
Eurycomanone Blocks TGF-β1-Induced Epithelial-to-Mesenchymal Transition, Migration, and Invasion Pathways in Human Non-Small Cell Lung Cancer Cells by Targeting Smad and Non-Smad Signaling

 ,
,  ,
, _Bonness.jpeg) and
and 
Abstract
1. Introduction
2. Results
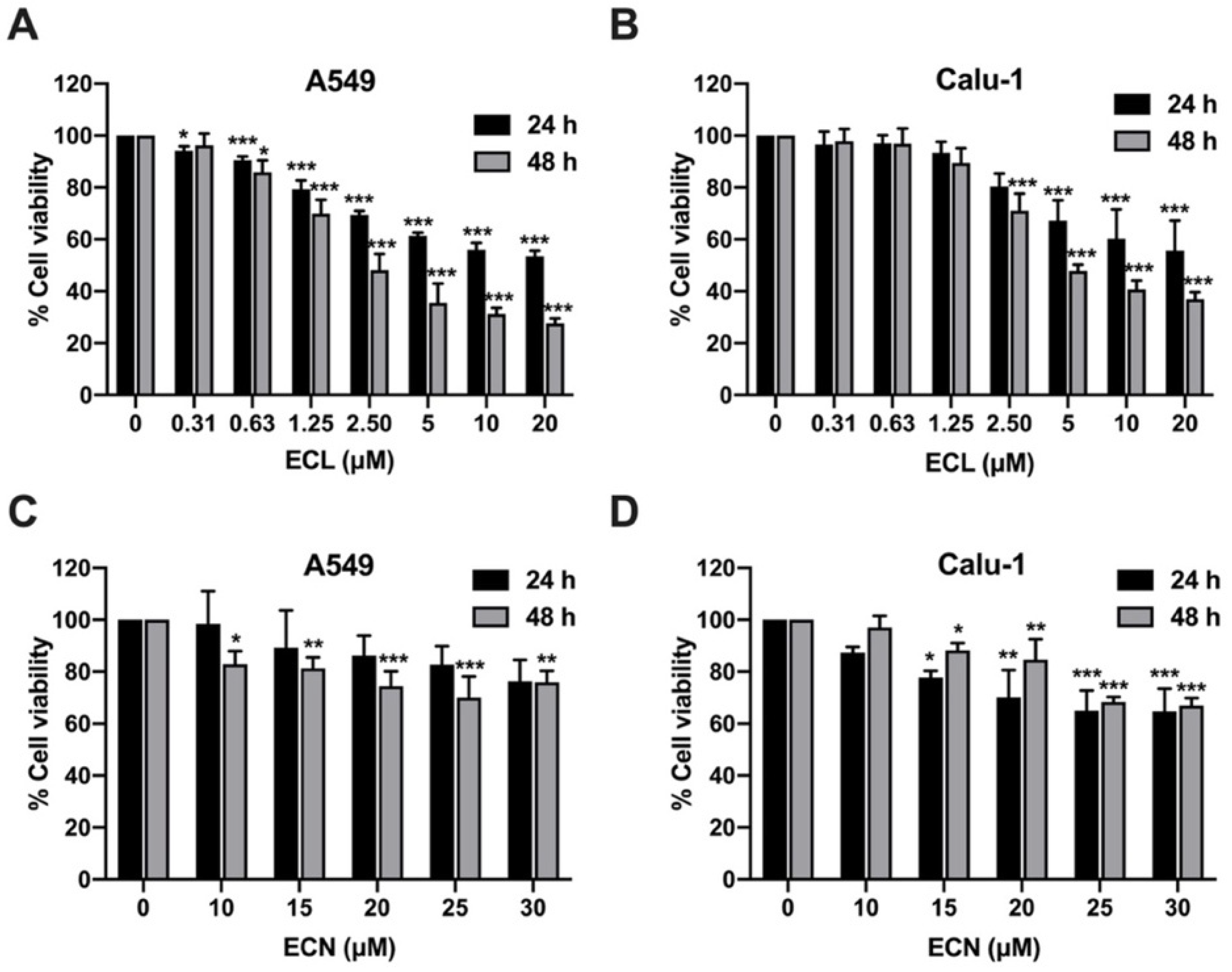
2.1. ECL and ECN Are Cytotoxic Against Human NSCLC Cells
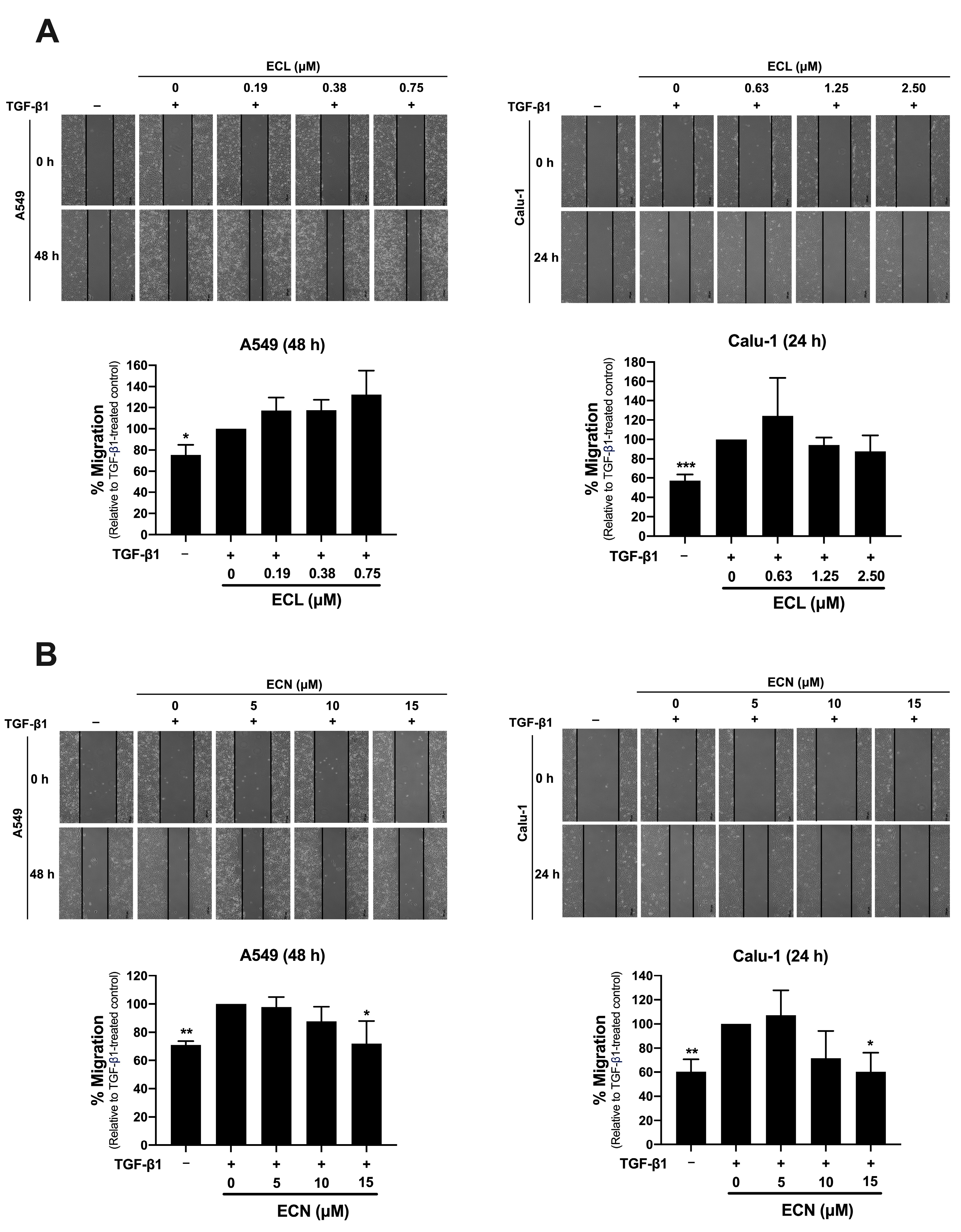
2.2. ECN, but Not ECL, Inhibits TGF-β1-Induced Migration of NSCLC Cells and MMP-2 Secretion
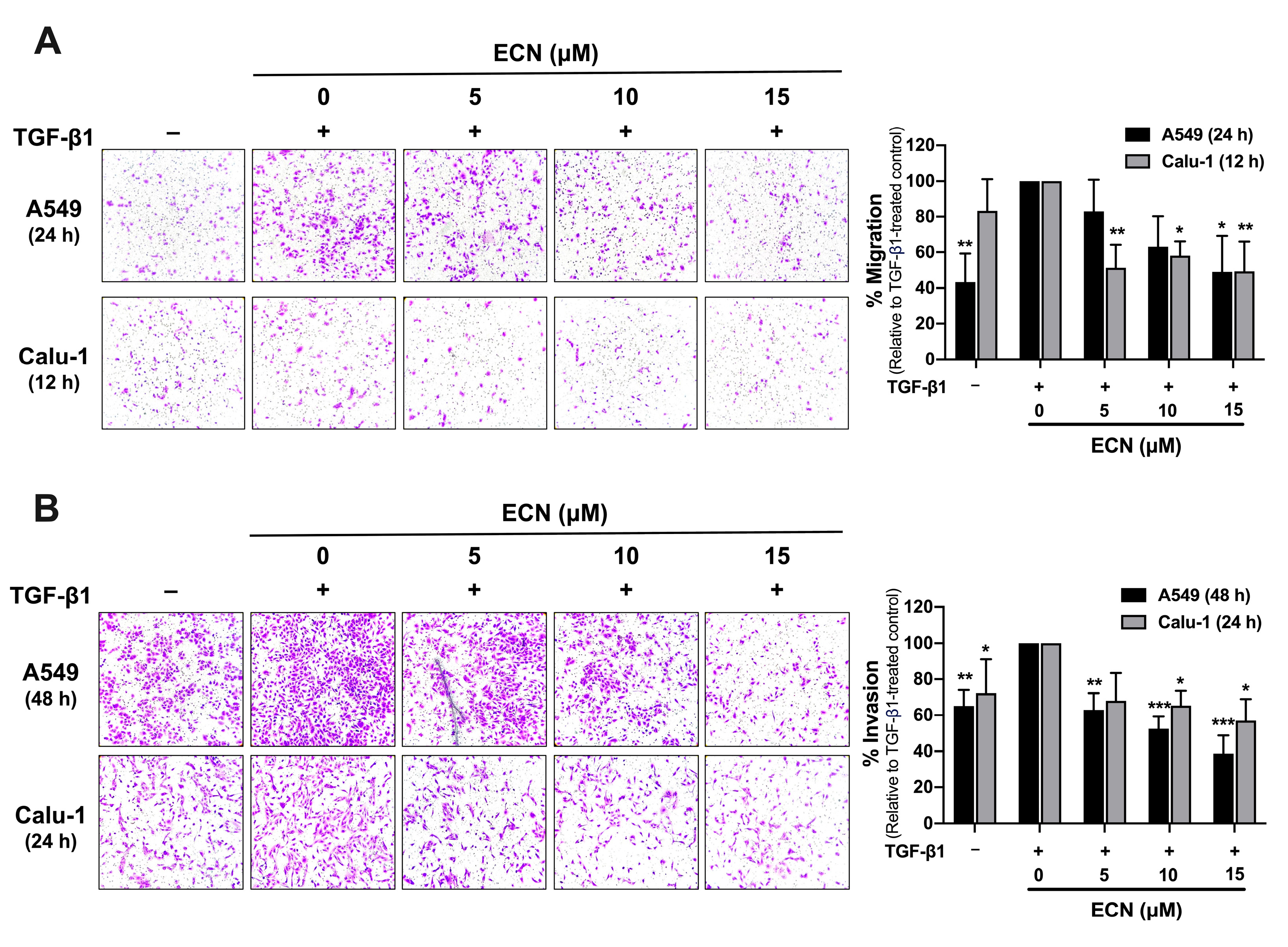
2.3. ECN Suppresses TGF-β1-Induced Migration and Invasion of NSCLC Cells
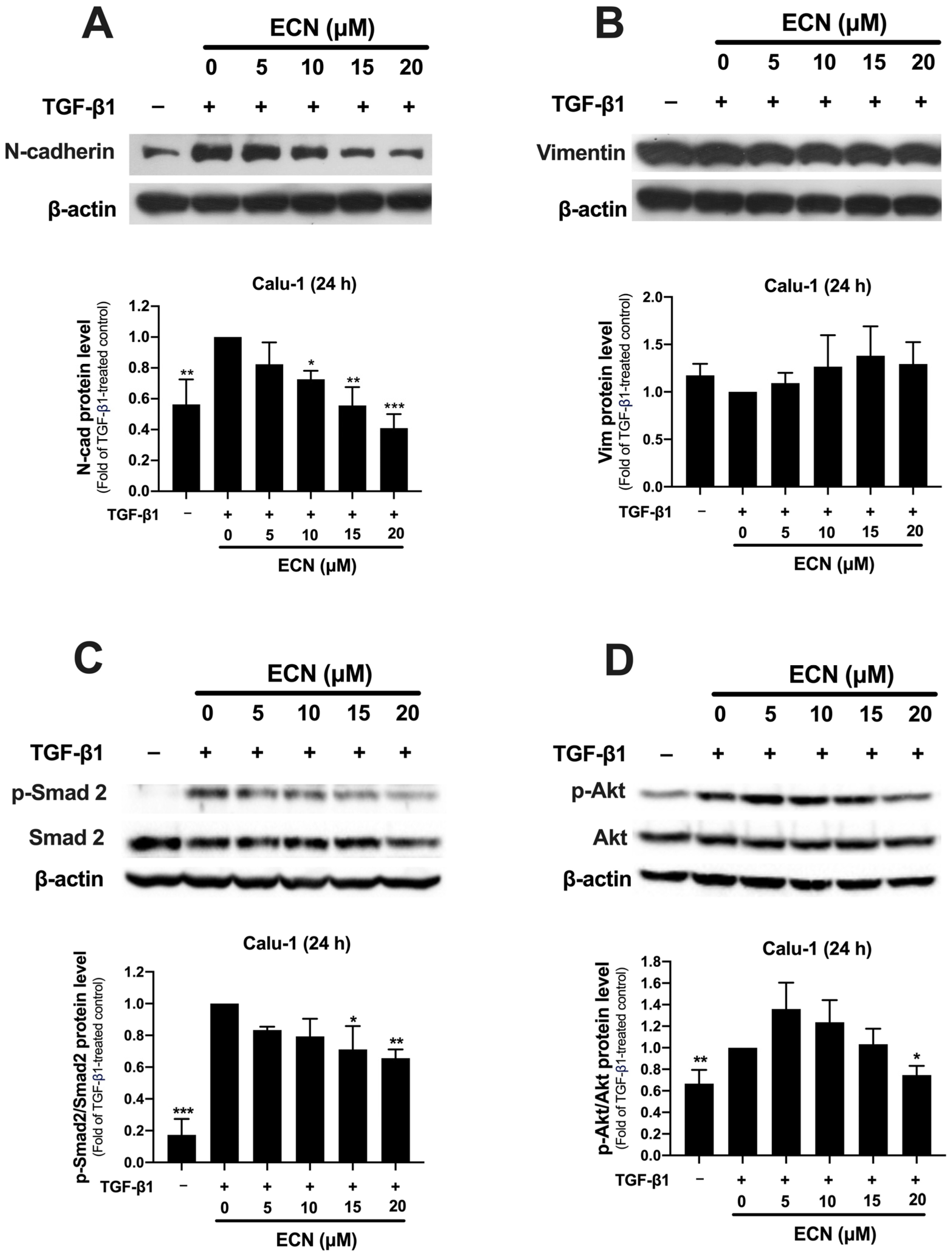
2.4. ECN Suppresses TGF-β1-Induced Epithelial-to-Mesenchymal Transition of NSCLC Cells
2.5. ECN Reduces the Aggressive Phenotype of TGF-β1-Stimulated Calu-1 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, and Antibodies
4.2. Cell Lines and Cell Cultures
4.3. Cytotoxicity Test by MTT Assay
4.4. Wound-Healing Assay
4.5. Gelatin Zymography Assay
4.6. Transwell Migration and Invasion Assay
4.7. Western Blotting
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana Davila, R.; Molina, J.R. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, H.; Zhu, L.; Feng, J.; Huang, X.; Baak, J.P.A. “How long have I got?” in stage IV NSCLC patients with at least 3 months up to 10 years survival, accuracy of long-, intermediate-, and short-term survival prediction is not good enough to answer this question. Front. Oncol. 2021, 11, 761042. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, F.; Cai, Q.; Deng, L.; Ouyang, Q.; Zhang, X.H.F.; Zheng, J. Invasion and metastasis in cancer: Molecular insights and therapeutic targets. Signal Transduct. Target. Ther. 2025, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, W.X.; Li, L.L.; Cao, Y.Z.; Geng, Y.D.; Feng, X.J.; Wang, A.Y.; Chen, Z.L.; Lu, Y.; Shen, A.Z. Paeonol suppresses proliferation and motility of non-small-cell lung cancer cells by disrupting STAT3/NF-κB signaling. Front. Pharmacol. 