, S.; Meirow, D. Dual suppression of follicle activation pathways completely prevents the cyclophosphamide-induced loss of ovarian reserve. Hum. Reprod. 2023, 38, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Bindels, J.; Squatrito, M.; Bernet, L.; Nisolle, M.; Munaut, C. Ovarian cryopreservation with rapamycin improves fertility restoration in a murine orthotopic transplantation model. Sci. Rep. 2025, 15, 9441. [Google Scholar] [CrossRef]
- Kalich-Philosoph, L.; Roness, H.; Carmely, A.; Fishel-Bartal, M.; Ligumsky, H.; Paglin, S.; Wolf, I.; Kanety, H.; Sredni, B.; Meirow, D. Cyclophosphamide triggers follicle activation and « burnout »; AS101 prevents follicle loss and preserves fertility. Sci. Transl. Med. 2013, 5, 185ra62. [Google Scholar] [CrossRef]
- Di Emidio, G.; Rossi, G.; Bonomo, I.; Alonso, G.L.; Sferra, R.; Vetuschi, A.; Artini, P.G.; Provenzani, A.; Falone, S.; Carta, G.; et al. The Natural Carotenoid Crocetin and the Synthetic Tellurium Compound AS101 Protect the Ovary against Cyclophosphamide by Modulating SIRT1 and Mitochondrial Markers. Oxid. Med. Cell. Longev. 2017, 2017, 8928604. [Google Scholar] [CrossRef]
- Gonfloni, S.; Di Tella, L.; Caldarola, S.; Cannata, S.M.; Klinger, F.G.; Di Bartolomeo, C.; Mattei, M.; Candi, E.; De Felici, M.; Melino, G.; et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy induced death. Nat. Med. 2009, 15, 1179–1185. [Google Scholar] [CrossRef]
- Morgan, S.; Lopes, F.; Gourley, C.; Anderson, R.A.; Spears, N. Cisplatin and doxorubicin induce distinct mechanisms of ovarian follicle loss; imatinib provides selective protection only against cisplatin. PLoS ONE 2013, 8, e70117. [Google Scholar] [CrossRef]
- Kim, S.Y.; Cordeiro, M.H.; Serna, V.A.; Ebbert, K.; Butler, L.M.; Sinha, S.; Mills, A.A.; Woodruff, T.K.; Kurita, T. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell Death Differ. 2013, 20, 987–997. [Google Scholar] [CrossRef]
- Bildik, G.; Acılan, C.; Sahin, G.N.; Karahuseyinoglu, S.; Oktem, O. C-Abl is not actıvated in DNA damage induced and Tap63-mediated oocyte apoptosıs in human ovary. Cell Death Dis. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.B.; Hutt, K.J.; Cook, M.; Speed, T.P.; Strasser, A.; Findlay, J.K.; Scott, C.L. Cisplatin-induced primordial follicle oocyte killing and loss of fertility are not prevented by imatinib. Nat. Med. 2012, 18, 1170–1172, author reply 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Kaya, H.; Desdicioglu, R.; Sezik, M.; Ulukaya, E.; Ozkaya, O.; Yilmaztepe, A.; Demirci, M. Does sphingosine-1-phosphate have a protective effect on cyclophosphamide- and irradiation-induced ovarian damage in the rat model? Fertil. Steril. 2008, 89, 732–735. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, S.; Chen, L.; Liu, X.; Su, H.; Chen, L.; Yang, L.; Zhang, H. Sphingosine 1-phosphate protects against radiation-induced ovarian injury in female rats—Impact on mitochondrial-related genes. Reprod. Biol. Endocrinol. 2020, 18, 99. [Google Scholar] [CrossRef]
- Zhao, J.; Tang, M.; Tang, H.; Wang, M.; Guan, H.; Tang, L.; Zhang, H. Sphingosine 1-phosphate alleviates radiation-induced ferroptosis in ovarian granulosa cells by upregulating glutathione peroxidase 4. Reprod Toxicol. 2023, 115, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Turan, V.; Lierman, S.; Cuvelier, C.; De Sutter, P.; Oktay, K. Sphingosine-1-phosphate prevents chemotherapy-induced human primordial follicle death. Hum. Reprod. 2014, 29, 107–113. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guzel, Y.; Bildik, G.; Dilege, E.; Oktem, O. Sphingosine-1-phosphate reduces atresia of primordial follicles occurring during slow-freezing and thawing of human ovarian cortical strips. Mol. Reprod. Dev. 2018, 85, 858–864. [Google Scholar] [CrossRef]
