
The Roles of Lactate and Lactylation in Diseases Related to Mitochondrial Dysfunction

Abstract
1. Introduction
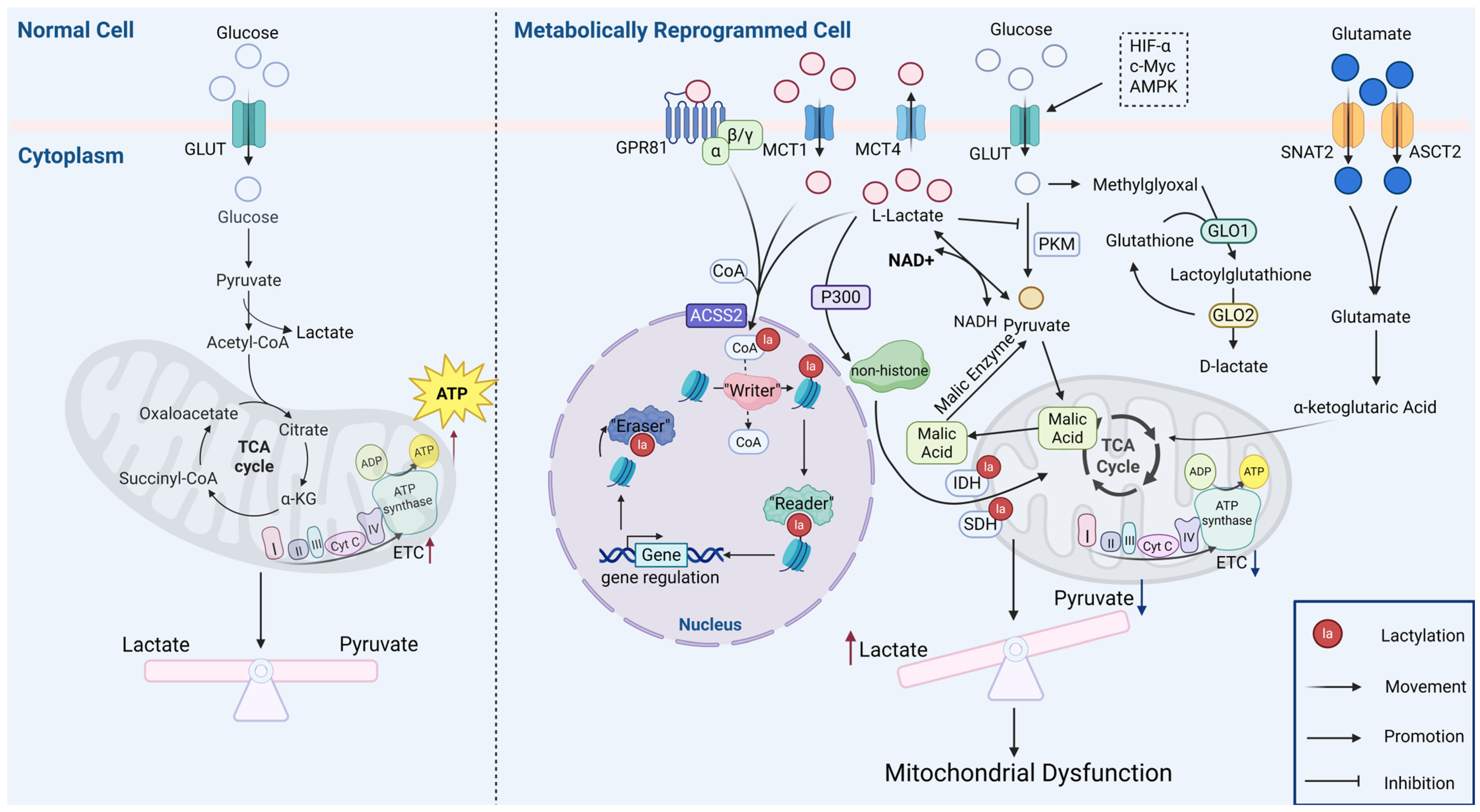
2. Mechanisms of Lactate Generation and Lactylation
3. Metabolic Reprogramming and Mitochondrial Dysfunction
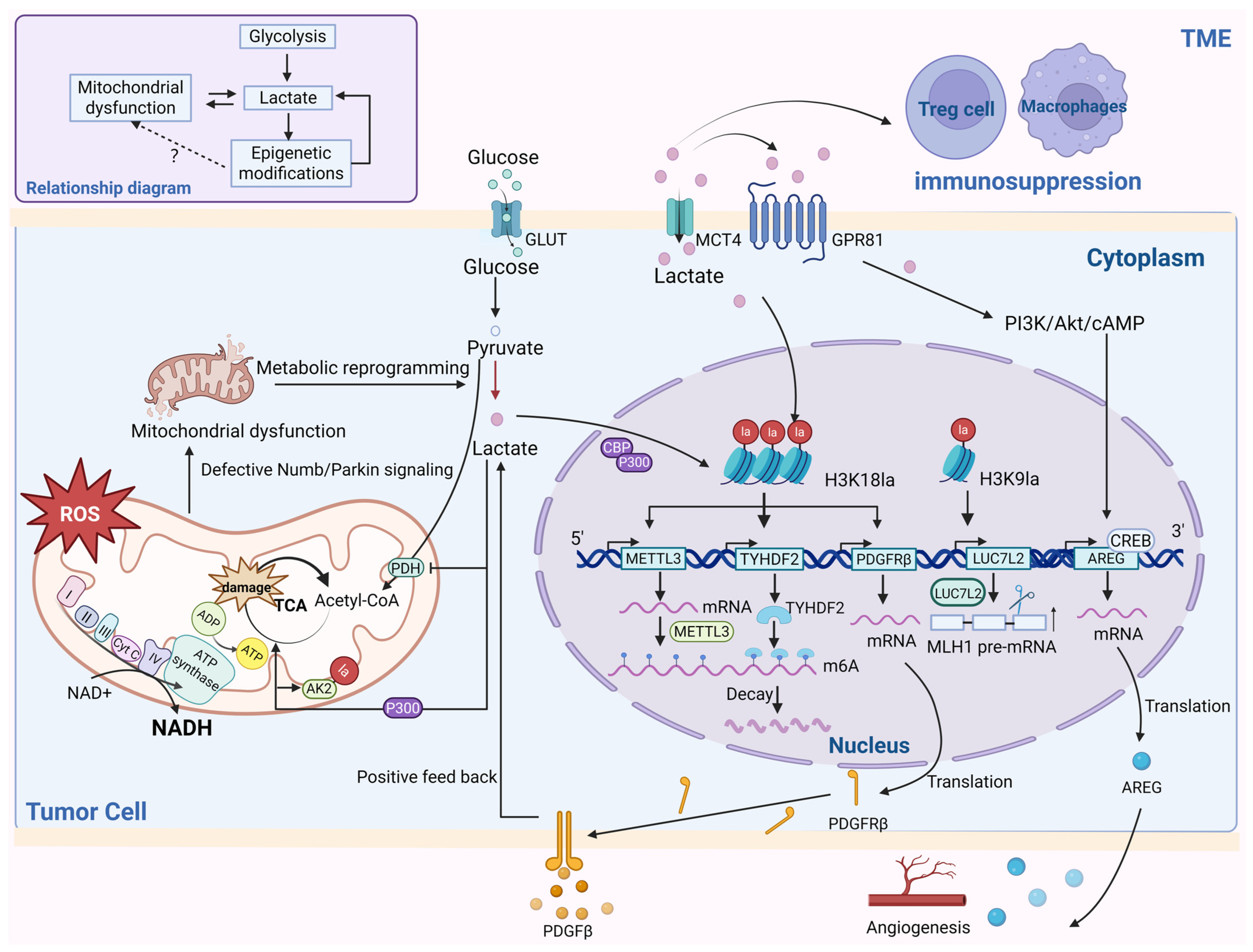
4. Cancer
5. Neurological Disorders
5.1. Ischemia–Reperfusion Injury
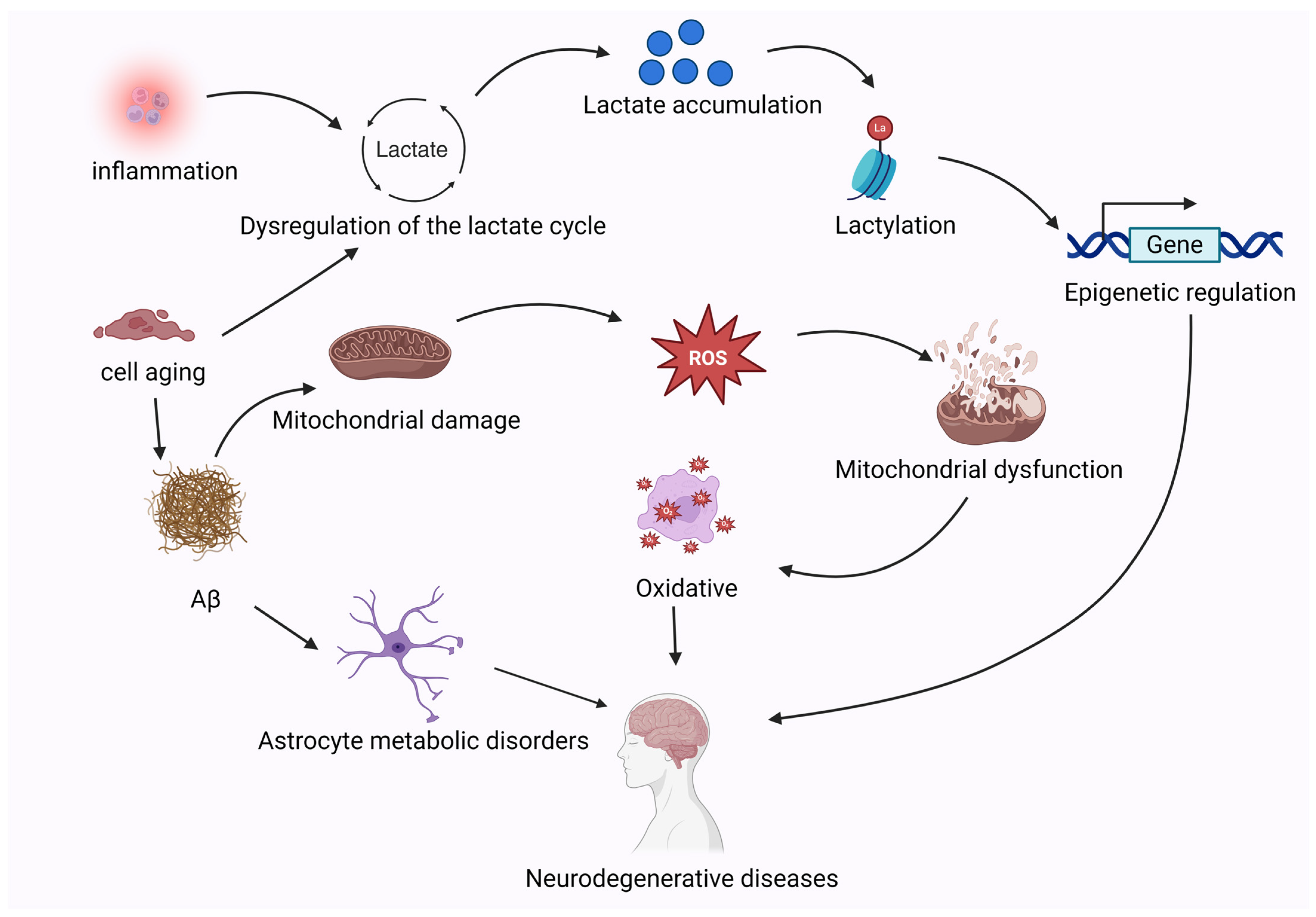
5.2. Neurodegenerative Diseases
6. Other Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Mechanism | Detail | Reference |
|---|---|---|---|
| APAP-induced liver injury | Inactivation of SIRT1\PGC-1α\LDHB | Decreasing SIRT1/PGC-1α/LDHB expression leads to metabolism reprogramming, and increased protein lactylation, mitochondrial lactate levels, and pathological damage in liver mitochondria. PGC-1α overexpression increased LDHB synthesis, reduced lactylation, and induced a switch from lactate to pyruvate production. | [119] |
| Hepatic ferroptosis | Inactivation of Parkin\OXSM | Lactate activates mitochondrial phosphoenolpyruvate carboxykinase 2 (PCK2) through KAT8-mediated lactylation modification. This activation suppresses Parkin-mediated ubiquitination degradation of 3-oxoacyl-ACP synthase (OXSM), leading to metabolic reprogramming of mitochondrial fatty acid synthesis (mtFAS). | [122] |
| Sepsis induced acute kidney injury | Activation of Fis 1 | Pathological stimulation leads to metabolism reprogramming, lactate accumulation mediates lysine 20 lactylation (K20la) of mitochondrial fission 1 protein (Fis1), and elevated Fis1 K20la promotes excessive mitochondrial fission, resulting in ATP depletion, overproduction of mitochondrial reactive oxygen species (ROS), and mitochondrial apoptosis. | [51] |
| Pulmonary fibrosis | Inactivation of ERK/DRP1 | Lactate produced by metabolic reprogramming could promote lung fibrosis by increasing mitochondrial fission-derived ROS via ERK/DRP1 signaling. | [123] |
| Inflammatory responses in macrophages | Overexpression of Arg 1 | Mitochondrial-fragmentation-caused metabolic reprogramming leads to increase pan-histone lactylation, which caused an increase in arginase 1 expression, which promotes Inflammation Resolution Responses. | [114] |
| Vascular calcification | Dysfunction of PARP1\POLG\UCP2 | Lactate induced the translocation of PARP1 from the nucleus to mitochondria, where it subsequently bound to DNA polymerase gamma catalytic subunit (POLG) and inhibited mitochondrial DNA synthesis. | [124] |
| Activation of NR4A\DNA-PKcs\p53 | Lactate accelerates vascular smooth muscle cell (VSMC) calcification by suppressing BCL2-interacting protein 3 (BNIP3)-mediated mitophagy. Lactate enhances mitochondrial fission through activation of the nuclear receptor subfamily 4 group A member 1 (NR4A1) pathway. | [125] | |
| Retinal degeneration | Lactate-mediated regulation | Lactate activated autophagy by upregulating the ratio of LC3II/I, and increased formation of LC3 puncta and autophagic vacuole. Lactate prevented H2O2-induced mitochondrial fission and maintained mitochondrial function by alleviating H2O2-induced mitochondrial membrane potential disruption and intracellular ROS generation. | [126] |
| Maintain skeletal muscle function | Activation of SIRT1\PGC-1α | MCT1 deficiency leads to lactate accumulation in the cytoplasm, which, in turn, activates the SIRT1\PGC-1α signaling pathway to regulate mitochondrial biogenesis. | [127] |
| Activation of Vps34 | ULK1-mediated metabolic reprogramming leads to lactate accumulation and, in turn, lactated Vps34 increases lipid kinase activity to enhance mitochondrial autophagy and endolysosomal degradation. | [128] |
7. Discussion and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Certo, M.; Tsai, C.-H.; Pucino, V.; Ho, P.-C.; Mauro, C. Lactate Modulation of Immune Responses in Inflammatory versus Tumour Microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Jiang, H.; Ren, Y.; Yu, J.; Hu, S.; Zhang, J. Analysis of Lactate Metabolism-Related Genes and Their Association with Immune Infiltration in Septic Shock via Bioinformatics Method. Front. Genet. 2023, 14, 1223243. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.F.; Ruminot, I.; Martín, A.S.; Lerchundi, R.; Fernández-Moncada, I.; Baeza-Lehnert, F. Aerobic Glycolysis in the Brain: Warburg and Crabtree Contra Pasteur. Neurochem. Res. 2021, 46, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-Y. Lactate: A Multifunctional Signaling Molecule. Yeungnam Univ. J. Med. 2021, 38, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate Metabolism in Human Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef]