2020, 11, 572616. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Feng, Y.; Zheng, X.; Sun, L.; Wasan, H.S.; Ruan, S.; Shen, M. Resveratrol and its analogs: Potent agents to reverse epithelial-to-mesenchymal transition in tumors. Front. Oncol. 2021, 11, 644134. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Choe, K.; Yoo, H.H. Review on a traditional herbal medicine, Eurycoma longifolia Jack (Tongkat Ali): Its traditional uses, chemistry, evidence-based pharmacology and toxicology. Molecules 2016, 21, 331. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Karim, A.A. Tongkat Ali (Eurycoma longifolia Jack): A review on its ethnobotany and pharmacological importance. Fitoterapia 2010, 81, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Li, F.; Tezuka, Y.; Awale, S.; Kadota, S. Cytotoxic activity of quassinoids from Eurycoma longifolia. Nat. Prod. Commun. 2010, 5, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Samiulla, D.S.; Teh, B.P.; Zainol, M.; Zolkifli, N.A.; Muhammad, A.; Matom, E.; Zulkapli, A.; Abdullah, N.R.; Ismail, Z.; et al. Bioavailability of Eurycomanone in Its Pure Form and in a Standardised Eurycoma longifolia Water Extract. Pharmaceutics 2018, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Sitthisak, C.; Nisoa, M.; Chunglok, W.; Prasopthum, A.; Phaisan, S.; Putalun, W.; Kanchanapoom, T.; Juengwatanatrakul, T.; Yusakul, G. Efficient extraction of quassinoids and alkaloids from Eurycoma longifolia Jack roots using natural deep eutectic solvents and microwave-assisted extraction. Microchem. J. 2024, 196, 109676. [Google Scholar] [CrossRef]
