
Comparative Transcriptomic Analysis for Identification of Environmental-Responsive Genes in Seven Species of Threadfin Breams (Nemipterus)
 , ,
, ,
Abstract
1. Introduction
2. Results
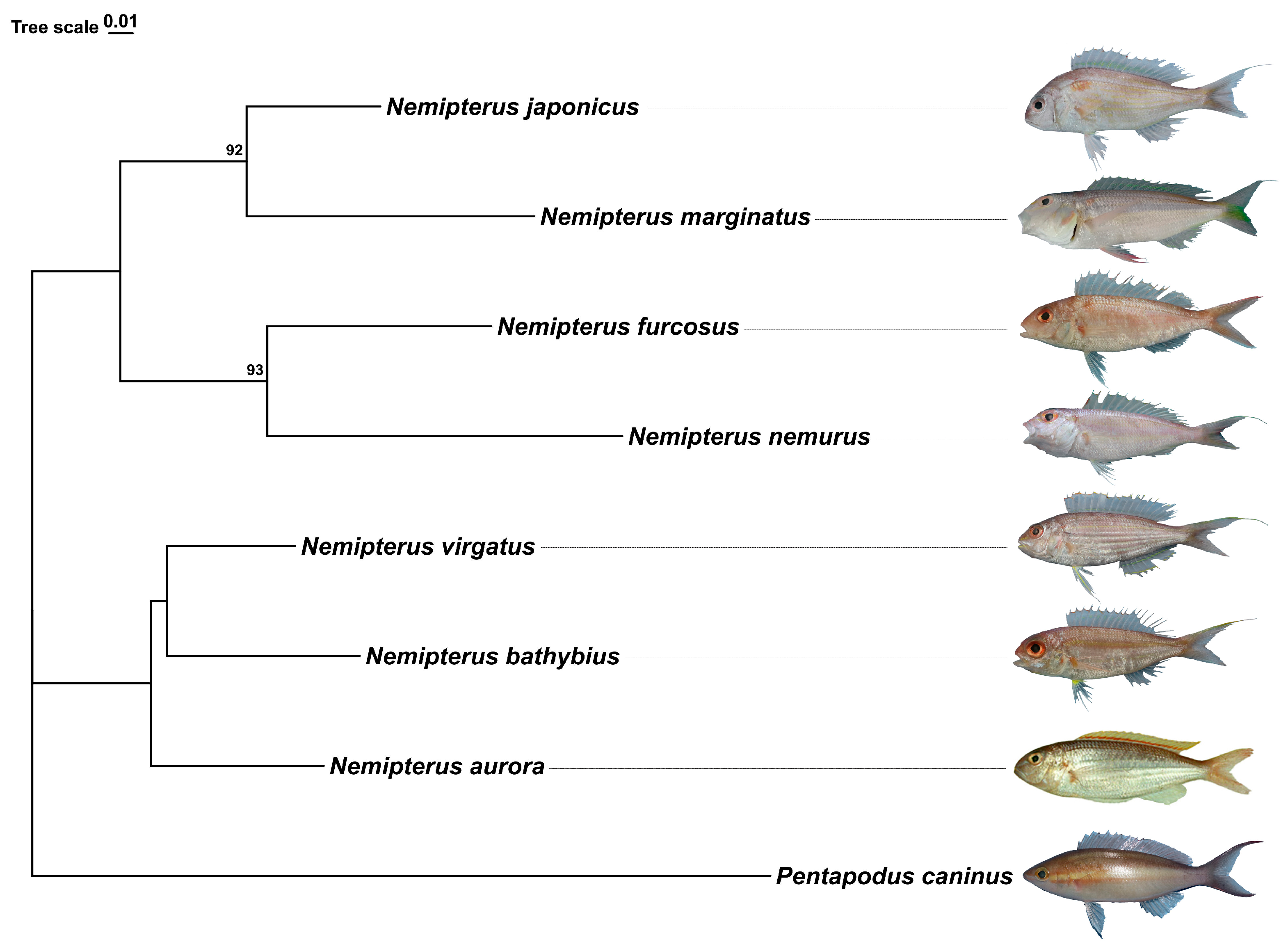
2.1. Species Identification and Phylogenetic Inferences
2.2. Transcriptome Sequencing and De Novo Assembly
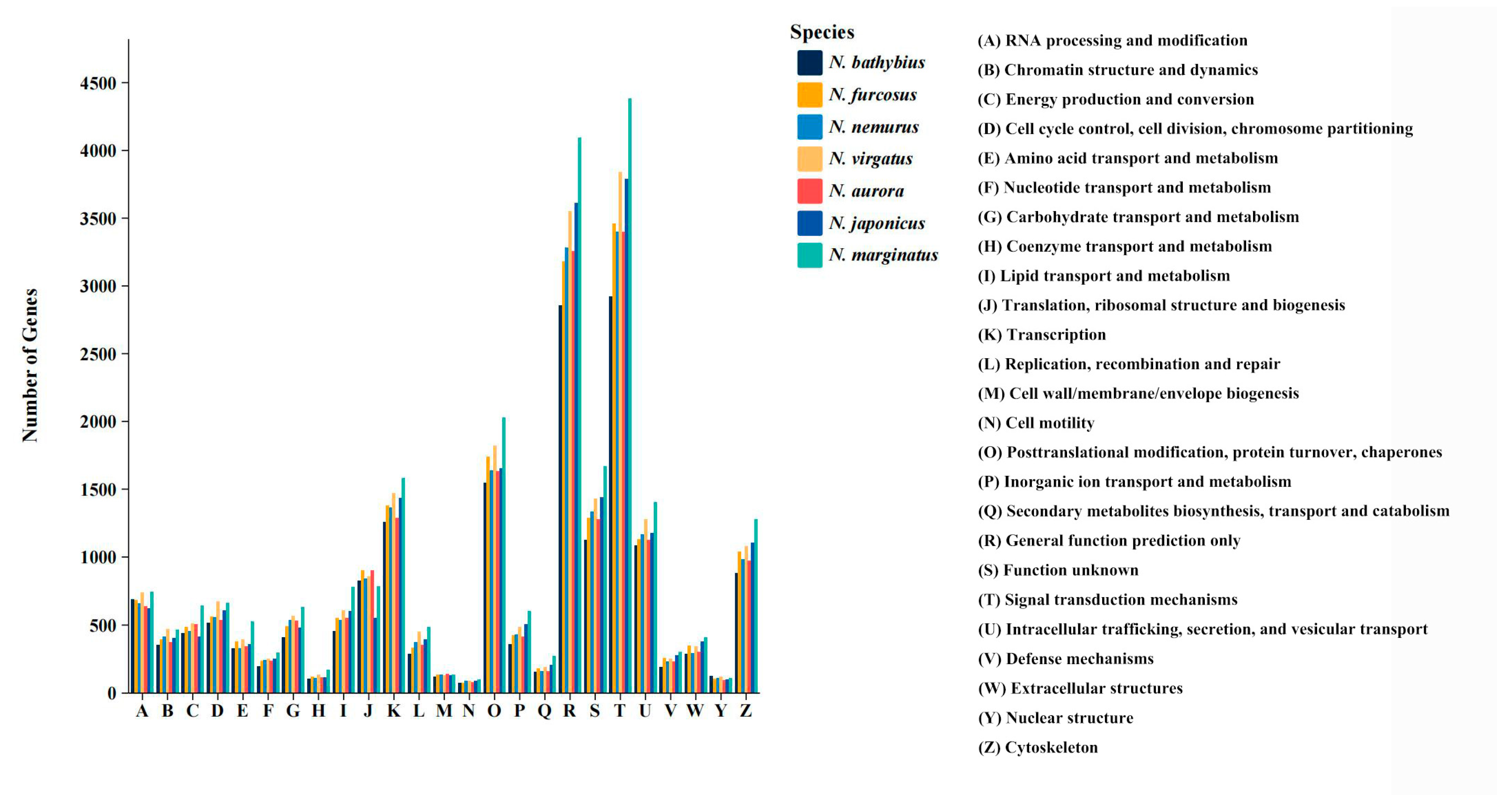
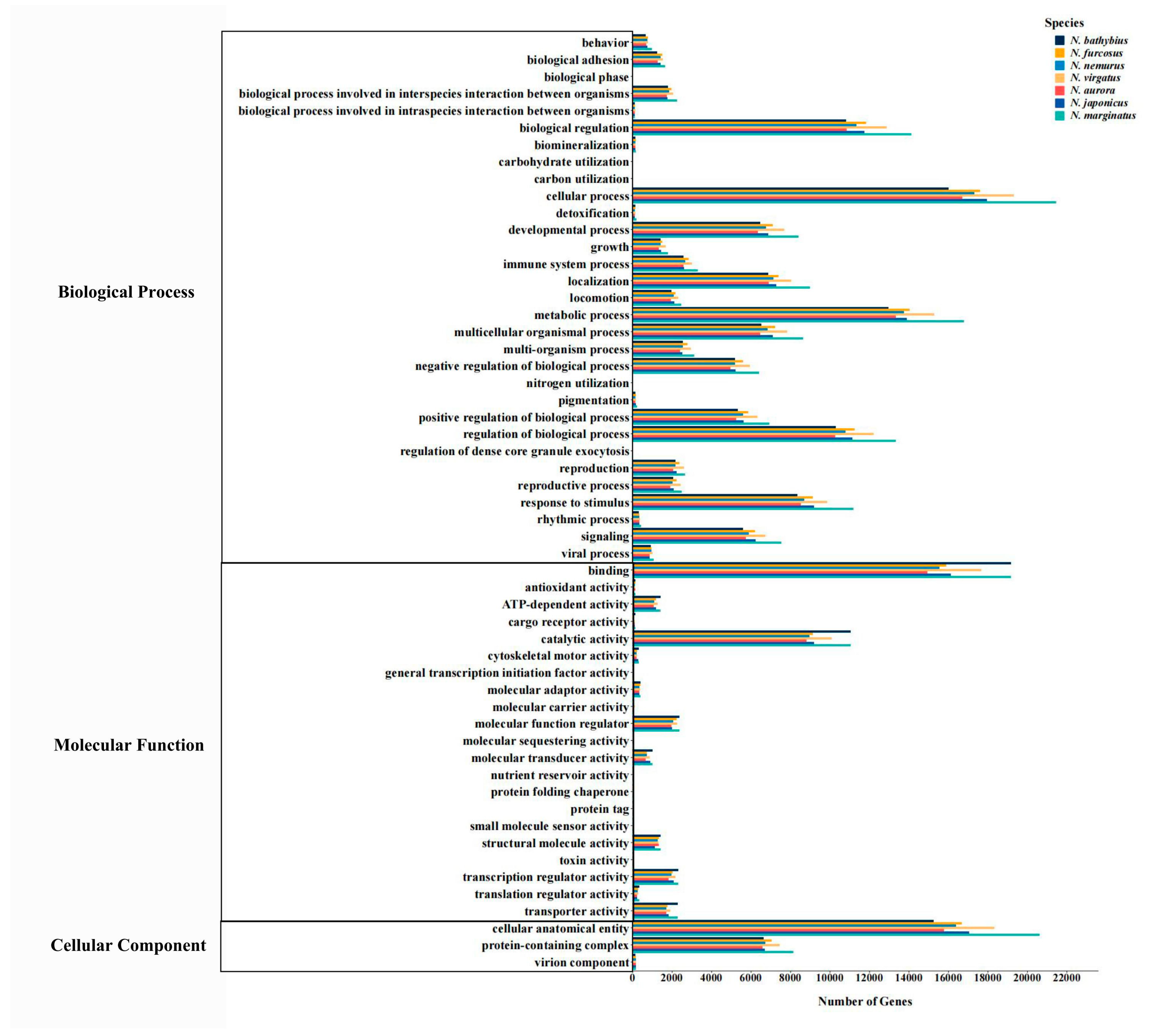
