
From Acute Injury to Chronic Neurodegeneration: Molecular Mechanisms Linking Secondary Brain Injury to Long-Term Pathology
 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Mechanisms of Secondary Brain Injury
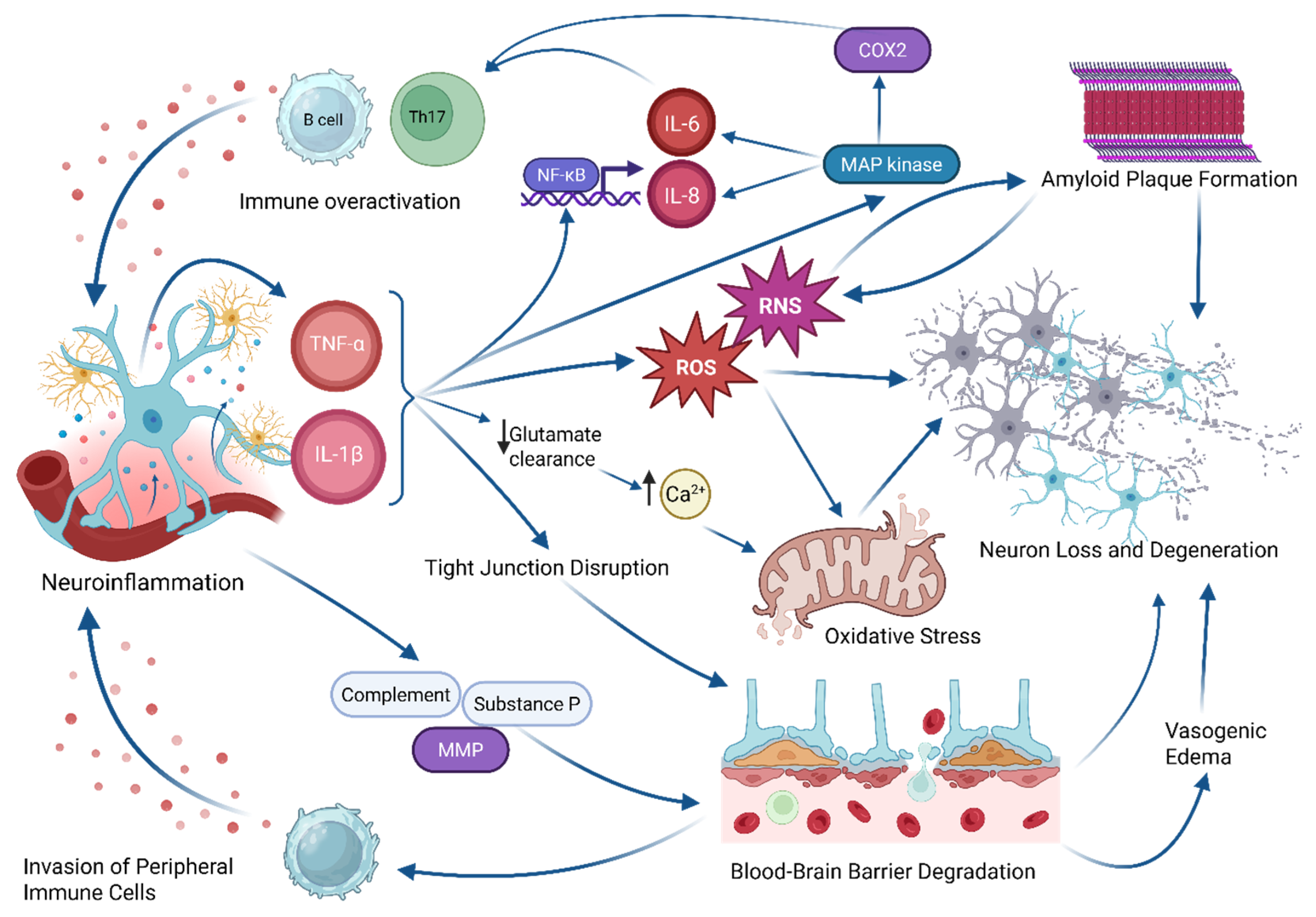
2.1. Neuroinflammation
2.2. Excitotoxicity and Neurotransmitter Dysregulation
2.3. BBB Disruption
2.4. Mitochondrial Dysfunction and Oxidative Stress
2.5. Cytoskeletal Damage and Axonal Pathology
2.6. Epigenetic Modifications
2.7. Memory and Cognitive Dysfunction
2.8. Psychiatric Sequalae
2.9. Preclinical Models of TBI
3. Diagnostic Approaches and Biomarkers
3.1. Molecular Biomarkers
3.2. Diagnostic Imaging
4. Therapeutic Strategies and Future Directions

{kind=link}
| Biomarker | Inclusion Criteria | Recruitment Status | Enrollment | Sponsor | Clinical Trial Identifier |
|---|---|---|---|---|---|
| GFAP | >18 years old, neurological symptoms | Unknown Status | 120 | Hospices Civils de Lyon | NCT05742087 |
| GFAP/UCH-L1 | >18 years old, GCS 13–15 | Recruiting | 1500 (estimated) | Centre Hospitalier Princesse Grace | NCT05885529 |
| S100B/GFAP | 18–100 years old, GCS 3–8 | Completed | 6 | University of Aarhus | NCT03062566 |
| S100B/GFAP | >18 years old, GCS 13–15 | Completed | 595 | University of Aarhus | NCT02867137 |
| GFAP/UCH-L1 | >18 years old, GCS 13–15 | Completed | 194 | Banyan Biomarkers, Inc. | NCT02541123 |
| GFAP/UCH-L1 | 0–20 years old, GCS <12 | Completed | 77 | Valleywise Health | NCT02609568 |
| S100B | >18 years old, GCS 15 | Completed | 1025 | University Hospital, Clermont-Ferrand | NCT04543162 |
| GFAP/UCH-L1 | >18 years old, GCS 13–15 | Completed | 1501 | University Hospital, Grenoble | NCT04032509 |
| GFAP/UCH-L1 | >18 years old, GCS 9–15 | Completed | 119 | Banyan Biomarkers, Inc. | NCT02439736 |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TBI | Traumatic Brain Injury |
| CTE | Chronic Traumatic Encephalopathy |
| GFAP | Glial Fibrillary Acidic Protein |
| NfL | Neurofilament Light Chain |
| UCH-L1 | Ubiquitin C-terminal Hydrolase L1 |
| BBB | Blood brain barrier |
| ICP | Intracranial pressure |
| DAMP | Damage-associated molecular pathogen |
| ROS | Reactive oxygen species |
| FADD | Fas-associated death domain |
| Aß | Amyloid beta |
| tau | Tubulin-associated unit |
| AD | Alzheimer’s Disease |
| PTE | Post-traumatic epilepsy |
| MMP | Matrix Metalloproteinase |
| MPTP | Mitochondrial Permeability Transition Pore |
| RNS | Reactive nitrogen species |
| MDD | Major Depressive Disorder |
| STM | Short-term memory |
| miRNA | Micro RNA |
| PTSD | Post-traumatic stress disorder |
| CCI | Controlled cortical impact model |
| FPI | Fluid percussion injury model |
| S100B | S100 Calcium Binding Protein |
| DTI | Diffusion Tensor Imaging |
| HDFT | High-definition fiber tracking |
| PET | Positron Emission Tomography |
| fMRI | Functional magnetic resonance imaging |
| DMN | Default mode network |
| MRS | Magnetic Resonance Imaging |
| DCE-MRI | Dynamic contrast-enhanced MRI |
| SWI | Susceptibility-weighted imaging |
| HDAC | Histone deacetylase |
| MSC | Mesenchymal stem cell |
| NPC | Neuroprogenitor cell |
References
- CDC. About Mild TBI and Concussion. Available online: https://www.cdc.gov/traumatic-brain-injury/about/index.html (accessed on 1 March 2025).