- Pascuali, N.; Scotti, L.; Di Pietro, M.; Oubiña, G.; Bas, D.; May, M.; Gómez Muñoz, A.; Cuasnicú, P.S.; Cohen, D.J.; Tesone, M.; et al. Ceramide-1-phosphate has protective properties against cyclophosphamide-induced ovarian damage in a mice model of premature ovarian failure. Hum. Reprod. 2018, 33, 844–859. [Google Scholar] [CrossRef]
- Mumusoglu, S.; Turan, V.; Uckan, H.; Suzer, A.; Sokmensuer, L.K.; Bozdag, G. The Impact of a Long-Acting Oral Sphingosine-1-Phosphate Analogue on Ovarian Aging in a Rat Model. Reprod. Sci. 2018, 25, 1330–1335. [Google Scholar] [CrossRef]
- Rossi, V.; Lispi, M.; Longobardi, S.; Mattei, M.; Di Rella, F.; Salustri, A.; De Felici, M.; Klinger, F.G. LH prevents cisplatin-induced apoptosis in oocytes and preserves female fertility in mouse. Cell Death Differ. 2017, 24, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Lamsira, H.K.; Marcozzi, S.; Vicenti, R.; Centonze, C.; Rella, F.D.; Felici, M.D.; Serracchioli, R.; Fabbri, R.; Klinger, F.G. O-265Luteinizing hormone is able to protect reproductive health in cancer patients. Hum. Reprod. 2022, 37 (Suppl. S1), deac106.047. [Google Scholar] [CrossRef]
- Horicks, F.; Van Den Steen, G.; Gervy, C.; Clarke, H.J.; Demeestere, I. Both in vivo FSH depletion and follicular exposure to gonadotrophin-releasing hormone analogues in vitro are not effective to prevent follicular depletion during chemotherapy in mice. Mol. Hum. Reprod. 2018, 24, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, Z. How to preserve fertility in young women exposed to chemotherapy? The role of GnRH agonist cotreatment in addition to cryopreservation of embrya, oocytes, or ovaries. Oncologist 2007, 12, 1044–1054. [Google Scholar] [CrossRef]
- Blumenfeld, Z. Fertility preservation using GnRH agonists: Rationale, possible mechanisms, and explanation of controversy. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119870163. [Google Scholar] [CrossRef]
- Poggio, F.; Lambertini, M.; Bighin, C.; Conte, B.; Blondeaux, E.; D’Alonzo, A.; Dellepiane, C.; Buzzatti, G.; Molinelli, C.; Boccardo, F.; et al. Potential mechanisms of ovarian protection with gonadotropin-releasing hormone agonist in breast Cancer patients: A review. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119864584. [Google Scholar] [CrossRef]
- Lambertini, M.; Horicks, F.; Del Mastro, L.; Partridge, A.H.; Demeestere, I. Ovarian protection with gonadotropinreleasing hormone agonists during chemotherapy in cancer patients: From biological evidence to clinical application. Cancer Treat. Rev. 2019, 72, 65–77. [Google Scholar] [CrossRef]
- Rodriguez-Wallberg, K.A.; Kieler, H.; Foukakis, T.; Li, J.; Gissler, M.; Oberg, A.S.; Bergh, J.; Lundberg, F.E. Gonadotropin Releasing Hormone agonist (GnRHa) during chemotherapy and post-cancer childbirths—A Nationwide population-based cohort study of 24,922 women diagnosed with cancer in Sweden. EClinicalMedicine 2023, 67, 102335. [Google Scholar] [CrossRef]
- Rodriguez-Wallberg, K.A.; Bergh, J.; Foukakis, T. ProFertil study protocol for the investigation of gonadotropin-releasing hormone agonists (GnRHa) during chemotherapy aiming at fertility protection of young women and teenagers with cancer in Sweden—A phase III randomised double-blinded placebo-controlled study. BMJ Open 2023, 13, e078023. [Google Scholar] [CrossRef]
- Skaznik-Wikiel, M.E.; McGuire, M.M.; Sukhwani, M.; Donohue, J.; Chu, T.; Krivak, T.C.; Rajkovic, A.; Orwig, K.E. Granulocyte colony-stimulating factor with or without stem cell factor extends time to premature ovarian insuffciency in female mice treated with alkylating chemotherapy. Fertil. Steril. 2013, 99, 2045–2054. [Google Scholar] [CrossRef]
- Akdemir, A.; Zeybek, B.; Akman, L.; Ergenoglu, A.M.; Yeniel, A.O.; Erbas, O.; Yavasoglu, A.; Terek, M.C.; Taskiran, D. Granulocyte-colony stimulating factor decreases the extent of ovarian damage caused by cisplatin in an experimental rat model. J. Gynecol. Oncol. 2014, 25, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, C.; Dimakopoulou, V.; Rotas, E. Primary ovarian insufficiency associated with imatinib therapy. N. Engl. J. Med. 2008, 358, 1079–1080. [Google Scholar] [CrossRef]