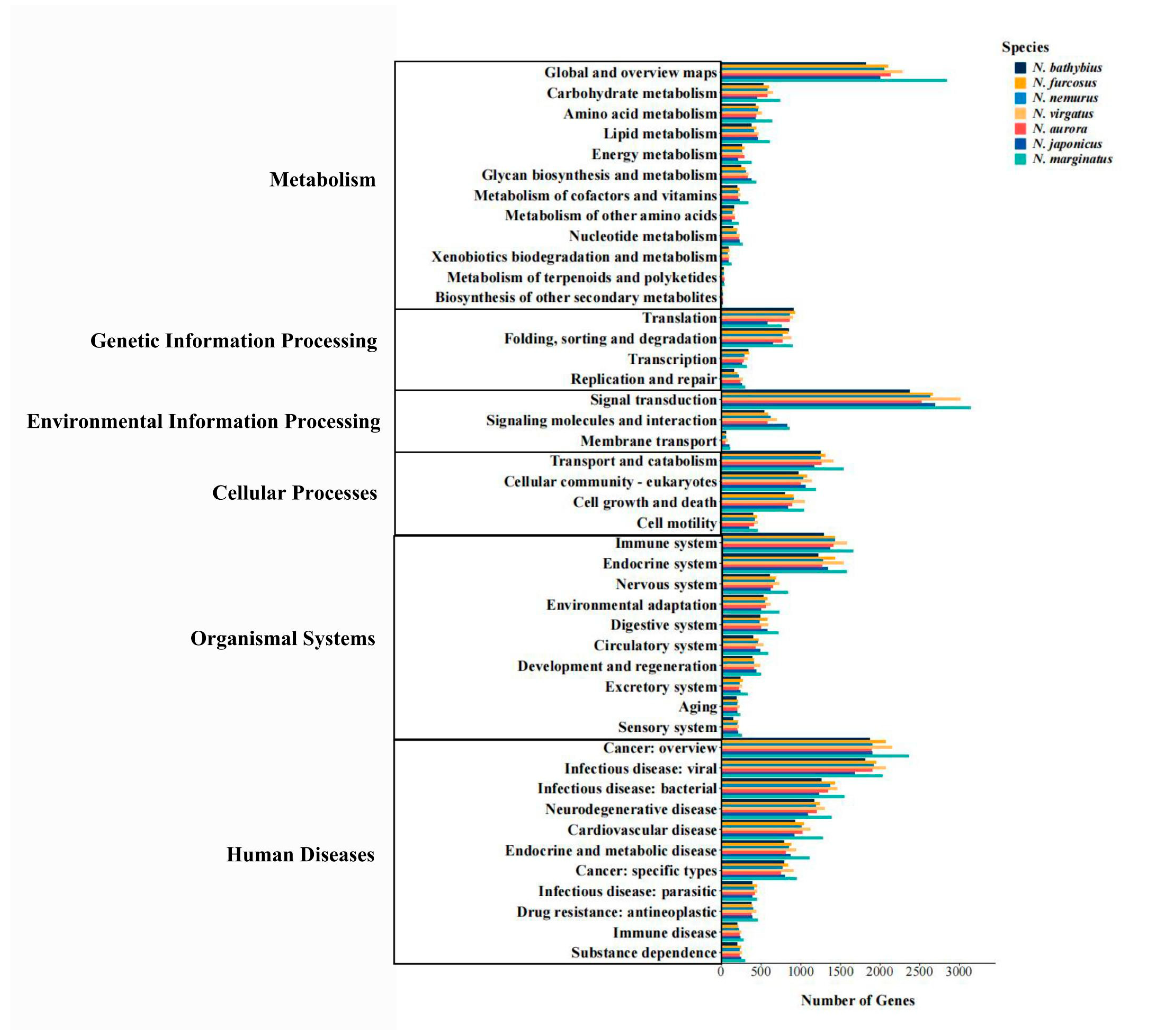
2.3. Functional Annotation
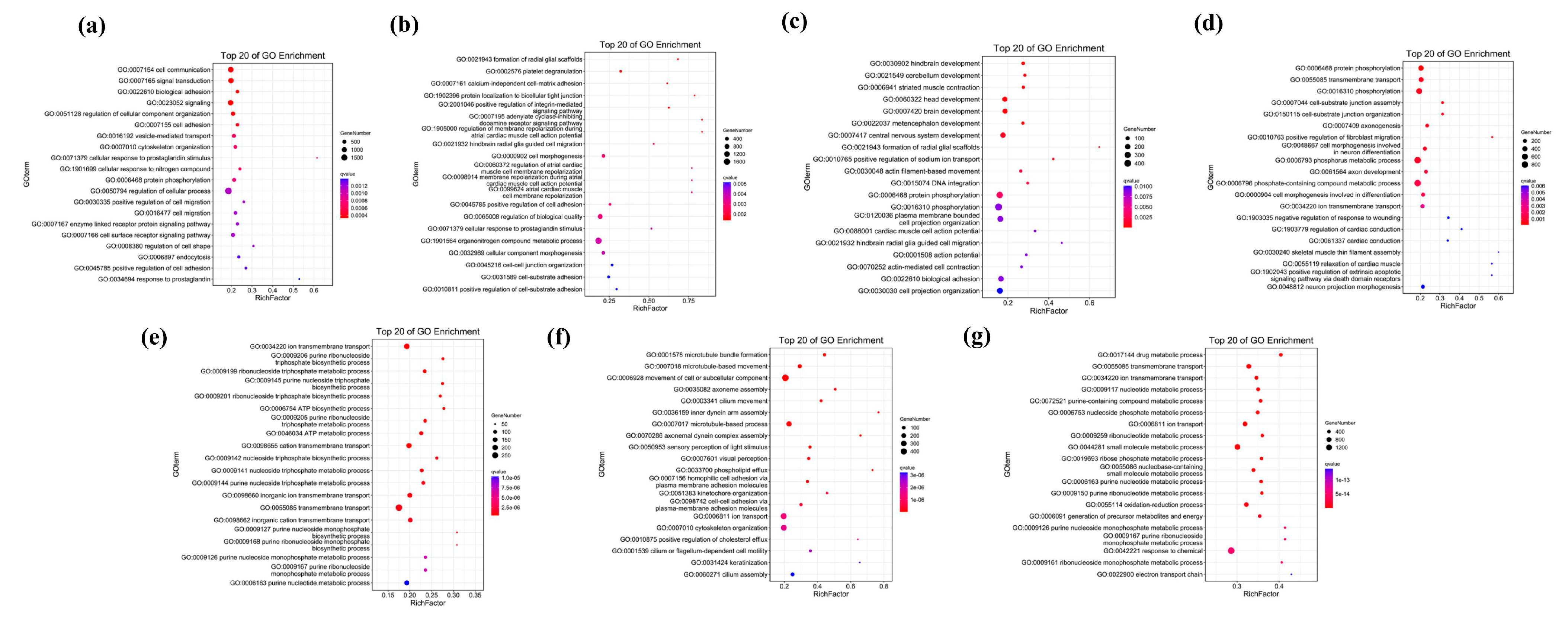
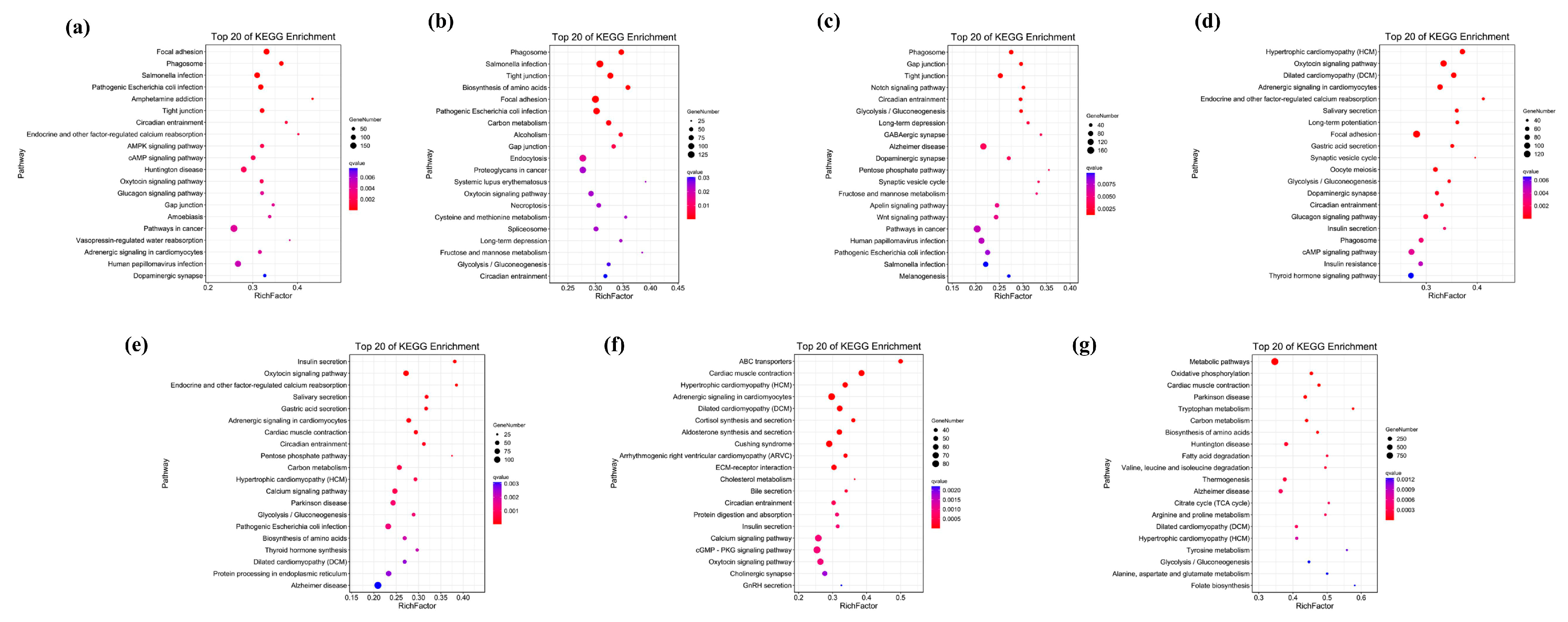
2.4. Functional Enrichment Analysis of Unique Genes
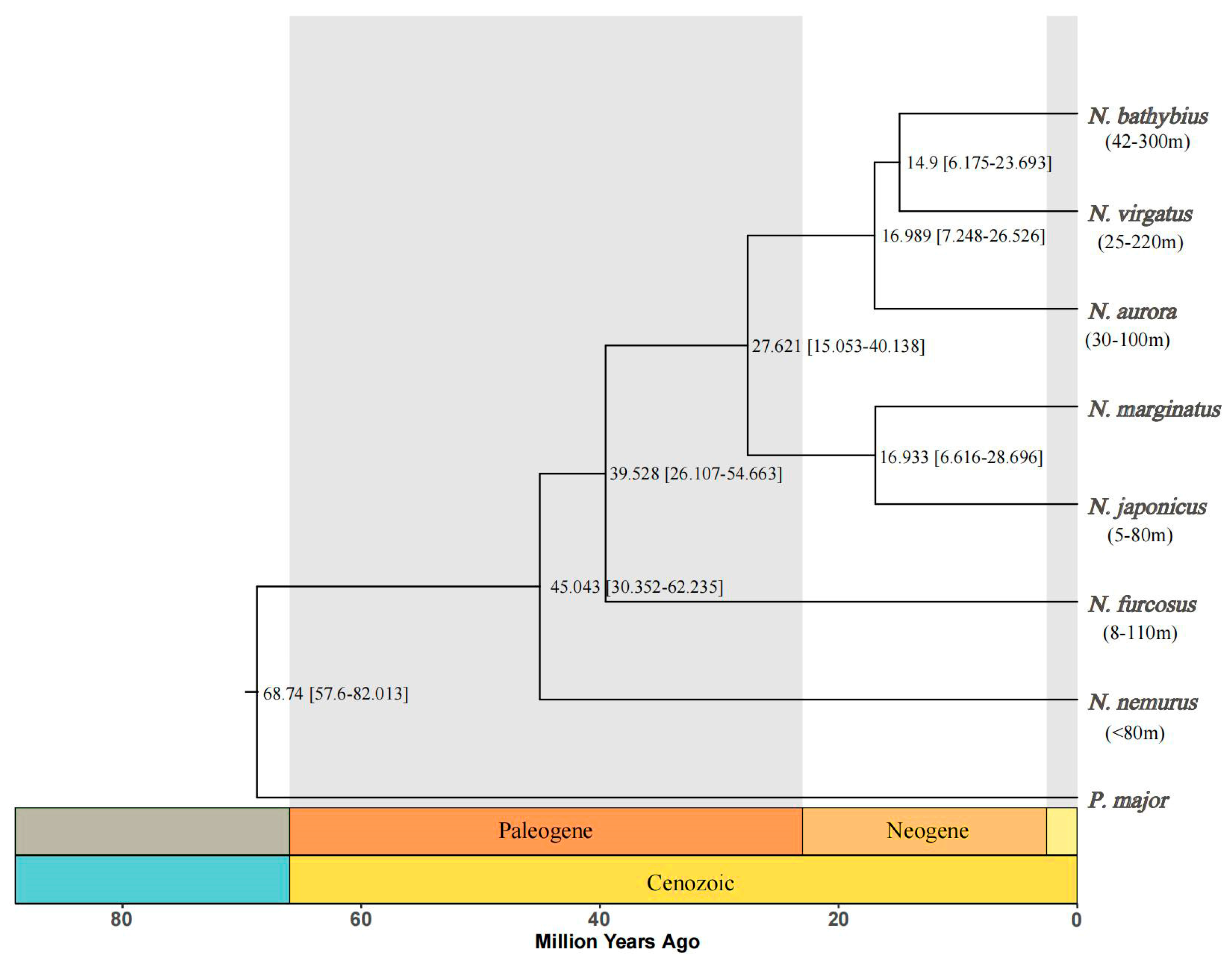
2.5. Phylogenetic and Divergence Time Estimations
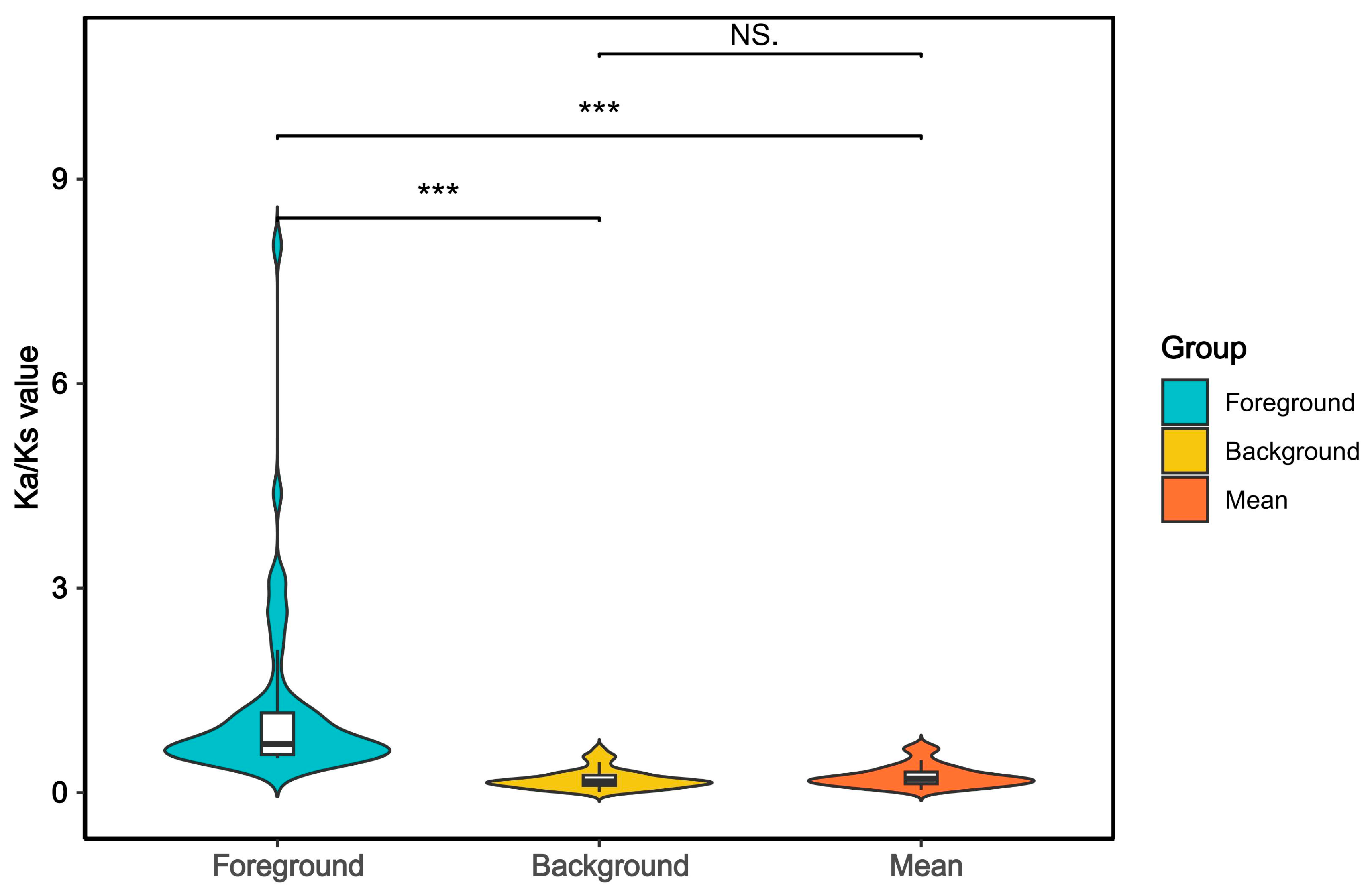
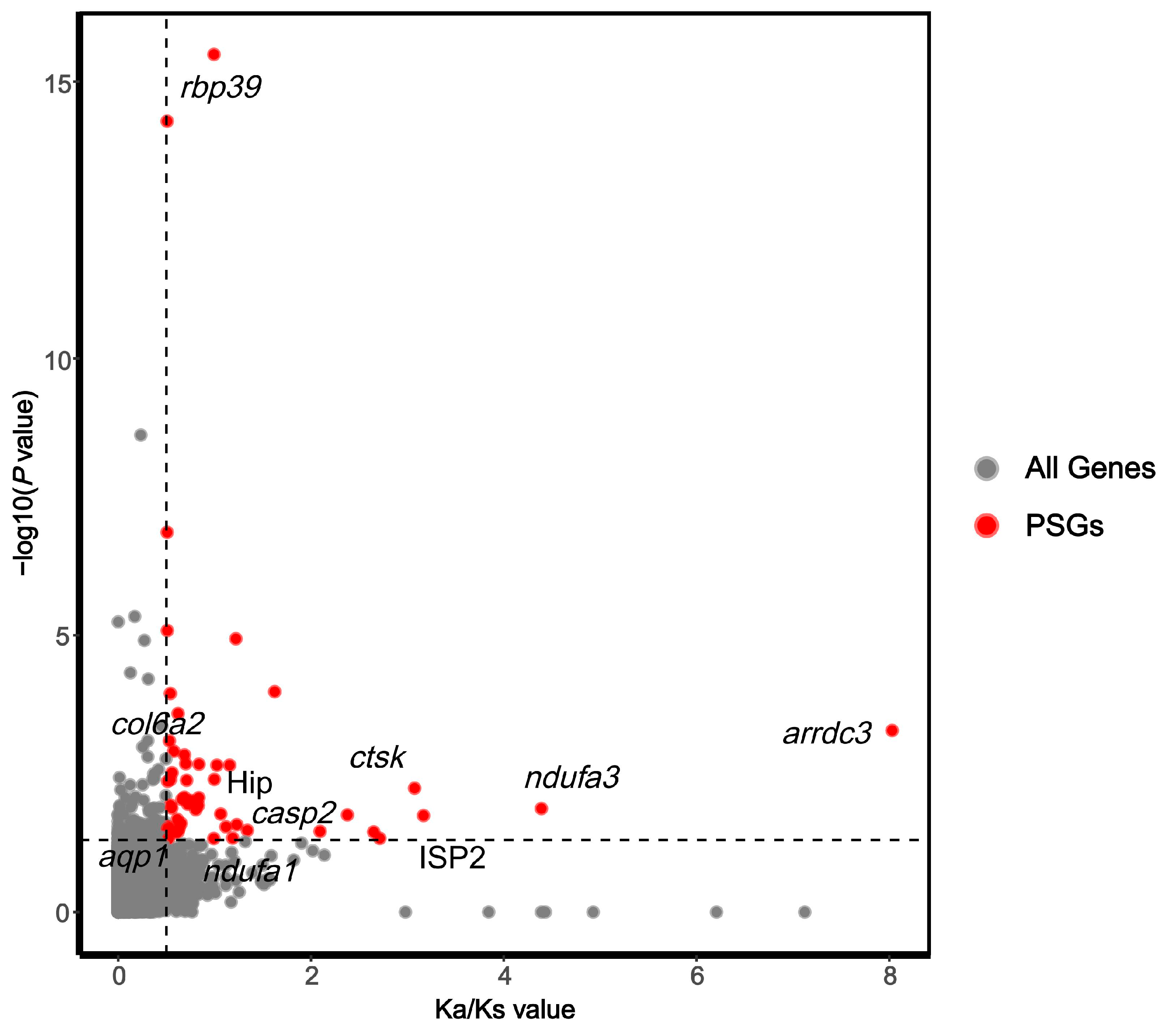
2.6. Selection Pressure Analysis