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of Microglia and Astrocytes: A Roadway to Neuroinflammation and Alzheimer’s Disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Maas, A.I. Traumatic Intracranial Hypertension. N. Engl. J. Med. 2014, 370, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the TBI Brain: The Good, the Bad, and the Dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia as Neuroprotective, Immunocompetent Cells of the CNS. Glia 2002, 40, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Walton, N.M.; Sutter, B.M.; Laywell, E.D.; Levkoff, L.H.; Kearns, S.M.; Marshall II, G.P.; Scheffler, B.; Steindler, D.A. Microglia Instruct Subventricular Zone Neurogenesis. Glia 2006, 54, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Marín-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; van Rooijen, N.; Mallat, M. Microglia Promote the Death of Developing Purkinje Cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active Sensor and Versatile Effector Cells in the Normal and Pathologic Brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of Pro- and Anti-Inflammatory Cytokines and Chemokines in the Pathophysiology of Traumatic Brain Injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Liu, W.; Dickson, D.W.; Brosnan, C.F.; Berman, J.W. Cytokine Production by Human Fetal Microglia and Astrocytes. Differential Induction by Lipopolysaccharide and IL-1 Beta. J. Immunol. 1993, 150, 2659–2667. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.D.; Iannotti, F.; Pringle, A.K.; Trikkas, C.; Clough, G.F.; Church, M.K. A Microdialysis Method for the Recovery of IL-1β, IL-6 and Nerve Growth Factor from Human Brain In Vivo. J. Neurosci. Methods 2002, 119, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Brown, G.C. Inflammatory Neurodegeneration Mediated by Nitric Oxide from Activated Glia-Inhibiting Neuronal Respiration, Causing Glutamate Release and Excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Loddick, S.A.; Rothwell, N.J. Neuroprotective Effects of Human Recombinant Interleukin-1 Receptor Antagonist in Focal Cerebral Ischaemia in the Rat. J. Cereb. Blood Flow Metab. 1996, 16, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Hu, S.X.; Ehrlich, L.; Peterson, P.K. Interleukin-1 and Tumor Necrosis Factor-α Synergistically Mediate Neurotoxicity: Involvement of Nitric Oxide and of N-Methyl-D-Aspartate Receptors. Brain Behav. Immun. 1995, 9, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The Role and Consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-Dependent Recruitment of Endogenous FADD and Caspase-8 to Death Receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The Non-Canonical NF-κB Pathway in Immunity and Inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, S.; Rivest, S. Effect of Acute Systemic Inflammatory Response and Cytokines on the Transcription of the Genes Encoding Cyclooxygenase Enzymes (COX-1 and COX-2) in the Rat Brain. J. Neurochem. 1998, 70, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Stalder, A.K.; Ermini, F.; Bondolfi, L.; Krenger, W.; Burbach, G.J.; Deller, T.; Coomaraswamy, J.; Staufenbiel, M.; Landmann, R.; Jucker, M. Invasion of Hematopoietic Cells into the Brain of Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 2005, 25, 11125–11132. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Liu, Q.; Sayre, J.; Pop, V.; Brahmandam, V.; Graves, M.; Vinters, H. Cyclooxygenase-2-positive Macrophages Infiltrate the Alzheimer’s Disease Brain and Damage the Blood–Brain Barrier. Eur. J. Clin. Investig. 2002, 32, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Baud, O.; Vartanian, T.; Volpe, J.J.; Rosenberg, P.A. Peroxynitrite Generated by Inducible Nitric Oxide Synthase and NADPH Oxidase Mediates Microglial Toxicity to Oligodendrocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 9936–9941. [Google Scholar] [CrossRef] [PubMed]
- Soltani Khaboushan, A.; Yazdanpanah, N.; Rezaei, N. Neuroinflammation and Proinflammatory Cytokines in Epileptogenesis. Mol. Neurobiol. 2022, 59, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gentleman, S.M.; Leclercq, P.D.; Murray, L.S.; Griffin, W.S.T.; Graham, D.I.; Nicoll, J.A.R. The Neuroinflammatory Response in Humans after Traumatic Brain Injury. Neuropathol. Appl. Neurobiol. 2013, 39, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and White Matter Degeneration Persist for Years after a Single Traumatic Brain Injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular Regulation in the Normal Brain and in Alzheimer’s Disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and Neuron Numbers, but Not Amyloid Load, Predict Cognitive Status in Alzheimer’s Disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; Wang, C.; Bao, X.; Finn, M.B.; Hu, H.; et al. Microglia-Mediated T Cell Infiltration Drives Neurodegeneration in Tauopathy. Nature 2023, 615, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three Dimensions of the Amyloid Hypothesis: Time, Space and “Wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.-L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic Traumatic Encephalopathy in Blast-Exposed Military Veterans and a Blast Neurotrauma Mouse Model. Sci. Transl. Med. 2012, 4, 134ra60. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stein, T.D.; Huber, B.R.; Crary, J.F.; Bieniek, K.; Dickson, D.; Alvarez, V.E.; Cherry, J.D.; Farrell, K.; Butler, M.; et al. Chronic Traumatic Encephalopathy (CTE): Criteria for Neuropathological Diagnosis and Relationship to Repetitive Head Impacts. Acta Neuropathol. 2023, 145, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.D.; Alvarez, V.E.; McKee, A.C. Chronic Traumatic Encephalopathy: A Spectrum of Neuropathological Changes Following Repetitive Brain Trauma in Athletes and Military Personnel. Alzheimer’s Res. Ther. 2014, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of Microglia and Inhibition of Exosome Synthesis Halt Tau Propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, M.M.; Parks, E.A.; Dedman, J.R.; Kaetzel, M.A.; Lyeth, B.G.; Berman, R.F. Phosphorylation of Calcium Calmodulin—Dependent Protein Kinase II Following Lateral Fluid Percussion Brain Injury in Rats. J. Neurotrauma 2007, 24, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of Extracellular Glutamate Measured by Cerebral Microdialysis in Severe Traumatic Brain Injury: Clinical Article. J. Neurosurg. 2010, 113, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Bowen, D.M.; DeKosky, S.T. Increased Transmitter Amino Acid Concentration in Human Ventricular CSF after Brain Trauma. NeuroReport 1994, 6, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Thapa, K.; Khan, H.; Singh, T.G.; Kaur, A. Traumatic Brain Injury: Mechanistic Insight on Pathophysiology and Potential Therapeutic Targets. J. Mol. Neurosci. 