- Li, L.; Chen, K.; Wang, T.; Wu, Y.; Xing, G.; Chen, M.; Hao, Z.; Zhang, C.; Zhang, J.; Ma, B.; et al. Glis1 Facilitates Induction of Pluripotency via an Epigenome-Metabolome-Epigenome Signalling Cascade. Nat. Metab. 2020, 2, 882–892. [Google Scholar] [CrossRef]
- Irizarry-Caro, R.A.; McDaniel, M.M.; Overcast, G.R.; Jain, V.G.; Troutman, T.D.; Pasare, C. TLR Signaling Adapter BCAP Regulates Inflammatory to Reparatory Macrophage Transition by Promoting Histone Lactylation. Proc. Natl. Acad. Sci. USA 2020, 117, 30628–30638. [Google Scholar] [CrossRef]
- Yu, J.; Chai, P.; Xie, M.; Ge, S.; Ruan, J.; Fan, X.; Jia, R. Histone Lactylation Drives Oncogenesis by Facilitating m6A Reader Protein YTHDF2 Expression in Ocular Melanoma. Genome Biol. 2021, 22, 85. [Google Scholar] [CrossRef]
- Wan, N.; Wang, N.; Yu, S.; Zhang, H.; Tang, S.; Wang, D.; Lu, W.; Li, H.; Delafield, D.G.; Kong, Y.; et al. Cyclic Immonium Ion of Lactyllysine Reveals Widespread Lactylation in the Human Proteome. Nat. Methods 2022, 19, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Bonnay, F.; Veloso, A.; Steinmann, V.; Köcher, T.; Abdusselamoglu, M.D.; Bajaj, S.; Rivelles, E.; Landskron, L.; Esterbauer, H.; Zinzen, R.P.; et al. Oxidative Metabolism Drives Immortalization of Neural Stem Cells during Tumorigenesis. Cell 2020, 182, 1490–1507.e19. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, F.; Li, C.; Nan, P.; Song, Y.; Wan, X.; Mo, H.; Wang, J.; Zhou, Y.; Guo, Y.; et al. MTA1, a Novel ATP Synthase Complex Modulator, Enhances Colon Cancer Liver Metastasis by Driving Mitochondrial Metabolism Reprogramming. Adv. Sci. 2023, 10, e2300756. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Feng, F.; Wu, J.; Fan, S.; Han, J.; Wang, S.; Yang, L.; Liu, W.; Wang, C.; Xu, K. Demethylzeylasteral Targets Lactate by Inhibiting Histone Lactylation to Suppress the Tumorigenicity of Liver Cancer Stem Cells. Pharmacol. Res. 2022, 181, 106270. [Google Scholar] [CrossRef]
- Vander Linden, C.; Corbet, C.; Bastien, E.; Martherus, R.; Guilbaud, C.; Petit, L.; Wauthier, L.; Loriot, A.; De Smet, C.; Feron, O. Therapy-Induced DNA Methylation Inactivates MCT1 and Renders Tumor Cells Vulnerable to MCT4 Inhibition. Cell Rep. 2021, 35, 109202. [Google Scholar] [CrossRef]
- He, Y.; Ji, Z.; Gong, Y.; Fan, L.; Xu, P.; Chen, X.; Miao, J.; Zhang, K.; Zhang, W.; Ma, P.; et al. Numb/Parkin-Directed Mitochondrial Fitness Governs Cancer Cell Fate via Metabolic Regulation of Histone Lactylation. Cell Rep. 2023, 42, 112033. [Google Scholar] [CrossRef]
- Gu, L.; Ding, X.; Wang, Y.; Gu, M.; Zhang, J.; Yan, S.; Li, N.; Song, Z.; Yin, J.; Lu, L.; et al. Spexin Alleviates Insulin Resistance and Inhibits Hepatic Gluconeogenesis via the FoxO1/PGC-1α Pathway in High-Fat-Diet-Induced Rats and Insulin Resistant Cells. Int. J. Biol. Sci. 2019, 15, 2815–2829. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; van Hout, G.P.J.; Timmers, L.; de Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 23–34. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Brooks, G.A. Lactate as a Fulcrum of Metabolism. Redox Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Dou, X.; Fu, Q.; Long, Q.; Liu, S.; Zou, Y.; Fu, D.; Xu, Q.; Jiang, Z.; Ren, X.; Zhang, G.; et al. PDK4-Dependent Hypercatabolism and Lactate Production of Senescent Cells Promotes Cancer Malignancy. Nat. Metab. 2023, 5, 1887–1910. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Lee, I.-K.; Suk, K. Metabolic Reprogramming by the Pyruvate Dehydrogenase Kinase-Lactic Acid Axis: Linking Metabolism and Diverse Neuropathophysiologies. Neurosci. Biobehav. Rev. 2016, 68, 1–19. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Zhao, R.; Yi, Y.; Liu, H.; Xu, J.; Chen, S.; Wu, D.; Wang, L.; Li, F. RHOF Promotes Snail1 Lactylation by Enhancing PKM2-Mediated Glycolysis to Induce Pancreatic Cancer Cell Endothelial-Mesenchymal Transition. Cancer Metab. 2024, 12, 32. [Google Scholar] [CrossRef]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 Signaling and Proton Motive Force in Cancer: Role in Angiogenesis, Immune Escape, Nutrition, and Warburg Phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Bæk, M.; Liu, W.; Gao, J.; Danková, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I Histone Deacetylases (HDAC1-3) Are Histone Lysine Delactylases. Sci. Adv. 2022, 8, eabi6696. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, H.; Shoji, H.; Otabi, H.; Toyoda, A.; Katoh, K.; Namihira, M.; Miyakawa, T. Protein Lactylation Induced by Neural Excitation. Cell Rep. 2021, 37, 109820. [Google Scholar] [CrossRef]