3. Discussion
3.1. Transcriptomic Characteristics of the Genus Nemipterus
3.2. Phylogenetic and Divergence Time Analysis of the Genus Nemipterus
3.3. Adaptive Evolution of the Genus Nemipterus
4. Material and Methods
4.1. Sample Collection and Species Identification
4.2. RNA Extraction, cDNA Library Building and Illumina Sequencing
4.3. Transcriptome De Novo Assembly and Functional Annotation
4.4. Differential Gene Expression Analysis
4.5. Single Copy Orthologous Gene Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Eschmeyer, W.N.; Fong, J.D. Species of Fishes by Family/Subfamily. Available online: http://research.calacademy.org/research/ichthyology/catalog/SpeciesBYFamily.asp (accessed on 15 September 2024).
- Russell, B.C. Nemipterid fishes of the world. FAO Fish. Synop. 1990, 12, I. [Google Scholar]
- Masuda, H.; Uyeno, T.; Yoshino, T. The Fishes of the Japanese Archipelago; Tokai University Press: Tokyo, Japan, 1984. [Google Scholar]
- Russell, B.C. A review of the threadfin breams of the genus Nemipterus (Nemipteridae) from Japan and Taiwan, with description of a new species. Jpn. J. Ichthyol. 1993, 39, 295–310. [Google Scholar] [CrossRef]
- Menezes, M.R.; Arolkar, U.; Rathod, V. Genetic relationships among five nemipterid