2021, 71, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Kermer, P.; Klöcker, N.; Bähr, M. Neuronal Death after Brain Injury. Cell Tissue Res. 1999, 298, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Spruston, N. Pyramidal Neurons: Dendritic Structure and Synaptic Integration. Nat. Rev. Neurosci. 2008, 9, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Prince, D.A. Synaptic Activity in Chronically Injured, Epileptogenic Sensory-Motor Neocortex. J. Neurophysiol. 2002, 88, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.K.; Gu, F.; Parada, I.; Vyas, S.; Prince, D.A. Aberrant Excitatory Rewiring of Layer V Pyramidal Neurons Early after Neocortical Trauma. Neurobiol. Dis. 2016, 91, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Avramescu, S.; Nita, D.A.; Timofeev, I. Neocortical Post-Traumatic Epileptogenesis Is Associated with Loss of GABAergic Neurons. J. Neurotrauma 2009, 26, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Faria, L.C.; Prince, D.A. Presynaptic Inhibitory Terminals Are Functionally Abnormal in a Rat Model of Posttraumatic Epilepsy. J. Neurophysiol. 2010, 104, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, S.N.; Salin, P.A.; Prince, D.A. Chronic Neocortical Epileptogenesis In Vitro. J. Neurophysiol. 1994, 71, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Sulhan, S.; Lyon, K.A.; Shapiro, L.A.; Huang, J.H. Neuroinflammation and Blood–Brain Barrier Disruption Following Traumatic Brain Injury: Pathophysiology and Potential Therapeutic Targets. J. Neurosci. Res. 2018, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The Role of Neurogenic Inflammation in Blood-Brain Barrier Disruption and Development of Cerebral Oedema Following Acute Central Nervous System (CNS) Injury. Int. J. Mol. Sci. 2017, 18, 1788. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–Endothelial Interactions at the Blood–Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.H.; Anderson, A.J.; Taylor, S.M.; Woodruff, T.M.; Ruitenberg, M.J. Complement Activation in the Injured Central Nervous System: Another Dual-Edged Sword? J. Neuroinflamm. 2012, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Bellander, B.-M.; Singhrao, S.K.; Ohlsson, M.; Mattsson, P.; Svensson, M. Complement Activation in the Human Brain after Traumatic Head Injury. J. Neurotrauma 2001, 18, 1295–1311. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Li, S.; Chung, S.H.; Zhu, L.; Stayt, J.; Su, T.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Gillies, M.C. Tyrosine Phosphorylation of VE-Cadherin and Claudin-5 Is Associated with TGF-Β1-Induced Permeability of Centrally Derived Vascular Endothelium. Eur. J. Cell Biol. 2011, 90, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Mori, T.; Maeda, T.; Kawamata, T. Pathogenesis of the Mass Effect of Cerebral Contusions: Rapid Increase in Osmolality within the Contusion Necrosis. In Intracranial Pressure and Neuromonitoring in Brain Injury, Proceedings of the Tenth International ICP Symposium, Williamsburg, VA, USA, 25–29 May 1997; Marmarou, A., Bullock, R., Avezaat, C., Baethmann, A., Becker, D., Brock, M., Hoff, J., Nagai, H., Reulen, H.-J., Teasdale, G., Eds.; Springer: Vienna, Austria, 1998; pp. 289–292. [Google Scholar]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and Treatment of Cerebral Edema in Traumatic Brain Injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Nichols, P.; Urriola, J.; Miller, S.; Bjorkman, T.; Mahady, K.; Vegh, V.; Nasrallah, F.; Winter, C. Blood-Brain Barrier Dysfunction Significantly Correlates with Serum Matrix Metalloproteinase-7 (MMP-7) Following Traumatic Brain Injury. NeuroImage Clin. 2021, 31, 102741. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.-C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.-L.B.; et al. Tau Depletion Prevents Progressive Blood-Brain Barrier Damage in a Mouse Model of Tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Sullivan, P.G.; Deng, Y.; Mbye, L.H.; Hall, E.D. Time Course of Post-Traumatic Mitochondrial Oxidative Damage and Dysfunction in a Mouse Model of Focal Traumatic Brain Injury: Implications for Neuroprotective Therapy. J. Cereb. Blood Flow Metab. 2006, 26, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Dadsena, S.; García-Sáez, A.J. Mitochondrial Pores at the Crossroad between Cell Death and Inflammatory Signaling. Mol. Cell 2023, 83, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Permeability Transition Pore Contributes to Mitochondrial Dysfunction in Fibroblasts of Patients with Sporadic Alzheimer’s Disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Thompson, B.M.; Gao, X.; Hall, E.D. Temporal Relationship of Peroxynitrite-Induced Oxidative Damage, Calpain-Mediated Cytoskeletal Degradation and Neurodegeneration after Traumatic Brain Injury. Exp. Neurol. 2007, 205, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- VanItallie, T.B. Traumatic Brain Injury (TBI) in Collision Sports: Possible Mechanisms of Transformation into Chronic Traumatic Encephalopathy (CTE). Metabolism 2019, 100, 153943. [Google Scholar] [CrossRef] [PubMed]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.-Y.; Li, C.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-Tau Is Induced in Clinical and Preclinical Brain Injury and Contributes to Post-Injury Sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess Brain Protein Oxidation and Enzyme Dysfunction in Normal Aging and in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Howlett, J.R.; Nelson, L.D.; Stein, M.B. Mental Health Consequences of Traumatic Brain Injury. Biol. Psychiatry 2022, 91, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.; Chiba, A.A. The Amygdala and Emotion. Curr. Opin. Neurobiol. 1996, 6, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S.; et al. Nitration of Tyrosine 10 Critically Enhances Amyloid β Aggregation and Plaque Formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Ibi, M.; Liu, J.; Arakawa, N.; Kitaoka, S.; Kawaji, A.; Matsuda, K.; Iwata, K.; Matsumoto, M.; Katsuyama, M.; Zhu, K.; et al. Depressive-Like Behaviors Are Regulated by NOX1/NADPH Oxidase by Redox Modification of NMDA Receptor 1. J. Neurosci. 2017, 37, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pinto-Duarte, A.; Sejnowski, T.J.; Behrens, M.M. How Nox2-Containing NADPH Oxidase Affects Cortical Circuits in the NMDA Receptor Antagonist Model of Schizophrenia. Antioxid. Redox Signal. 2013, 18, 1444–1462. [Google Scholar] [CrossRef] [PubMed]
- Hervy, J.; Bicout, D.J. Dynamical Decoration of Stabilized-Microtubules by Tau-Proteins. Sci. Rep. 2019, 9, 12473. [Google Scholar] [CrossRef] [PubMed]
- Tang-Schomer, M.D.; Patel, A.R.; Baas, P.W.; Smith, D.H. Mechanical Breaking of Microtubules in Axons during Dynamic Stretch Injury Underlies Delayed Elasticity, Microtubule Disassembly, and Axon Degeneration. FASEB J. 2010, 24, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Méphon-Gaspard, A.; Boca, M.; Pioche-Durieu, C.; Desforges, B.; Burgo, A.; Hamon, L.; Piétrement, O.; Pastré, D. Role of Tau in the Spatial Organization of Axonal Microtubules: Keeping Parallel Microtubules Evenly Distributed despite Macromolecular Crowding. Cell. Mol. Life Sci. 2016, 73, 3745–3760. [Google Scholar] [CrossRef] [PubMed]