- Gao, X.; Pang, C.; Fan, Z.; Wang, Y.; Duan, Y.; Zhan, H. Regulation of Newly Identified Lysine Lactylation in Cancer. Cancer Lett. 2024, 587, 216680. [Google Scholar] [CrossRef]
- Gaffney, D.O.; Jennings, E.Q.; Anderson, C.C.; Marentette, J.O.; Shi, T.; Schou Oxvig, A.-M.; Streeter, M.D.; Johannsen, M.; Spiegel, D.A.; Chapman, E.; et al. Non-Enzymatic Lysine Lactoylation of Glycolytic Enzymes. Cell Chem. Biol. 2020, 27, 206–213.e6. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, J.; Zhu, Z.; Mao, Q.; Xu, Z.; Singh, P.K.; Rimayi, C.C.; Moreno-Yruela, C.; Xu, S.; Li, G.; et al. Lysine L-Lactylation Is the Dominant Lactylation Isomer Induced by Glycolysis. Nat. Chem. Biol. 2025, 21, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, M.; Baur, R.; Stoll, A.; Mackensen, A.; Mougiakakos, D. Linking Immunoevasion and Metabolic Reprogramming in B-Cell-Derived Lymphomas. Front. Oncol. 2020, 10, 594782. [Google Scholar] [CrossRef]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem Cells. Asymmetric Apportioning of Aged Mitochondria between Daughter Cells Is Required for Stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of Metabolic Enzymes Coordinates Carbon Source Utilization and Metabolic Flux. Science 2010, 327, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Pongsuwan, K.; Kusirisin, P.; Narongkiattikhun, P.; Chattipakorn, S.C.; Chattipakorn, N. Mitochondria and Vascular Calcification in Chronic Kidney Disease: Lessons Learned from the Past to Improve Future Therapy. J. Cell. Physiol. 2022, 237, 4369–4396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, S.-N.; Yuan, S.-T.; Lei, X.; Sun, X.; Xing, L.; Li, H.-J.; He, C.-X.; Qin, W.; Zhao, D.; et al. Multiple Functions of Autophagy in Vascular Calcification. Cell Biosci. 2021, 11, 159. [Google Scholar] [CrossRef]
- Ma, W.; Jia, K.; Cheng, H.; Xu, H.; Li, Z.; Zhang, H.; Xie, H.; Sun, H.; Yi, L.; Chen, Z.; et al. Orphan Nuclear Receptor NR4A3 Promotes Vascular Calcification via Histone Lactylation. Circ. Res. 2024, 134, 1427–1447. [Google Scholar] [CrossRef]
- Shao, G.; Wang, L.; Wang, X.; Fu, C. Apaf-1/Caspase-4 Pyroptosome: A Mediator of Mitochondrial Permeability Transition-Triggered Pyroptosis. Signal Transduct. Target. Ther. 2021, 6, 116. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, J.; Zhou, Q.; He, X.; Zheng, Z.; Wei, Y.; Zhou, K.; Lin, Y.; Yu, H.; Zhang, H.; et al. Hypoxia Induces Mitochondrial Protein Lactylation to Limit Oxidative Phosphorylation. Cell Res. 2024, 34, 13–30. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, M.; Liu, Y.; Zhao, S.; Wang, Y.; Wang, M.; Niu, W.; Jin, F.; Li, Z. Histone Lactylation Driven by mROS-Mediated Glycolytic Shift Promotes Hypoxic Pulmonary Hypertension. J. Mol. Cell Biol. 2023, 14, mjac073. [Google Scholar] [CrossRef]
- Caielli, S.; Cardenas, J.; de Jesus, A.A.; Baisch, J.; Walters, L.; Blanck, J.P.; Balasubramanian, P.; Stagnar, C.; Ohouo, M.; Hong, S.; et al. Erythroid Mitochondrial Retention Triggers Myeloid-Dependent Type I Interferon in Human SLE. Cell 2021, 184, 4464–4479.e19. [Google Scholar] [CrossRef]
- Li, H.; Boulougoura, A.; Endo, Y.; Tsokos, G.C. Abnormalities of T Cells in Systemic Lupus Erythematosus: New Insights in Pathogenesis and Therapeutic Strategies. J. Autoimmun. 2022, 132, 102870. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, H.; Liu, M.; Zheng, M.; Wen, Z.; Shen, H. Mitochondrial DNA Programs Lactylation of cGAS to Induce IFN Responses in Patients with Systemic Lupus Erythematosus. J. Immunol. 2024, 213, 795–807. [Google Scholar] [CrossRef]
- She, H.; Hu, Y.; Zhao, G.; Du, Y.; Wu, Y.; Chen, W.; Li, Y.; Wang, Y.; Tan, L.; Zhou, Y.; et al. Dexmedetomidine Ameliorates Myocardial Ischemia-Reperfusion Injury by Inhibiting MDH2 Lactylation via Regulating Metabolic Reprogramming. Adv. Sci. 2024, 11, e2409499. [Google Scholar] [CrossRef] [PubMed]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised Acyl-CoA Metabolism and Roles in Chromatin Regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Breusegem, S.Y.; Larrieu, D.; Rubinsztein, D.C. P300 Nucleocytoplasmic Shuttling Underlies mTORC1 Hyperactivation in Hutchinson-Gilford Progeria Syndrome. Nat. Cell Biol. 2024, 26, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.-Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive Feedback Regulation of Microglial Glucose Metabolism by Histone H4 Lysine 12 Lactylation in Alzheimer’s Disease. Cell Metab. 2022, 34, 634–648.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, L.; Peng, J.; Zhang, X.; Zhang, F.; Wu, Y.; Huang, A.; Du, F.; Liao, Y.; He, Y.; et al. Astrocytic LRP1 Enables Mitochondria Transfer to Neurons and Mitigates Brain Ischemic Stroke by Suppressing ARF1 Lactylation. Cell Metab. 2024, 36, 2054–2068.e14. [Google Scholar] [CrossRef]