- Kuo, P.C.; Damu, A.G.; Lee, K.H.; Wu, T.S. Cytotoxic and antimalarial constituents from the roots of Eurycoma longifolia. Bioorg. Med. Chem. 2004, 12, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, Y.; Rahmat, A.; Pihie, A.H.L.; Abdullah, N.R.; Houghton, P.J. Eurycomanone induce apoptosis in HepG2 cells via up regulation of p53. Cancer Cell 2009, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Dukaew, N.; Chairatvit, K.; Pitchakarn, P.; Imsumran, A.; Karinchai, J.; Tuntiwechapikul, W.; Wongnoppavich, A. Inactivation of AKT/NF-κB signaling by eurycomalactone decreases human NSCLC cell viability and improves the chemosensitivity to cisplatin. Oncol. Rep. 2020, 44, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Hajjouli, S.; Chateauvieux, S.; Teiten, M.H.; Orlikova, B.; Schumacher, M.; Dicato, M.; Choo, C.Y.; Diederich, M. Eurycomanone and eurycomanol from Eurycoma longifolia Jack as regulators of signaling pathways involved in proliferation, cell death and inflammation. Molecules 2014, 19, 14649–14666. [Google Scholar] [CrossRef] [PubMed]
- Dukaew, N.; Konishi, T.; Chairatvit, K.; Autsavapromporn, N.; Soonthornchareonnon, N.; Wongnoppavich, A. Enhancement of radiosensitivity by eurycomalactone in human NSCLC cells through G2/M cell cycle arrest and delayed DNA double-strand break repair. Oncol. Res. 2020, 28, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.V.; Malainer, C.; Schwaiger, S.; Atanasov, A.G.; Heiss, E.H.; Dirsch, V.M.; Stuppner, H. NF-κB inhibitors from Eurycoma longifolia. J. Nat. Prod. 2014, 77, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.D.; He, S.Q.; Shen, L.Q.; Wei, J.C.; Li, X.S.; Wei, Y.Y.; Zhang, Y.M.; Wang, X.Y.; Lin, F.; He, Z.L.; et al. 14,15β-Dihydroxyklaineanone inhibits HepG2 cell proliferation and migration through p38MAPK pathway. J. Pharm. Pharmacol. 2020, 72, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yu, X.; Yan, Z. Ailanthone suppresses the activity of human colorectal cancer cells through the STAT3 signaling pathway. Int. J. Mol. Med. 2022, 49, 21. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.Y.; Su, C.Y.; Yan, Y.Y.; Zhang, L.L.; Guo, Q.R.; Chene, H.B.; Zhumabieke, S.; Danabek, Y.; Li, J.J.; Liu, Y.; et al. A study on the mechanism of bruceine D in the treatment of non-small cell lung cancer H1299 cells. World J. Tradit. Chin. Med. 2020, 6, 500–507. [Google Scholar] [CrossRef]
- Ye, R.; Dai, N.; He, Q.; Guo, P.; Xiang, Y.; Zhang, Q.; Hong, Z.; Zhang, Q.Y. Comprehensive anti tumor effect of Brusatol through inhibition of cell viability and promotion of apoptosis caused by autophagy via the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 105, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Li, J.; Zhong, X.; Zhang, Z.; Zhao, Y. Endothelial-to-mesenchymal transition in the tumor microenvironment: Roles of transforming growth factor-β and matrix metalloproteins. Heliyon 2024, 10, e40118. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhu, J.; Cai, T.; Du, W.; Zhang, Y.; Zhu, Q.; Liu, Z.; Huang, J.A. Suppression of non-small cell lung cancer migration and invasion by hsa-miR-486-5p via the TGF-β/SMAD2 signaling pathway. J. Cancer 2019, 10, 6014–6024. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.Y.; Wang, Q.M.; Mak, T.S.; Huang, X.R.; Yu, X.Q.; Lan, H.Y. Inhibition of tumor invasion and metastasis by targeting TGF-β-Smad-MMP2 pathway with Asiatic acid and Naringenin. Mol. Ther. Oncolytics 2021, 20, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.; Lalli, E. Targeting the cytoskeleton against metastatic dissemination. Cancer Metastasis Rev. 2021, 40, 89–140. [Google Scholar] [CrossRef] [PubMed]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix metalloproteinases shape the tumor microenvironment in cancer progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Puneeta, N.; Santosh, T.; Mishra, I.; Gaikwad, P.; Sahu, A. Evaluation of E-Cadherin and vimentin expression for different grades of oral epithelial dysplasia and oral squamous cell carcinoma—An immunohistochemical study. J. Oral Maxillofac. Pathol. 