- Collins-Praino, L.E.; Corrigan, F. Does Neuroinflammation Drive the Relationship between Tau Hyperphosphorylation and Dementia Development Following Traumatic Brain Injury? Brain Behav. Immun. 2017, 60, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Saatman, K.E.; Creed, J.; Raghupathi, R. Calpain as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics 2010, 7, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.M.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. Modulation of Microtubule Dynamics by Tau in Living Cells: Implications for Development and Neurodegeneration. Mol. Biol. Cell 2004, 15, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular Tau Oligomers as Intermediates of Tau Filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Ruan, K.; Zhai, R.G. NMNAT Suppresses Tau-Induced Neurodegeneration by Promoting Clearance of Hyperphosphorylated Tau Oligomers in a Drosophila Model of Tauopathy. Hum. Mol. Genet. 2012, 21, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Hanger, D.P.; Miller, C.C.; Lovestone, S. The Importance of Tau Phosphorylation for Neurodegenerative Diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Zima, L.; West, R.; Smolen, P.; Kobori, N.; Hergenroeder, G.; Choi, H.A.; Moore, A.N.; Redell, J.B.; Dash, P.K. Epigenetic Modifications and Their Potential Contribution to Traumatic Brain Injury Pathobiology and Outcome. J. Neurotrauma 2022, 39, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-S.; Lee, S.; Shin, J.-Y.; Hwang, Y.J.; Cho, H.; Yoo, S.-K.; Kim, Y.; Lim, S.; Kim, Y.K.; Hwang, E.M.; et al. Transcriptome Analyses of Chronic Traumatic Encephalopathy Show Alterations in Protein Phosphatase Expression Associated with Tauopathy. Exp. Mol. Med. 2017, 49, e333. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, F.; Ge, Y.; Chen, S.; Xin, Y.; Umali, M.U.; De Gasperi, R.; Gama Sosa, M.A.; Ahlers, S.T.; Elder, G.A. Neuronal DNA Methylation Profiling of Blast-Related Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P. miRNA Dysregulation in Traumatic Brain Injury and Epilepsy: A Systematic Review to Identify Putative Biomarkers for Post-Traumatic Epilepsy. Metab. Brain Dis. 2023, 38, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. [Google Scholar] [CrossRef] [PubMed]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of Fragile X Syndrome and Fragile X-Related Disorders. Dev. Med. Child Neurol. 2019, 61, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lalande, M.; Calciano, M.A. Molecular Epigenetics of Angelman Syndrome. Cell. Mol. Life Sci. 2007, 64, 947. [Google Scholar] [CrossRef] [PubMed]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s Disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Milman, A.; Rosenberg, A.; Weizman, R.; Pick, C.G. Mild Traumatic Brain Injury Induces Persistent Cognitive Deficits and Behavioral Disturbances in Mice. J. Neurotrauma 2005, 22, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.E.S.; Smith, D.H.; Trojanowski, J.Q.; McIntosh, T.K. Enduring Cognitive, Neurobehavioral and Histopathological Changes Persist for up to One Year Following Severe Experimental Brain Injury in Rats. Neuroscience 1998, 87, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Kobori, N.; Dash, P.K. Reversal of Brain Injury-Induced Prefrontal Glutamic Acid Decarboxylase Expression and Working Memory Deficits by D1Receptor Antagonism. J. Neurosci. 2006, 26, 4236–4246. [Google Scholar] [CrossRef] [PubMed]
- Hoskison, M.M.; Moore, A.N.; Hu, B.; Orsi, S.; Kobori, N.; Dash, P.K. Persistent Working Memory Dysfunction Following Traumatic Brain Injury: Evidence for a Time-Dependent Mechanism. Neuroscience 2009, 159, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.; Moore, A.; Dixon, C. Spatial Memory Deficits, Increased Phosphorylation of the Transcription Factor CREB, and Induction of the AP-1 Complex Following Experimental Brain Injury. J. Neurosci. 1995, 15, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.B.; Fan, L.; LeVASSEUR, R.A.; Faden, A.I. Effect of Traumatic Brain Injury on Mouse Spatial and Nonspatial Learning in the Barnes Circular Maze. J. Neurotrauma 1998, 15, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Lowenstein, D.H.; Gennarelli, T.A.; McIntosh, T.K. Persistent Memory Dysfunction Is Associated with Bilateral Hippocampal Damage Following Experimental Brain Injury. Neurosci. Lett. 1994, 168, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Gurkoff, G.G.; Gahan, J.D.; Ghiasvand, R.T.; Hunsaker, M.R.; Van, K.; Feng, J.; Shahlaie, K.; Berman, R.F.; Lyeth, B.G.; Folkerts, M.M. Evaluation of Metric, Topological, and Temporal Ordering Memory Tasks after Lateral Fluid Percussion Injury. J. Neurotrauma 2013, 30, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Paterno, R.; Folweiler, K.A.; Cohen, A.S. Pathophysiology and Treatment of Memory Dysfunction After Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2017, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Bohbot, V.D.; Allen, J.J.B.; Nadel, L. Memory Deficits Characterized by Patterns of Lesions to the Hippocampus and Parahippocampal Cortex. Ann. N. Y. Acad. Sci. 2000, 911, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Rempel-Clower, N.L.; Zola, S.M.; Squire, L.R.; Amaral, D.G. Three Cases of Enduring Memory Impairment after Bilateral Damage Limited to the Hippocampal Formation. J. Neurosci. 1996, 16, 5233–5255. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.H.; Doyle, D.; Ford, I.; Gennarelli, T.A.; Graham, D.I.; Mclellan, D.R. Diffuse Axonal Injury in Head Injury: Definition, Diagnosis and Grading. Histopathology 1989, 15, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S.; Pignatelli, M.; Roy, D.S.; Ryan, T.J. Memory Engram Storage and Retrieval. Curr. Opin. Neurobiol. 2015, 35, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Witgen, B.M.; Lifshitz, J.; Smith, M.L.; Schwarzbach, E.; Liang, S.-L.; Grady, M.S.; Cohen, A.S. Regional Hippocampal Alteration Associated with Cognitive Deficit Following Experimental Brain Injury: A Systems, Network and Cellular Evaluation. Neuroscience 2005, 133, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Leutgeb, J.K.; Leutgeb, S.; Moser, M.-B.; Moser, E.I. Pattern Separation in the Dentate Gyrus and CA3 of the Hippocampus. Science 2007, 315, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kesner, R.P. Differential Contributions of Dorsal Hippocampal Subregions to Memory Acquisition and Retrieval in Contextual Fear-conditioning. Hippocampus 2004, 14, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.; Thomas, M.; Smith, D.; McIntosh, T. Selective Vulnerability of Dentate Hilar Neurons Following Traumatic Brain Injury: A Potential Mechanistic Link between Head Trauma and Disorders of the Hippocampus. J. Neurosci. 1992, 12, 4846–4853. [Google Scholar] [CrossRef] [PubMed]