- Baumeister, J.; Chatain, N.; Hubrich, A.; Maié, T.; Costa, I.G.; Denecke, B.; Han, L.; Küstermann, C.; Sontag, S.; Seré, K.; et al. Hypoxia-Inducible Factor 1 (HIF-1) Is a New Therapeutic Target in JAK2V617F-Positive Myeloproliferative Neoplasms. Leukemia 2020, 34, 1062–1074. [Google Scholar] [CrossRef]
- Yao, Y.; Bade, R.; Li, G.; Zhang, A.; Zhao, H.; Fan, L.; Zhu, R.; Yuan, J. Global-Scale Profiling of Differential Expressed Lysine-Lactylated Proteins in the Cerebral Endothelium of Cerebral Ischemia–Reperfusion Injury Rats. Cell. Mol. Neurobiol. 2022, 43, 1989–2004. [Google Scholar] [CrossRef]
- An, S.; Yao, Y.; Hu, H.; Wu, J.; Li, J.; Li, L.; Wu, J.; Sun, M.; Deng, Z.; Zhang, Y.; et al. PDHA1 Hyperacetylation-Mediated Lactate Overproduction Promotes Sepsis-Induced Acute Kidney Injury via Fis1 Lactylation. Cell Death Dis. 2023, 14, 457. [Google Scholar] [CrossRef]
- Li, J.; Shi, X.; Hou, F.; Luan, X.; Chen, L. #1225 Lactate Aggravates Mitochondrial Dysfunction via ALDH2 Lactylation in Acute Kidney Injury. Nephrol. Dial. Transplant. 2024, 39, gfae069-1090-1225. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, J.; Sun, Y.; Chen, T.; Wang, J.; Yu, S. Mitochondria in Cancer Stem Cells: Achilles Heel or Hard Armor. Trends Cell Biol. 2023, 33, 708–727. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA Variation and Cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg Effect in Non-Tumor Diseases Processes. J. Cell. Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, A.; Zou, Y.; Su, N.; Loscalzo, J.; Yang, Y. In Vivo Monitoring of Cellular Energy Metabolism Using SoNar, a Highly Responsive Sensor for NAD(+)/NADH Redox State. Nat. Protoc. 2016, 11, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Xiong, N.; Yan, R.; Li, S.-T.; Liu, H.; Mao, Q.; Sun, Y.; Shen, S.; Ye, L.; Gao, P.; et al. PDHX Acetylation Facilitates Tumor Progression by Disrupting PDC Assembly and Activating Lactylation-Mediated Gene Expression. Protein Cell 2025, 16, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Jin, J.; Duan, Y.; Xie, Z.; Li, Y.; Gao, A.; Gu, M.; Zhang, X.; Peng, C.; Xia, C.; et al. Mitochondrial Uncoupling Coordinated With PDH Activation Safely Ameliorates Hyperglycemia via Promoting Glucose Oxidation. Diabetes 2019, 68, 2197–2209. [Google Scholar] [CrossRef]
- Zhu, H.; Chan, K.T.; Huang, X.; Cerra, C.; Blake, S.; Trigos, A.S.; Anderson, D.; Creek, D.J.; De Souza, D.P.; Wang, X.; et al. Cystathionine-β-Synthase Is Essential for AKT-Induced Senescence and Suppresses the Development of Gastric Cancers with PI3K/AKT Activation. eLife 2022, 11, e71929. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shin, K.J.; Park, S.-A.; Park, K.S.; Park, S.; Heo, K.; Seo, Y.-K.; Noh, D.-Y.; Ryu, S.H.; Suh, P.-G. G-Protein-Coupled Receptor 81 Promotes a Malignant Phenotype in Breast Cancer through Angiogenic Factor Secretion. Oncotarget 2016, 7, 70898–70911. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, Y.; Yang, B.; Sun, S.; Zhang, P.; Luo, Z.; Feng, T.; Cui, Z.; Zhu, T.; Li, Y.; et al. Lactylation of METTL16 Promotes Cuproptosis via m6A-Modification on FDX1 mRNA in Gastric Cancer. Nat. Commun. 2023, 14, 6523. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Luo, L.; Zhao, C.; Li, X.; Wang, Z.; Zeng, Z.; Yang, X.; Zheng, X.; Jie, H.; Kang, L.; et al. A Positive Feedback Loop between Inactive VHL-Triggered Histone Lactylation and PDGFRβ Signaling Drives Clear Cell Renal Cell Carcinoma Progression. Int. J. Biol. Sci. 2022, 18, 3470–3483. [Google Scholar] [CrossRef] [PubMed]
- Qiao, K.; Chen, C.; Liu, H.; Qin, Y.; Liu, H. Pinin Induces Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma by Regulating m6A Modification. J. Oncol. 2021, 2021, 7529164. [Google Scholar] [CrossRef]
- Yue, Q.; Wang, Z.; Shen, Y.; Lan, Y.; Zhong, X.; Luo, X.; Yang, T.; Zhang, M.; Zuo, B.; Zeng, T.; et al. Histone H3K9 Lactylation Confers Temozolomide Resistance in Glioblastoma via LUC7L2-Mediated MLH1 Intron Retention. Adv. Sci. 2024, 11, e2309290. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, Z.; Yu, Y.; Zhang, P. HIF1α Lactylation Enhances KIAA1199 Transcription to Promote Angiogenesis and Vasculogenic Mimicry in Prostate Cancer. Int. J. Biol. Macromol. 2022, 222, 2225–2243. [Google Scholar] [CrossRef]