2022, 26, 285. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhang, X.; Li, D.; He, P.; Tian, W.; Zeng, B. Expression Analysis and Clinical Significance of EIF4E, VEGF-C, E-Cadherin and MMP-2 in Colorectal Adenocarcinoma. Oncotarget 2016, 7, 85502–85514. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Lee, E.J.; Ji, M.; Park, S.M. HDAC inhibitors, trichostatin A and valproic acid, increase E-cadherin and vimentin expression but inhibit migration and invasion of cholangiocarcinoma cells. Oncol. Rep. 2018, 40, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, M.; Tanaka, K.; Kikuchi-Yanoshita, R.; Muraoka, M.; Konishi, M.; Takeichi, M. Increased cell-substratum adhesion, and decreased gelatinase secretion and cell growth, induced by E-cadherin transfection of human colon carcinoma cells. Oncogene 1995, 11, 2547–2552. [Google Scholar] [PubMed]
- Luo, J.; Lubaroff, D.M.; Hendrix, M.J. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999, 59, 3552–3556. [Google Scholar] [PubMed]
- Nawrocki-Raby, B.; Gilles, C.; Polette, M.; Martinella-Catusse, C.; Bonnet, N.; Puchelle, E.; Foidart, J.-M.; van Roy, F.; Birembaut, P. E-Cadherin mediates MMP down-regulation in highly invasive bronchial tumor cells. Am. J. Pathol. 2003, 163, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, S.N.; Zhitnyak, I.Y.; Gloushankova, N.A. Dual Role of E-Cadherin in Cancer Cells. Tissue Barrier 2021, 10, 2005420. [Google Scholar] [CrossRef] [PubMed]
- Rudisch, A.; Dewhurst, M.R.; Horga, L.G.; Kramer, N.; Harrer, N.; Dong, M.; van der Kuip, H.; Wernitznig, A.; Bernthaler, A.; Dolznig, H.; et al. High EMT signature score of invasive non-small cell lung cancer (NSCLC) cells correlates with NFκB driven colony-stimulating factor 2 (CSF2/GM-CSF) secretion by neighboring stromal fibroblasts. PLoS ONE 2015, 10, e0124283. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Pützer, B.M. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Kusumastuti, E.H.; Djuanda, S.N.; Ariani, G.; Mastutik, G. Correlation of N-cadherin and MMP-9 expression with regional nodal metastasis in laryngeal squamous cell carcinoma. Pharmacogn. J. 2024, 16, 679–683. [Google Scholar] [CrossRef]
- Hsu, C.C.; Huang, S.F.; Wang, J.S.; Chu, W.K.; Nien, J.E.; Chen, W.S.; Chow, S.E. Interplay of N-Cadherin and matrix metalloproteinase 9 enhances human nasopharyngeal carcinoma cell invasion. BMC Cancer 2016, 16, 800. [Google Scholar] [CrossRef] [PubMed]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef] [PubMed]
- Sravani, A.N.K.V.; Thomas, J. Targeting epithelial-mesenchymal transition signaling pathways with dietary phytocompounds and repurposed drug combinations for overcoming drug resistance in various cancers. Heliyon 2025, 11, e41964. [Google Scholar] [CrossRef] [PubMed]
- Ciołczyk-Wierzbicka, D.; Laidler, P. The inhibition of invasion of human melanoma cells through N-cadherin knock-down. Med. Oncol. 2018, 35, 42. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.G.; Castillo-Martin, M.; Bonal, D.M.; Jia, A.J.; Rybicki, B.A.; Christiano, A.M.; Cordon-Cardo, C. PI3K/AKT pathway regulates E-cadherin and Desmoglein 2 in aggressive prostate cancer. Cancer Med. 2015, 4, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Barrette, K.; Van Kelst, S.; Wouters, J.; Marasigan, V.; Fieuws, S.; Agostinis, P.; Oord, J.; Garmyn, M. Epithelial-Mesenchymal Transition during invasion of cutaneous squamous cell carcinoma is paralleled by akt activation. Br. J. Dermatol. 