- Treves, A.; Rolls, E.T. Computational Analysis of the Role of the Hippocampus in Memory. Hippocampus 1994, 4, 374–391. [Google Scholar] [CrossRef] [PubMed]
- Dunning, D.L.; Westgate, B.; Adlam, A.-L.R. A Meta-Analysis of Working Memory Impairments in Survivors of Moderate-to-Severe Traumatic Brain Injury. Neuropsychology 2016, 30, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Lennon, M.J.; Brooker, H.; Creese, B.; Thayanandan, T.; Rigney, G.; Aarsland, D.; Hampshire, A.; Ballard, C.; Corbett, A.; Raymont, V. Lifetime Traumatic Brain Injury and Cognitive Domain Deficits in Late Life: The PROTECT-TBI Cohort Study. J. Neurotrauma 2023, 40, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Weis, C.N.; Webb, E.K.; deRoon-Cassini, T.A.; Larson, C.L. Emotion Dysregulation Following Trauma: Shared Neurocircuitry of Traumatic Brain Injury and Trauma-Related Psychiatric Disorders. Biol. Psychiatry 2022, 91, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Esagoff, A.I.; Stevens, D.A.; Kosyakova, N.; Woodard, K.; Jung, D.; Richey, L.N.; Daneshvari, N.O.; Luna, L.P.; Bray, M.J.C.; Bryant, B.R.; et al. Neuroimaging Correlates of Post-Traumatic Stress Disorder in Traumatic Brain Injury: A Systematic Review of the Literature. J. Neurotrauma 2023, 40, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.A.; Dorr, N.P.; De Gasperi, R.; Gama Sosa, M.A.; Shaughness, M.C.; Maudlin-Jeronimo, E.; Hall, A.A.; McCarron, R.M.; Ahlers, S.T. Blast Exposure Induces Post-Traumatic Stress Disorder-Related Traits in a Rat Model of Mild Traumatic Brain Injury. J. Neurotrauma 2012, 29, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Beamer, M.; Tummala, S.R.; Gullotti, D.; Kopil, C.; Gorka, S.; Abel, T.; “Dale” Bass, C.R.; Morrison, B., III; Cohen, A.S.; Meaney, D.F. Primary Blast Injury Causes Cognitive Impairments and Hippocampal Circuit Alterations. Exp. Neurol. 2016, 283, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Arun, P.; Abu-Taleb, R.; Oguntayo, S.; Wang, Y.; Valiyaveettil, M.; Long, J.B.; Nambiar, M.P. Acute Mitochondrial Dysfunction after Blast Exposure: Potential Role of Mitochondrial Glutamate Oxaloacetate Transaminase. J. Neurotrauma 2013, 30, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.B.; Leite-Morris, K.A.; Wang, L.; Rumbika, K.K.; Heinrichs, S.C.; Zeng, X.; Wu, L.; Arena, D.T.; Teng, Y.D. Pathophysiological Bases of Comorbidity: Traumatic Brain Injury and Post-Traumatic Stress Disorder. J. Neurotrauma 2018, 35, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R.; Avants, B.B.; Tustison, N.J.; Wassermann, E.M.; Gill, J.; Polejaeva, E.; Dell, K.C.; Carr, W.; Yarnell, A.M.; LoPresti, M.L.; et al. Functional and Structural Neuroimaging Correlates of Repetitive Low-Level Blast Exposure in Career Breachers. J. Neurotrauma 2020, 37, 2468–2481. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The Stressed Synapse: The Impact of Stress and Glucocorticoids on Glutamate Transmission. Nat. Rev. Neurosci. 2012, 13, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA Imbalance Following Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Marklund, N. Rodent Models of Traumatic Brain Injury: Methods and Challenges. In Injury Models of the Central Nervous System; Kobeissy, F.H., Dixon, C.E., Hayes, R.L., Mondello, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1462, pp. 29–46. ISBN 978-1-4939-3814-8. [Google Scholar]
- Dixon, C.E.; Lyeth, B.G.; Povlishock, J.T.; Findling, R.L.; Hamm, R.J.; Marmarou, A.; Young, H.F.; Hayes, R.L. A Fluid Percussion Model of Experimental Brain Injury in the Rat. J. Neurosurg. 1987, 67, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; Maris, D.O.; McCALL, T.; Grady, M.S. Adaptation of the Fluid Percussion Injury Model to the Mouse. J. Neurotrauma 1998, 15, 217–229. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, T.K.; Vink, R.; Noble, L.; Yamakami, I.; Fernyak, S.; Soares, H.; Faden, A.L. Traumatic Brain Injury in the Rat: Characterization of a Lateral Fluid-Percussion Model. Neuroscience 1989, 28, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Clausen, F.; Hillered, L. Intracranial Pressure Changes during Fluid Percussion, Controlled Cortical Impact and Weight Drop Injury in Rats. Acta Neurochir. 2005, 147, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef] [PubMed]
- Lighthall, J.W. Controlled Cortical Impact: A New Experimental Brain Injury Model. J. Neurotrauma 1988, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Soares, H.D.; Pierce, J.S.; Perlman, K.G.; Saatman, K.E.; Meaney, D.F.; Dixon, C.E.; McIntosh, T.K. A Model of Parasagittal Controlled Cortical Impact in the Mouse: Cognitive and Histopathologic Effects. J. Neurotrauma 1995, 12, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Bryant, Y.D.; Cho, W.; Sullivan, P.G. Evolution of Post-Traumatic Neurodegeneration after Controlled Cortical Impact Traumatic Brain Injury in Mice and Rats as Assessed by the De Olmos Silver and Fluorojade Staining Methods. J. Neurotrauma 2008, 25, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Feeney, D.M.; Boyeson, M.G.; Linn, R.T.; Murray, H.M.; Dail, W.G. Responses to Cortical Injury: I. Methodology and Local Effects of Contusions in the Rat. Brain Res. 1981, 211, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Cai, J.; Shields, L.B.E.; Liu, N.; Xu, X.-M.; Shields, C.B. Traumatic Brain Injury Using Mouse Models. Transl. Stroke Res. 2014, 5, 454–471. [Google Scholar] [CrossRef] [PubMed]
- Vink, R. Large Animal Models of Traumatic Brain Injury. J. Neurosci. Res. 2018, 96, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Bonnier, C.; Mesplès, B.; Carpentier, S.; Henin, D.; Gressens, P. Delayed White Matter Injury in a Murine Model of Shaken Baby Syndrome. Brain Pathol. 2002, 12, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Asif, S.; Pandey, D.K.; Chaudhary, A.; Kapoor, V.; Verma, P.K. Biomarkers in Acute Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Cureus 2024, 16, e63020. [Google Scholar] [CrossRef] [PubMed]