- Yang, Z.; Yan, C.; Ma, J.; Peng, P.; Ren, X.; Cai, S.; Shen, X.; Wu, Y.; Zhang, S.; Wang, X.; et al. Lactylome Analysis Suggests Lactylation-Dependent Mechanisms of Metabolic Adaptation in Hepatocellular Carcinoma. Nat. Metab. 2023, 5, 61–79. [Google Scholar] [CrossRef]
- Kong, D.; Liu, C.; Miao, X.; Wang, Y.; Ding, X.; Gong, W. Current Statuses of Molecular Targeted and Immune Checkpoint Therapies in Hepatocellular Carcinoma. Am. J. Cancer Res. 2020, 10, 1522–1533. [Google Scholar]
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; et al. Lactate Modulates Cellular Metabolism Through Histone Lactylation-Mediated Gene Expression in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 647559. [Google Scholar] [CrossRef]
- Jia, L.; Liao, M.; Mou, A.; Zheng, Q.; Yang, W.; Yu, Z.; Cui, Y.; Xia, X.; Qin, Y.; Chen, M.; et al. Rheb-Regulated Mitochondrial Pyruvate Metabolism of Schwann Cells Linked to Axon Stability. Dev. Cell 2021, 56, 2980–2994.e6. [Google Scholar] [CrossRef]
- Huang, M.; Wu, Y.; Cheng, L.; Fu, L.; Yan, H.; Ru, H.; Mo, X.; Yan, L.; Su, Z. Multi-Omics Analyses of Glucose Metabolic Reprogramming in Colorectal Cancer. Front. Immunol. 2023, 14, 1179699. [Google Scholar] [CrossRef]
- Wei, Y.; Miao, Q.; Zhang, Q.; Mao, S.; Li, M.; Xu, X.; Xia, X.; Wei, K.; Fan, Y.; Zheng, X.; et al. Aerobic Glycolysis Is the Predominant Means of Glucose Metabolism in Neuronal Somata, Which Protects against Oxidative Damage. Nat. Neurosci. 2023, 26, 2081–2089. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s Disease: Current Evidence and Future Directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Yoon, N.A.; Jin, S.; Kim, J.D.; Liu, Z.W.; Sun, Q.; Cardone, R.; Kibbey, R.; Diano, S. UCP2-Dependent Redox Sensing in POMC Neurons Regulates Feeding. Cell Rep. 2022, 41, 111894. [Google Scholar] [CrossRef]
- Wei, L.; Yang, X.; Wang, J.; Wang, Z.; Wang, Q.; Ding, Y.; Yu, A. H3K18 Lactylation of Senescent Microglia Potentiates Brain Aging and Alzheimer’s Disease through the NFκB Signaling Pathway. J. Neuroinflamm. 2023, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wang, D.; Qu, Y.; Li, J.; An, K.; Mao, Z.; Li, J.; Xiong, Y.; Min, Z.; Xue, Z. Enhanced Glycolysis-Derived Lactate Promotes Microglial Activation in Parkinson’s Disease via Histone Lactylation. NPJ Park. Dis. 2025, 11, 3. [Google Scholar] [CrossRef]
- Wang, Z.; Hao, D.; Zhao, S.; Zhang, Z.; Zeng, Z.; Wang, X. Lactate and Lactylation: Clinical Applications of Routine Carbon Source and Novel Modification in Human Diseases. Mol. Cell. Proteom. 2023, 22, 100641. [Google Scholar] [CrossRef]
- Xue, X.; Wang, H.; Su, J. Inhibition of MiR-122 Decreases Cerebral Ischemia-Reperfusion Injury by Upregulating DJ-1-Phosphatase and Tensin Homologue Deleted on Chromosome 10 (PTEN)/Phosphonosinol-3 Kinase (PI3K)/AKT. Med. Sci. Monit. 2020, 26, e915825. [Google Scholar] [CrossRef]
- Liu, C.; Wang, G.; Han, W.; Tian, Q.; Li, M. Ferroptosis: A Potential Therapeutic Target for Stroke. Neural Regen. Res. 2023, 19, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Shao, G.; Zhang, Q.; Wang, X.; Meng, Y.; Wang, L.; Huang, F.; Yang, T.; Jin, Y.; Fu, C. The Antimicrobial Peptide PFR Induces Necroptosis Mediated by ER Stress and Elevated Cytoplasmic Calcium and Mitochondrial ROS Levels: Cooperation with Ara-C to Act against Acute Myeloid Leukemia. Signal Transduct. Target. Ther. 2019, 4, 38. [Google Scholar] [CrossRef]
- Nagase, M.; Takahashi, Y.; Watabe, A.M.; Kubo, Y.; Kato, F. On-Site Energy Supply at Synapses through Monocarboxylate Transporters Maintains Excitatory Synaptic Transmission. J. Neurosci. 2014, 34, 2605–2617. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.-H.; Cao, Y.-T.; Zhang, L.-L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef]
- Guerreiro, S.; Privat, A.-L.; Bressac, L.; Toulorge, D. CD38 in Neurodegeneration and Neuroinflammation. Cells 2020, 9, 471. [Google Scholar] [CrossRef]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinós, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef]
- Magistretti, P.J. Neuron–Glia Metabolic Coupling and Plasticity. Exp. Physiol. 2011, 96, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Arevalo, J.A.; Osmond, A.D.; Leija, R.G.; Curl, C.C.; Tovar, A.P. Lactate in Contemporary Biology: A Phoenix Risen. J. Physiol. 2022, 600, 1229–1251. [Google Scholar] [CrossRef]