- Zamah, A.M.; Mauro, M.J.; Druker, B.J.; Oktay, K.; Egorin, M.J.; Cedars, M.I.; Rosen, M.P. Will imatinib compromise reproductive capacity? Oncologist 2011, 16, 1422–1427. [Google Scholar] [CrossRef]
- Ohki, Y.; Heissig, B.; Sato, Y.; Akiyama, H.; Zhu, Z.; Hicklin, D.J.; Shimada, K.; Ogawa, H.; Daida, H.; Hattori, K.; et al. Granulocyte colony-stimulating factor promotes neovascularization by releasing vascular endothelial growth factor from neutrophils. FASEB J. 2005, 19, 2005–2007. [Google Scholar] [CrossRef]
- Pourtaji, A.; Jahani, V.; Moallem, S.M.H.; Karimani, A.; Mohammadpour, A.H. Application of G-CSF in Congestive Heart Failure Treatment. Curr. Cardiol. Rev. 2019, 15, 83–90. [Google Scholar] [CrossRef]
- Morita, Y.; Perez, G.I.; Paris, F.; Miranda, S.R.; Ehleiter, D.; Haimovitz-Friedman, A.; Fuks, Z.; Xie, Z.; Reed, J.C.; Schuchman, E.H.; et al. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat. Med. 2000, 6, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Hayun, M.; Naor, Y.; Weil, M.; Albeck, M.; Peled, A.; Don, J.; Haran-Ghera, N.; Sredni, B. The immunomodulator AS101 induces growth arrest and apoptosis in multiple myeloma: Association with the Akt/survivin pathway. Biochem. Pharmacol. 2006, 72, 1423–1431. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fertoprotective Mechanism | Fertoprotective Agents | Mechanism of Action | Model | References |
|---|---|---|---|---|
| Inhibition of primordial follicle recruitment | Anti-Müllerian hormone (AMH) | Ovary hormone | Rodent | [52,53,54,55,56,57,58,59,60] |
| Melatonin | Pineal hormone | Rodent | [61,62,63,64,65,66] | |
| Everolimus | mTOR inhibitor | Rodent | [44,67] | |
| Rapamycin | mTOR inhibitor | Rodent | [68,69,70] | |
| AS101 | PI3K-Pten-Akt pathway modulator | Rodent | [71,72] | |
| Inhibition of primordial follicular apoptosis | Imatinib | Competitive tyrosine kinase inhibitor | Rodent | [73,74,75,76,77] |
| Sphingosine-1-phosphate (S1P) | Membrane sphingolipid | Rodent | [78,79,80,81] | |
| Human ovarian cortical strips (in vitro) | [82] | |||
| Ceramide-1-phosphate (C1P) | Membrane sphingolipid | Rodent | [83,84] | |
| Luteinizing hormone (LH) | Gonadotropin | Rodent | [85] | |
| Human ovarian cortical strips (in vitro) | [86] | |||
| Vascular effect Upregulation of anti-apoptotic molecule | Gonadotropin-releasing hormone agonists (GnRHa) | Inhibition of the pituitary-gonadal-axis | Human (clinical trials) | [87,88,89,90,91,92,93] |
| Rodent | ||||
| Vascular effect | Granulocyte colony-stimulating factor (G-CSF) | Granulocyte colony-stimulating factor | Rodent | [94,95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajączkowska, W.; Buda, M.; Kędzia, W.; Kapczuk, K. Fertility Protection in Female Cancer Patients: From Molecular Mechanisms of Gonadotoxic Therapies to Pharmacotherapeutic Possibilities. Int. J. Mol. Sci. 2025, 26, 7314. https://doi.org/10.3390/ijms26157314
Zajączkowska W, Buda M, Kędzia W, Kapczuk K. Fertility Protection in Female Cancer Patients: From Molecular Mechanisms of Gonadotoxic Therapies to Pharmacotherapeutic Possibilities. International Journal of Molecular Sciences. 2025; 26(15):7314. https://doi.org/10.3390/ijms26157314
Chicago/Turabian StyleZajączkowska, Weronika, Maria Buda, Witold Kędzia, and Karina Kapczuk. 2025. "Fertility Protection in Female Cancer Patients: From Molecular Mechanisms of Gonadotoxic Therapies to Pharmacotherapeutic Possibilities" International Journal of Molecular Sciences 26, no. 15: 7314. https://doi.org/10.3390/ijms26157314
APA StyleZajączkowska, W., Buda, M., Kędzia, W., & Kapczuk, K. (2025). Fertility Protection in Female Cancer Patients: From Molecular Mechanisms of Gonadotoxic Therapies to Pharmacotherapeutic Possibilities. International Journal of Molecular Sciences, 26(15), 7314. https://doi.org/10.3390/ijms26157314