- Magnoni, S.; Esparza, T.J.; Conte, V.; Carbonara, M.; Carrabba, G.; Holtzman, D.M.; Zipfel, G.J.; Stocchetti, N.; Brody, D.L. Tau Elevations in the Brain Extracellular Space Correlate with Reduced Amyloid-β Levels and Predict Adverse Clinical Outcomes after Severe Traumatic Brain Injury. Brain 2012, 135, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, S.; Andriolo, M.; Boccagni, C.; Sant’Angelo, A.; D’Ippolito, M.E.; Galardi, G. Dissociation of Cerebrospinal Fluid Amyloid-β and Tau Levels in Patients with Prolonged Posttraumatic Disorders of Consciousness. Brain Inj. 2018, 32, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Pattinson, C.L.; Shahim, P.; Taylor, P.; Dunbar, K.; Guedes, V.; Motamedi, V.; Lai, C.; Devoto, C.; Peyer, J.; Roy, M.J.; et al. Elevated Tau in Military Personnel Relates to Chronic Symptoms Following Traumatic Brain Injury. J. Head Trauma Rehabil. 2020, 35, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Butt, A.; Li, B.; Illes, P.; Zorec, R.; Semyanov, A.; Tang, Y.; Sofroniew, M.V. Astrocytes in Human Central Nervous System Diseases: A Frontier for New Therapies. Signal Transduct. Target. Ther. 2023, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K.W. Glial Fibrillary Acidic Protein: From Intermediate Filament Assembly and Gliosis to Neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, E.-J.; Lim, Y.-M.; Kim, K.-K. Glial Fibrillary Acidic Protein in Blood as a Disease Biomarker of Neuromyelitis Optica Spectrum Disorders. Front. Neurol. 2022, 13, 865730. [Google Scholar] [CrossRef] [PubMed]
- Fitch, M.T.; Silver, J. CNS Injury, Glial Scars, and Inflammation. Exp. Neurol. 2008, 209, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Kouchaki, E.; Dashti, F.; Mirazimi, S.M.A.; Alirezaei, Z.; Jafari, S.H.; Hamblin, M.R.; Mirzaei, H. Neurofilament Light Chain as a Biomarker for Diagnosis of Multiple Sclerosis. EXCLI J. 2021, 20, 1308–1325. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, Z.; Pourhanifeh, M.H.; Borran, S.; Nejati, M.; Mirzaei, H.; Hamblin, M.R. Neurofilament Light Chain as a Biomarker, and Correlation with Magnetic Resonance Imaging in Diagnosis of CNS-Related Disorders. Mol. Neurobiol. 2020, 57, 469–491. [Google Scholar] [CrossRef] [PubMed]
- Shahim, P.; Politis, A.; van der Merwe, A.; Moore, B.; Chou, Y.-Y.; Pham, D.L.; Butman, J.A.; Diaz-Arrastia, R.; Gill, J.M.; Brody, D.L.; et al. Neurofilament Light as a Biomarker in Traumatic Brain Injury. Neurology 2020, 95, e610–e622. [Google Scholar] [CrossRef] [PubMed]
- Kartau, M.; Melkas, S.; Kartau, J.; Arola, A.; Laakso, H.; Pitkänen, J.; Lempiäinen, J.; Koikkalainen, J.; Lötjönen, J.; Korvenoja, A.; et al. Neurofilament Light Level Correlates with Brain Atrophy, and Cognitive and Motor Performance. Front. Aging Neurosci. 2023, 14, 939155. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.; Rubin, P.; Thomson, A.R.; Rocca, D.; Henley, J.M. The Ubiquitin C-Terminal Hydrolase L1 (UCH-L1) C Terminus Plays a Key Role in Protein Stability, but Its Farnesylation Is Not Required for Membrane Association in Primary Neurons. J. Biol. Chem. 2014, 289, 36140–36149. [Google Scholar] [CrossRef] [PubMed]
- Brophy, G.M.; Mondello, S.; Papa, L.; Robicsek, S.A.; Gabrielli, A.; Tepas, J.; Buki, A.; Robertson, C.; Tortella, F.C.; Hayes, R.L.; et al. Biokinetic Analysis of Ubiquitin C-Terminal Hydrolase-L1 (UCH-L1) in Severe Traumatic Brain Injury Patient Biofluids. J. Neurotrauma 2011, 28, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Kobeissy, F.; Arja, R.D.; Munoz, J.C.; Shear, D.A.; Gilsdorf, J.; Zhu, J.; Yadikar, H.; Haskins, W.; Tyndall, J.A.; Wang, K.K. The Game Changer: UCH-L1 and GFAP-Based Blood Test as the First Marketed in Vitro Diagnostic Test for Mild Traumatic Brain Injury. Expert Rev. Mol. Diagn. 2024, 24, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Michetti, F.; Clementi, M.E.; Di Liddo, R.; Valeriani, F.; Ria, F.; Rende, M.; Di Sante, G.; Romano Spica, V. The S100B Protein: A Multifaceted Pathogenic Factor More Than a Biomarker. Int. J. Mol. Sci. 2023, 24, 9605. [Google Scholar] [CrossRef] [PubMed]
- Gayger-Dias, V.; Vizuete, A.F.; Rodrigues, L.; Wartchow, K.M.; Bobermin, L.; Leite, M.C.; Quincozes-Santos, A.; Kleindienst, A.; Gonçalves, C.-A. How S100B Crosses Brain Barriers and Why It Is Considered a Peripheral Marker of Brain Injury. Exp. Biol. Med. 2023, 248, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Thelin, E.P.; Nelson, D.W.; Bellander, B.-M. A Review of the Clinical Utility of Serum S100B Protein Levels in the Assessment of Traumatic Brain Injury. Acta Neurochir. 2017, 159, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. Fluid Transport in the Brain. Physiol. Rev. 2022, 102, 1025–1151. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.J.; Westin, C.-F. An Introduction to Diffusion Tensor Image Analysis. Neurosurg. Clin. N. Am. 2011, 22, 185. [Google Scholar] [CrossRef] [PubMed]
- Winklewski, P.J.; Sabisz, A.; Naumczyk, P.; Jodzio, K.; Szurowska, E.; Szarmach, A. Understanding the Physiopathology Behind Axial and Radial Diffusivity Changes—What Do We Know? Front. Neurol. 2018, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Dodd, W.S.; Panther, E.J.; Pierre, K.; Hernandez, J.S.; Patel, D.; Lucke-Wold, B. Traumatic Brain Injury and Secondary Neurodegenerative Disease. Trauma Care 2022, 2, 510–522. [Google Scholar] [CrossRef]
- Rosenkrantz, A.B.; Padhani, A.R.; Chenevert, T.L.; Koh, D.-M.; De Keyzer, F.; Taouli, B.; Le Bihan, D. Body Diffusion Kurtosis Imaging: Basic Principles, Applications, and Considerations for Clinical Practice. J. Magn. Reson. Imaging 2015, 42, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Abhinav, K.; Yeh, F.-C.; Mansouri, A.; Zadeh, G.; Fernandez-Miranda, J.C. High-Definition Fiber Tractography for the Evaluation of Perilesional White Matter Tracts in High-Grade Glioma Surgery. Neuro-Oncology 2015, 17, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.-C.; Irimia, A.; de Bastos, D.C.A.; Golby, A.J. Tractography Methods and Findings in Brain Tumors and Traumatic Brain Injury. NeuroImage 2021, 245, 118651. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-X.; Li, Y.-H.; Lu, W.; Huang, S.-H.; Li, M.-J.; Xiao, L.-Z.; Liu, J. Positron Emission Tomography Imaging for the Assessment of Mild Traumatic Brain Injury and Chronic Traumatic Encephalopathy: Recent Advances in Radiotracers. Neural Regen. Res. 2021, 17, 74–81. [Google Scholar] [CrossRef]
- Gogola, A.; Minhas, D.S.; Villemagne, V.L.; Cohen, A.D.; Mountz, J.M.; Pascoal, T.A.; Laymon, C.M.; Mason, N.S.; Ikonomovic, M.D.; Mathis, C.A.; et al. Direct Comparison of the Tau PET Tracers 18F-Flortaucipir and 18F-MK-6240 in Human Subjects. J. Nucl. Med. 2022, 63, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Lesman-Segev, O.H.; La Joie, R.; Stephens, M.L.; Sonni, I.; Tsai, R.; Bourakova, V.; Visani, A.V.; Edwards, L.; O’Neil, J.P.; Baker, S.L.; et al. Tau PET and Multimodal Brain Imaging in Patients at Risk for Chronic Traumatic Encephalopathy. Neuroimage Clin. 2019, 24, 102025. [Google Scholar] [CrossRef] [PubMed]