- Cai, M.; Wang, H.; Song, H.; Yang, R.; Wang, L.; Xue, X.; Sun, W.; Hu, J. Lactate Is Answerable for Brain Function and Treating Brain Diseases: Energy Substrates and Signal Molecule. Front. Nutr. 2022, 9, 800901. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Hollnagel, J.-O.; Cesetti, T.; Schneider, J.; Vazetdinova, A.; Valiullina-Rakhmatullina, F.; Lewen, A.; Rozov, A.; Kann, O. Lactate Attenuates Synaptic Transmission and Affects Brain Rhythms Featuring High Energy Expenditure. iScience 2020, 23, 101316. [Google Scholar] [CrossRef]
- Hsieh, C.-F.; Liu, C.-K.; Lee, C.-T.; Yu, L.-E.; Wang, J.-Y. Acute Glucose Fluctuation Impacts Microglial Activity, Leading to Inflammatory Activation or Self-Degradation. Sci. Rep. 2019, 9, 840. [Google Scholar] [CrossRef]
- Ishihara, Y.; Itoh, K. Microglial Inflammatory Reactions Regulated by Oxidative Stress. J. Clin. Biochem. Nutr. 2023, 72, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mai, W.; Chen, L.; Cao, K.; Zhang, B.; Zhang, Z.; Liu, Y.; Lou, H.; Duan, S.; Gao, Z. mTOR-Mediated Metabolic Reprogramming Shapes Distinct Microglia Functions in Response to Lipopolysaccharide and ATP. Glia 2020, 68, 1031–1045. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate Metabolism and Recycling at the Excitatory Synapse in Health and Neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Halim, N.D.; Mcfate, T.; Mohyeldin, A.; Okagaki, P.; Korotchkina, L.G.; Patel, M.S.; Jeoung, N.H.; Harris, R.A.; Schell, M.J.; Verma, A. Phosphorylation Status of Pyruvate Dehydrogenase Distinguishes Metabolic Phenotypes of Cultured Rat Brain Astrocytes and Neurons. Glia 2010, 58, 1168–1176. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic Glycolysis and Tau Deposition in Preclinical Alzheimer’s Disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar] [CrossRef]
- Fu, J.; Lu, Z.-T.; Wu, G.; Yang, Z.-C.; Wu, X.; Wang, D.; Nie, Z.-M.; Sheng, Q. Gastrodia Elata Specific miRNA Attenuates Neuroinflammation via Modulating NF-κB Signaling Pathway. Int. J. Neurosci. 2024, 134, 1652–1662. [Google Scholar] [CrossRef]
- Tauffenberger, A.; Fiumelli, H.; Almustafa, S.; Magistretti, P.J. Lactate and Pyruvate Promote Oxidative Stress Resistance through Hormetic ROS Signaling. Cell Death Dis. 2019, 10, 653. [Google Scholar] [CrossRef]
- Hadzic, A.; Nguyen, T.D.; Hosoyamada, M.; Tomioka, N.H.; Bergersen, L.H.; Storm-Mathisen, J.; Morland, C. The Lactate Receptor HCA1 Is Present in the Choroid Plexus, the Tela Choroidea, and the Neuroepithelial Lining of the Dorsal Part of the Third Ventricle. Int. J. Mol. Sci. 2020, 21, 6457. [Google Scholar] [CrossRef]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 Improves the Supportive Ability of Astrocytes to Neurons by Promoting Aerobic Glycolysis in Alzheimer’s Disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Kettunen, M.I.; Hu, D.; Witney, T.H.; McLaughlin, R.; Gallagher, F.A.; Bohndiek, S.E.; Day, S.E.; Brindle, K.M. Magnetization Transfer Measurements of Exchange between Hyperpolarized [1-13C]Pyruvate and [1-13C]Lactate in a Murine Lymphoma. Magn. Reson. Med. 2010, 63, 872–880. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, I.A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Xi, Y.; Tao, K.; Wen, X.; Feng, D.; Mai, Z.; Ding, H.; Mao, H.; Wang, M.; Yang, Q.; Xiang, J.; et al. SIRT3-Mediated Deacetylation of DRP1K711 Prevents Mitochondrial Dysfunction in Parkinson’s Disease. Adv. Sci. 2025, 12, e2411235. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, L.; Qin, Q.; Wang, D.; Zhao, J.; Gao, H.; Yuan, X.; Zhang, J.; Zou, Y.; Mao, Z.; et al. Upregulated Hexokinase 2 Expression Induces the Apoptosis of Dopaminergic Neurons by Promoting Lactate Production in Parkinson’s Disease. Neurobiol. Dis. 2022, 163, 105605. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Wang, Y.; Peng, W.-W.; Zheng, Y.-J.; Zhang, T.-N.; Wang, P.-J.; Huang, J.-D.; Zeng, Q.-Y. Effects of Interleukin-6 and IL-6/AMPK Signaling Pathway on Mitochondrial Biogenesis and Astrocytes Viability under Experimental Septic Condition. Int. Immunopharmacol. 2018, 59, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, M.; Lin, Z.; Zhuang, Y.; Wang, H.; Jia, J.; Lu, Y.; Wang, Z.; Zou, H.; Zhao, H. Buyang Huanwu Decoction Promotes Neurovascular Remodeling by Modulating Astrocyte and Microglia Polarization in Ischemic Stroke Rats. J. Ethnopharmacol. 2024, 323, 117620. [Google Scholar] [CrossRef]