2014, 171, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gao, M.; Shen, Z.; Luo, B.; Li, R.; Jiang, X.; Ding, R.; Ha, Y.; Wang, Z.; Jie, W. Blocking PI3K/Akt signaling attenuates metastasis of nasopharyngeal carcinoma cells through induction of mesenchymal-epithelial reverting transition. Oncol. Rep. 2014, 32, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Chea, C.; Miyauchi, M.; Inubushi, T.; Okamoto, K.; Haing, S.; Nguyen, P.T.; Imanaka, H.; Takata, T. Bovine lactoferrin reverses programming of epithelial-to-mesenchymal transition to mesenchymal-to-epithelial transition in oral squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2018, 507, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Wu, H.; Jin, Y.; Hou, J.; Liu, J.; Zhang, X.; Hu, W.; Sun, G.; Zhang, Z. Fruquintinib inhibits the migration and invasion of colorectal cancer cells by modulating epithelial-mesenchymal transition via TGF-β/Smad signaling pathway. Front. Oncol. 2025, 15, 1503133. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zhao, J.; Lou, A.; Huang, Q.; OuYang, Q.; Zhu, J.; Fan, M.; He, Y.; Ren, H.; Yang, M. Transforming growth factor β1 promotes fibroblast-like synoviocytes migration and invasion via TGF-β1/Smad signaling in rheumatoid arthritis. Mol. Cell Biochem. 2019, 459, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR Kinase Inhibition Causes Feedback-Dependent Biphasic Regulation of AKT Signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.T.; Liao, H.B.; Ye, Z.Q.; Jiang, H.S.; Li, J.X.; Ke, L.M.; Hua, J.Y.; Wei, B.; Wu, X.; Cui, L. Eurycomanone stimulates bone mineralization in zebrafish larvae and promotes osteogenic differentiation of mesenchymal stem cells by upregulating AKT/GSK-3β/β-catenin signaling. J. Orthop. Transl. 2023, 40, 132–146. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Types of Cells | Eurycomalactone (ECL) | Eurycomanone (ECN) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC20 (μM) | IC50 (μM) | IC20 (μM) | IC50 (μM) | ||||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | ||