- Varlow, C.; Vasdev, N. Evaluation of Tau Radiotracers in Chronic Traumatic Encephalopathy. J. Nucl. Med. 2023, 64, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Palleis, C.; Sauerbeck, J.; Beyer, L.; Harris, S.; Schmitt, J.; Morenas-Rodriguez, E.; Finze, A.; Nitschmann, A.; Ruch-Rubinstein, F.; Eckenweber, F.; et al. In Vivo Assessment of Neuroinflammation in 4-Repeat Tauopathies. Mov. Disord. 2021, 36, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, F.; Becker, G.; Boutin, H. Have (R)-[11C]PK11195 Challengers Fulfilled the Promise? A Scoping Review of Clinical TSPO PET Studies. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 201–220. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Chaney, A.M.; Carlson, M.L.; Jackson, I.M.; Rao, A.; James, M.L. Neuroinflammation PET Imaging: Current Opinion and Future Directions. J. Nucl. Med. 2020, 61, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Glover, G.H. Overview of Functional Magnetic Resonance Imaging. Neurosurg. Clin. N. Am. 2011, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Scheibel, R.S. Functional Magnetic Resonance Imaging of Cognitive Control Following Traumatic Brain Injury. Front. Neurol. 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.J.; Gardner, A.J.; Wojtowicz, M.; Williams, W.H.; Iverson, G.L.; Stanwell, P. Task-Related Functional Magnetic Resonance Imaging Activations in Patients with Acute and Subacute Mild Traumatic Brain Injury: A Coordinate-Based Meta-Analysis. Neuroimage Clin. 2019, 25, 102129. [Google Scholar] [CrossRef] [PubMed]
- Bickart, K.; Sheridan, C.; Frees, D.; Kang, K.; Fischer, J.; Parks, C.; Kashou, A. A Systematic Review of Resting-State fMRI in Traumatic Brain Injury Across Injury Age, Severity, Mechanism, Chronicity, and Imaging Methods (P8-1.009). Neurology 2023, 100, 4146. [Google Scholar] [CrossRef]
- Joyce, J.M.; La, P.L.; Walker, R.; Harris, A.D. Magnetic Resonance Spectroscopy of Traumatic Brain Injury and Subconcussive Hits: A Systematic Review and Meta-Analysis. J. Neurotrauma 2022, 39, 1455–1476. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, N.; Matthews, P.M.; Arnold, D.L. Reversible Decreases in N-Acetylaspartate after Acute Brain Injury. Magn. Reason. Med. 1995, 34, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.L.H.; Jalloh, I.; Hutchinson, P.J. Glycolysis and the Significance of Lactate in Traumatic Brain Injury. Front. Neurosci. 2015, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Traumatic Brain Injury: Progress and Challenges in Prevention, Clinical Care, and Research—PMC. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC10427240/ (accessed on 8 May 2025).
- Ware, J.B.; Sinha, S.; Morrison, J.; Walter, A.E.; Gugger, J.J.; Schneider, A.L.C.; Dabrowski, C.; Zamore, H.; Wesley, L.; Magdamo, B.; et al. Dynamic Contrast Enhanced MRI for Characterization of Blood-Brain-Barrier Dysfunction after Traumatic Brain Injury. Neuroimage Clin. 2022, 36, 103236. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.; Hof, J.; Nihom, J. Susceptibility-Weighted MRI and Microbleeds in Mild Traumatic Brain Injury: Prediction of Posttraumatic Complaints? Eur. Neurol. 2022, 85, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Wen, Q.; Thukral, R.; Yang, H.-C.; Gill, J.M.; Gao, S.; Lane, K.A.; Meier, T.B.; Riggen, L.D.; Harezlak, J.; et al. Longitudinal Associations Between Blood Biomarkers and White Matter MRI in Sport-Related Concussion. Neurology 2023, 101, e189–e201. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Arun, P.; Ariyannur, P.S.; Namboodiri, A.M.A. N-Acetylaspartate Reductions in Brain Injury: Impact on Post-Injury Neuroenergetics, Lipid Synthesis, and Protein Acetylation. Front. Neuroenergetics 2013, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Winzeck, S.; Correia, M.M.; Czeiter, E.; Whitehouse, D.; Kornaropoulos, E.N.; Williams, G.B.; Verheyden, J.; Das, T.; Tenovuo, O.; et al. Predicting Recovery in Patients with Mild Traumatic Brain Injury and a Normal CT Using Serum Biomarkers and Diffusion Tensor Imaging (CENTER-TBI): An Observational Cohort Study. eClinicalMedicine 2024, 75, 102751. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, F.; Bellapart, J.; Walsham, J.; Jacobson, E.; To, X.V.; Manzanero, S.; Brown, N.; Meyer, J.; Stuart, J.; Evans, T.; et al. PREdiction and Diagnosis Using Imaging and Clinical Biomarkers Trial in Traumatic Brain Injury (PREDICT-TBI) Study Protocol: An Observational, Prospective, Multicentre Cohort Study for the Prediction of Outcome in Moderate-to-Severe TBI. BMJ Open 2023, 13, e067740. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.H.; Habig, K.; Wright, C.; Hughes, A.; Davies, G.; Imray, C.H.E. Pre-Hospital Emergency Medicine. Lancet 2015, 386, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Unterberg, A.; Kiening, K.; Schmiedek, P.; Lanksch, W. Long-Term Observations of Intracranial Pressure after Severe Head Injury. The Phenomenon of Secondary Rise of Intracranial Pressure. Neurosurgery 1993, 32, 17. [Google Scholar] [CrossRef] [PubMed]
- Hossain, I.; Rostami, E.; Marklund, N. The Management of Severe Traumatic Brain Injury in the Initial Postinjury Hours—Current Evidence and Controversies. Curr. Opin. Crit. Care 2023, 29, 650. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.J.; Veenith, T.; Scomparin, C.; Wilson, M.H.; Hutchinson, P.J.; Kolias, A.G.; Lall, R.; Regan, S.; Mason, J.; Andrews, P.; et al. Sugar or Salt (“SOS”): A Protocol for a UK Multicentre Randomised Trial of Mannitol and Hypertonic Saline in Severe Traumatic Brain Injury and Intracranial Hypertension. J. Intensive Care Soc. 2022, 23, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Popal, Z.; Bossers, S.M.; Terra, M.; Schober, P.; de Leeuw, M.A.; Bloemers, F.W.; Giannakopoulos, G.F. Effect of Physician-Staffed Emergency Medical Services (P-EMS) on the Outcome of Patients with Severe Traumatic Brain Injury: A Review of the Literature. Prehosp. Emerg. Care 2019, 23, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, G.W.J.; Rubiano, A.M.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; Shutter, L.; et al. Guidelines for the Management of Severe Traumatic Brain Injury: 2020 Update of the Decompressive Craniectomy Recommendations. Neurosurgery 2020, 87, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bar-Joseph, G.; Guilburd, Y.; Tamir, A.; Guilburd, J.N. Effectiveness of Ketamine in Decreasing Intracranial Pressure in Children with Intracranial Hypertension. J. Neurosurg. Pediatr. 2009, 4, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; McCue, J.J.; DiTuro, P.; Galpin, A.J.; Wood, T.R. Mitigating Traumatic Brain Injury: A Narrative Review of Supplementation and Dietary Protocols. Nutrients 2024, 16, 2430. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Liu, S.; Chen, J.; Wang, H.; Wang, Z. Microglial Polarization Pathways and Therapeutic Drugs Targeting Activated Microglia in Traumatic Brain Injury. Neural Regen. Res. 2026, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Cuzzocrea, S.; Esposito, E.; Campolo, M.; Niehoff, M.L.; Doyle, T.M.; Salvemini, D. Adenosine A3 Receptor as a Novel Therapeutic Target to Reduce Secondary Events and Improve Neurocognitive Functions Following Traumatic Brain Injury. J. Neuroinflamm. 2020, 17, 339. [Google Scholar] [CrossRef] [PubMed]
- Treating Traumatic Brain Injury with Minocycline—Neurotherapeutics. Available online: https://www.neurotherapeuticsjournal.org/article/S1878-7479(23)01997-9/fulltext (accessed on 8 May 2025).