- Chen, C.; Chen, Y.; Liu, T.; Song, D.; Ma, D.; Cheng, O. Dexmedetomidine Can Enhance PINK1/Parkin-Mediated Mitophagy in MPTP-Induced PD Mice Model by Activating AMPK. Oxidative Med. Cell. Longev. 2022, 2022, 7511393. [Google Scholar] [CrossRef]
- Obsilova, V.; Obsil, T. Structural Insights into the Functional Roles of 14-3-3 Proteins. Front. Mol. Biosci. 2022, 9, 1016071. [Google Scholar] [CrossRef]
- Gao, W.; Yang, J.; Liu, W.; Wang, Y.; Shao, F. Site-Specific Phosphorylation and Microtubule Dynamics Control Pyrin Inflammasome Activation. Proc. Natl. Acad. Sci. USA 2016, 113, E4857–E4866. [Google Scholar] [CrossRef] [PubMed]
- Structure of Human PINK1 at a Mitochondrial TOM-VDAC Array. Available online: https://www.science.org/doi/10.1126/science.adu6445 (accessed on 26 March 2025).
- Li, X.; Fu, X.; Li, H.; Gao, Y.; Wang, W.; Shen, Y. Leptin Differentially Regulate Cell Apoptosis and Cycle by Histone Acetylation in Tibial and Vertebral Epiphyseal Plates. Cell Biol. Int. 2023, 47, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Susser, L.I.; Nguyen, M.-A.; Geoffrion, M.; Emerton, C.; Ouimet, M.; Khacho, M.; Rayner, K.J. Mitochondrial Fragmentation Promotes Inflammation Resolution Responses in Macrophages via Histone Lactylation. Mol. Cell. Biol. 2023, 43, 531–546. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.-H. Epigenetic Regulation of Aging: Implications for Interventions of Aging and Diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef]
- Im, C.-N.; Seo, J.-S. Overexpression of Tumor Necrosis Factor Receptor-Associated Protein 1 (TRAP1), Leads to Mitochondrial Aberrations in Mouse Fibroblast NIH/3T3 Cells. BMB Rep. 2014, 47, 280–285. [Google Scholar] [CrossRef]
- Li, X.; Chen, M.; Chen, X.; He, X.; Li, X.; Wei, H.; Tan, Y.; Min, J.; Azam, T.; Xue, M.; et al. TRAP1 Drives Smooth Muscle Cell Senescence and Promotes Atherosclerosis via HDAC3-Primed Histone H4 Lysine 12 Lactylation. Eur. Heart J. 2024, 45, 4219–4235. [Google Scholar] [CrossRef]
- Lv, M.; Zhou, W.; Hao, Y.; Li, F.; Zhang, H.; Yao, X.; Shi, Y.; Zhang, L. Structural Insights into the Specific Recognition of Mitochondrial Ribosome-Binding Factor hsRBFA and 12 S rRNA by Methyltransferase METTL15. Cell Discov. 2024, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Zeng, X.; Wang, H.; Tan, X.; Tian, Y.; Hu, H.; Ashrafizadeh, M.; Sethi, G.; Huang, H.; Duan, C. PGC-1α Loss Promotes Mitochondrial Protein Lactylation in Acetaminophen-Induced Liver Injury via the LDHB-Lactate Axis. Pharmacol. Res. 2024, 205, 107228. [Google Scholar] [CrossRef]
- Li, J.; Shi, X.; Xu, J.; Wang, K.; Hou, F.; Luan, X.; Chen, L. Aldehyde Dehydrogenase 2 Lactylation Aggravates Mitochondrial Dysfunction by Disrupting PHB2 Mediated Mitophagy in Acute Kidney Injury. Adv. Sci. 2025, 12, e2411943. [Google Scholar] [CrossRef]
- Ye, F.; Huang, J.; Wang, H.; Luo, C.; Zhao, K. Targeting Epigenetic Machinery: Emerging Novel Allosteric Inhibitors. Pharmacol. Ther. 2019, 204, 107406. [Google Scholar] [CrossRef]
- Yuan, J.; Yang, M.; Wu, Z.; Wu, J.; Zheng, K.; Wang, J.; Zeng, Q.; Chen, M.; Lv, T.; Shi, Y.; et al. The Lactate-Primed KAT8-PCK2 Axis Exacerbates Hepatic Ferroptosis During Ischemia/Reperfusion Injury by Reprogramming OXSM-Dependent Mitochondrial Fatty Acid Synthesis. Adv. Sci. 2025, 12, e2414141. [Google Scholar] [CrossRef]
- Sun, Z.; Ji, Z.; Meng, H.; He, W.; Li, B.; Pan, X.; Zhou, Y.; Yu, G. Lactate Facilitated Mitochondrial Fission-Derived ROS to Promote Pulmonary Fibrosis via ERK/DRP-1 Signaling. J. Transl. Med. 2024, 22, 479. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, J.-L.; Yan, X.-J.; Ji, Y.; Wang, F.-F. Exploring a New Mechanism between Lactate and VSMC Calcification: PARP1/POLG/UCP2 Signaling Pathway and Imbalance of Mitochondrial Homeostasis. Cell Death Dis. 2023, 14, 598. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ji, J.-J.; Yang, R.; Han, X.-Q.; Sun, X.-J.; Ma, W.-Q.; Liu, N.-F. Lactate Accelerates Calcification in VSMCs through Suppression of BNIP3-Mediated Mitophagy. Cell. Signal. 2019, 58, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.-P.; Wang, T.; Xiao, J.-X.; Wang, X.-Y.; Jiang, L.-P.; Tou, F.-F.; Chen, Z.-P.; Qu, X.-H.; Han, X.-J. Lactate Protects against Oxidative Stress-Induced Retinal Degeneration by Activating Autophagy. Free. Radic. Biol. Med. 2023, 194, 209–219. [Google Scholar] [CrossRef]
- Zhang, L.; Xin, C.; Wang, S.; Zhuo, S.; Zhu, J.; Li, Z.; Liu, Y.; Yang, L.; Chen, Y. Lactate Transported by MCT1 Plays an Active Role in Promoting Mitochondrial Biogenesis and Enhancing TCA Flux in Skeletal Muscle. Sci. Adv. 2024, 10, eadn4508. [Google Scholar] [CrossRef]