| A549 | Adenocarcinoma | 1.29 ± 0.27 | 0.87 ± 0.16 | >20 | 2.61 ± 0.64 | 25.31 ± 12.51 | 14.77 ± 2.98 | >30 | >30 |
| Calu-1 | Squamous cell carcinoma | 2.86 ± 0.74 | 1.90 ± 0.47 | >20 | 4.84 ± 0.43 | 15.86 ± 4.74 | 20.84 ± 2.33 | >30 | >30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soddaen, P.; Chairatvit, K.; Pitchakarn, P.; Laowanitwattana, T.; Imsumran, A.; Wongnoppavich, A. Eurycomanone Blocks TGF-β1-Induced Epithelial-to-Mesenchymal Transition, Migration, and Invasion Pathways in Human Non-Small Cell Lung Cancer Cells by Targeting Smad and Non-Smad Signaling. Int. J. Mol. Sci. 2025, 26, 7120. https://doi.org/10.3390/ijms26157120
Soddaen P, Chairatvit K, Pitchakarn P, Laowanitwattana T, Imsumran A, Wongnoppavich A. Eurycomanone Blocks TGF-β1-Induced Epithelial-to-Mesenchymal Transition, Migration, and Invasion Pathways in Human Non-Small Cell Lung Cancer Cells by Targeting Smad and Non-Smad Signaling. International Journal of Molecular Sciences. 2025; 26(15):7120. https://doi.org/10.3390/ijms26157120
Chicago/Turabian StyleSoddaen, Pratchayanon, Kongthawat Chairatvit, Pornsiri Pitchakarn, Tanongsak Laowanitwattana, Arisa Imsumran, and Ariyaphong Wongnoppavich. 2025. "Eurycomanone Blocks TGF-β1-Induced Epithelial-to-Mesenchymal Transition, Migration, and Invasion Pathways in Human Non-Small Cell Lung Cancer Cells by Targeting Smad and Non-Smad Signaling" International Journal of Molecular Sciences 26, no. 15: 7120. https://doi.org/10.3390/ijms26157120
APA StyleSoddaen, P., Chairatvit, K., Pitchakarn, P., Laowanitwattana, T., Imsumran, A., & Wongnoppavich, A. (2025). Eurycomanone Blocks TGF-β1-Induced Epithelial-to-Mesenchymal Transition, Migration, and Invasion Pathways in Human Non-Small Cell Lung Cancer Cells by Targeting Smad and Non-Smad Signaling. International Journal of Molecular Sciences, 26(15), 7120. https://doi.org/10.3390/ijms26157120