- Anderson, G.D.; Peterson, T.C.; Vonder Haar, C.; Farin, F.M.; Bammler, T.K.; MacDonald, J.W.; Kantor, E.D.; Hoane, M.R. Effect of Traumatic Brain Injury, Erythropoietin, and Anakinra on Hepatic Metabolizing Enzymes and Transporters in an Experimental Rat Model. AAPS J. 2015, 17, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Balu, R. Inflammation and Immune System Activation After Traumatic Brain Injury. Curr. Neurol Neurosci. Rep. 2014, 14, 484. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Nyanzu, M.; Yang, S.; Zhu, X.; Wang, K.; Ru, J.; Yu, E.; Zhang, H.; Wang, Z.; Shen, J.; et al. VX765 Attenuates Pyroptosis and HMGB1/TLR4/NF-κB Pathways to Improve Functional Outcomes in TBI Mice. Oxidative Med. Cell. Longev. 2020, 2020, 7879629. [Google Scholar] [CrossRef] [PubMed]
- Susanto, M.; Pangihutan Siahaan, A.M.; Wirjomartani, B.A.; Setiawan, H.; Aryanti, C. Michael The Neuroprotective Effect of Statin in Traumatic Brain Injury: A Systematic Review. World Neurosurg. X 2023, 19, 100211. [Google Scholar] [CrossRef] [PubMed]
- Alderson, P.; Roberts, I. Corticosteroids for Acute Traumatic Brain Injury. Cochrane Database Syst. Rev. 2005, 1, CD000196. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeier, F.; Schinzel, R.; Beer, R.; Bulters, D.; LeFrant, J.-Y.; Sahuquillo, J.; Unterberg, A.; Andrews, P.; Belli, A.; Ibanez, J.; et al. Efficacy of Ronopterin (VAS203) in Patients with Moderate and Severe Traumatic Brain Injury (NOSTRA Phase III Trial): Study Protocol of a Confirmatory, Placebo-Controlled, Randomised, Double Blind, Multi-Centre Study. Trials 2020, 21, 80. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.S.; Langley, B. Epigenetic Changes Following Traumatic Brain Injury and Their Implications for Outcome, Recovery and Therapy. Neurosci. Lett. 2016, 625, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, Y.; Xu, S.; Wei, L.; Zhang, Y.; Chen, W.; Liu, M.; Zhong, C. HDAC Inhibitor Attenuates Rat Traumatic Brain Injury Induced Neurological Impairments. Heliyon 2023, 9, e18485. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.-T.; Lei, P.; Wang, H.-C.; Zhang, A.-L.; Han, Z.-L.; Chen, X.; Li, S.-H.; Jiang, R.-C.; Kang, C.-S.; Zhang, J.-N. miR-21 Improves the Neurological Outcome after Traumatic Brain Injury in Rats. Sci. Rep. 2014, 4, 6718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shao, A.; Xu, W.; Wu, H.; Deng, Y. Advance of Stem Cell Treatment for Traumatic Brain Injury. Front. Cell. Neurosci. 2019, 13, 301. [Google Scholar] [CrossRef] [PubMed]
- Mesenchymal Stem Cell Therapy Modulates the Inflammatory Response in Experimental Traumatic Brain Injury—Galindo—2011—Neurology Research International—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1155/2011/564089 (accessed on 8 May 2025).
- Bioactive Scaffolds with Enhanced Supramolecular Motion Promote Recovery from Spinal Cord Injury. Available online: https://www.science.org/doi/10.1126/science.abh3602 (accessed on 8 May 2025).
- Li, Q.; Shao, X.; Dai, X.; Guo, Q.; Yuan, B.; Liu, Y.; Jiang, W. Recent Trends in the Development of Hydrogel Therapeutics for the Treatment of Central Nervous System Disorders. NPG Asia Mater. 2022, 14, 1–14. [Google Scholar] [CrossRef]
- Kou, Z.; Gattu, R.; Kobeissy, F.; Welch, R.D.; O’Neil, B.J.; Woodard, J.L.; Ayaz, S.I.; Kulek, A.; Kas-Shamoun, R.; Mika, V.; et al. Combining Biochemical and Imaging Markers to Improve Diagnosis and Characterization of Mild Traumatic Brain Injury in the Acute Setting: Results from a Pilot Study. PLoS ONE 2013, 8, e80296. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.K.; Trezise, J.; Kamnaksh, A.; Bekdash, R.; Johnston, L.A.; Ordidge, R.; Semple, B.D.; Gardner, A.J.; Stanwell, P.; O’Brien, T.J.; et al. Behavioral, Blood and Magnetic Resonance Imaging Biomarkers of Experimental Mild Traumatic Brain Injury. Sci. Rep. 2016, 6, 28713. [Google Scholar] [CrossRef] [PubMed]
- Massaad, E.; Shin, J.H.; Gibbs, W.N. The Prognostic Role of Magnetic Resonance Imaging Biomarkers in Mild Traumatic Injury. JAMA Netw. Open 2021, 4, e211824. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 6 June 2025).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaniuk, J.K.; Kumar, D.; Mazurek, C.; Khavari, S.; Sollenberger, C.; Ahuja, A.; Mossner, J.M.; Ahuja, C.S. From Acute Injury to Chronic Neurodegeneration: Molecular Mechanisms Linking Secondary Brain Injury to Long-Term Pathology. Int. J. Mol. Sci. 2025, 26, 7191. https://doi.org/10.3390/ijms26157191
Kaniuk JK, Kumar D, Mazurek C, Khavari S, Sollenberger C, Ahuja A, Mossner JM, Ahuja CS. From Acute Injury to Chronic Neurodegeneration: Molecular Mechanisms Linking Secondary Brain Injury to Long-Term Pathology. International Journal of Molecular Sciences. 2025; 26(15):7191. https://doi.org/10.3390/ijms26157191
Chicago/Turabian StyleKaniuk, Julia K., Divy Kumar, Christopher Mazurek, Sepehr Khavari, Christopher Sollenberger, Arun Ahuja, James M. Mossner, and Christopher S. Ahuja. 2025. "From Acute Injury to Chronic Neurodegeneration: Molecular Mechanisms Linking Secondary Brain Injury to Long-Term Pathology" International Journal of Molecular Sciences 26, no. 15: 7191. https://doi.org/10.3390/ijms26157191
APA StyleKaniuk, J. K., Kumar, D., Mazurek, C., Khavari, S., Sollenberger, C., Ahuja, A., Mossner, J. M., & Ahuja, C. S. (2025). From Acute Injury to Chronic Neurodegeneration: Molecular Mechanisms Linking Secondary Brain Injury to Long-Term Pathology. International Journal of Molecular Sciences, 26(15), 7191. https://doi.org/10.3390/ijms26157191