- Jia, M.; Yue, X.; Sun, W.; Zhou, Q.; Chang, C.; Gong, W.; Feng, J.; Li, X.; Zhan, R.; Mo, K.; et al. ULK1-Mediated Metabolic Reprogramming Regulates Vps34 Lipid Kinase Activity by Its Lactylation. Sci. Adv. 2023, 9, eadg4993. [Google Scholar] [CrossRef]
- Wood, I.S.; Trayhurn, P. Glucose Transporters (GLUT and SGLT): Expanded Families of Sugar Transport Proteins. Br. J. Nutr. 2003, 89, 3–9. [Google Scholar] [CrossRef]
- Barbosa, A.M.; Martel, F. Targeting Glucose Transporters for Breast Cancer Therapy: The Effect of Natural and Synthetic Compounds. Cancers 2020, 12, 154. [Google Scholar] [CrossRef]
- Senyilmaz, D.; Teleman, A.A. Chicken or the Egg: Warburg Effect and Mitochondrial Dysfunction. F1000Prime Rep. 2015, 7, 41. [Google Scholar] [CrossRef]
- Kee, H.J.; Cheong, J.-H. Tumor Bioenergetics: An Emerging Avenue for Cancer Metabolism Targeted Therapy. BMB Rep. 2014, 47, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhu, H.; Fu, C. Editorial: Clinical Therapeutic Development Against Cancers Resistant to Targeted Therapies. Front. Pharmacol. 2022, 12, 816896. [Google Scholar] [CrossRef] [PubMed]

| Target Proteins | Function | Effect of Lactylation | References |
|---|---|---|---|
| Histone H3K18 | Regulation of gene transcription. | Acute myocardial infarction via H3K18la mediated apoptosis and mitophagy imbalance. | [18] |
| Histone H3K56 | Regulates gene transcription and participates in DNA damage repair. | Mitochondrial pyruvate dehydrogenase inactivation shifts metabolism from OXPHOS to aerobic glycolysis, causing lactate accumulation that disrupts OXPHOS and enhances histone lactylation. | [46] |
| Histone H4K12 | Regulation of glycolysis and inflammation-related gene expression, involved in metabolic reprogramming. | Enhancing microglial glycolysis elevates histone lactylation, thereby upregulating pyruvate kinase M2 expression. | [47] |
| ARF1 | Small G proteins involved in vesicular trafficking and cytoskeletal dynamics. | K73la of ADP-ribosylation factor 1 (ARF1) modulates mitochondrial release and mitigates stroke-induced injury. | [48] |
| MDH2 | Mitochondrial Malate dehydrogenase catalyzes the redox of malate and oxalacetic acid in the TCA cycle to produce NADH, which is involved in ATP synthesis. | Lactylation inhibits enzyme activity, reduces NADH production, and improves mitochondrial function in myocardial ischemia–reperfusion injury. | [44] |
| AK2 | Mitochondrial Adenylate kinase 2 catalyzes the conversion of ADP to ATP and maintains the balance of mitochondrial energy metabolism. | K28la promotes the proliferation and metastasis of hepatocellular carcinoma cells and affects tumor progression by regulating mitochondrial metabolic pathways. | [49] |
| VDAC1 | Mitochondrial voltage-dependent anion channel 1, which controls metabolite transmembrane transport, regulates mitochondrial apoptosis pathways. | Decreased lactylation levels at K20 and K266 impair channel activity, thereby participating in the regulation of mitochondrial apoptosis and neuronal death. | [50] |
| Fis1 | Fis1 regulates mitochondrial fission by recruiting Drp1 to maintain mitochondrial morphology and function. | Elevated lactate at K20 promotes pathological mitochondrial excessive fission, triggering ATP depletion, mitochondrial ROS overproduction, and apoptotic signaling cascades. | [51] |
| ALDH2 | Mitochondrial Acetaldehyde dehydrogenase 2, which metabolizes acetaldehyde and reactive aldehydes, is involved in anti-oxidative stress and nitroglycerin metabolism. | Decreased lactylation at the K52 reduces enzyme activity, leading to a decline in mitochondrial membrane potential. | [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, F.; Yu, W. The Roles of Lactate and Lactylation in Diseases Related to Mitochondrial Dysfunction. Int. J. Mol. Sci. 2025, 26, 7149. https://doi.org/10.3390/ijms26157149
Ma F, Yu W. The Roles of Lactate and Lactylation in Diseases Related to Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2025; 26(15):7149. https://doi.org/10.3390/ijms26157149
Chicago/Turabian StyleMa, Fei, and Wei Yu. 2025. "The Roles of Lactate and Lactylation in Diseases Related to Mitochondrial Dysfunction" International Journal of Molecular Sciences 26, no. 15: 7149. https://doi.org/10.3390/ijms26157149
APA StyleMa, F., & Yu, W. (2025). The Roles of Lactate and Lactylation in Diseases Related to Mitochondrial Dysfunction. International Journal of Molecular Sciences, 26(15), 7149. https://doi.org/10.3390/ijms26157149