fish species from the Indian coast using allozyme analysis. J. Fish Biol. 2002, 60, 1326–1330. [Google Scholar] [CrossRef]
- Joshi, K.K. Biology and population dynamics of Nemipterus mesoprion (Bleeker) off Cochin. Indian J. Fish. 2005, 52, 315–322. [Google Scholar]
- Ning, P.; Wu, R.; Liu, J. First record of red filament threadfin bream, Nemipterus marginatus (Valenciennes, 1830) (Perciformes, Nemipteridae), from Chinese waters. Chin. J. Oceanol. Limnol. 2011, 29, 1306–1308. [Google Scholar] [CrossRef]
- ElHaweet, A.E.A. Biological studies of the invasive species Nemipterus japonicus (Bloch, 1791) as a Red Sea immigrant into the Mediterranean. Egypt. J. Aquat. Res. 2013, 39, 267–274. [Google Scholar] [CrossRef]
- Erguden, D.; Turan, C.; Gurlek, M.; Yaglioglu, D.; Gungor, M. Age and growth of the Randall’s threadfin bream Nemipterus randalli (Russell, 1986), a recent Lessepsian migrant in Iskenderun Bay, northeastern Mediterranean. J. Appl. Ichthyol. 2010, 26, 441–444. [Google Scholar] [CrossRef]
- Russell, B.C.; Allen, G.R. Nemipteridae. In FAO Species Identification Guide for Fishery Purposes. The Living Marine Resources of the Western Central Pacific; FAO: Rome, Italy, 2001; Volume 5, pp. 3051–3089. [Google Scholar]
- Imtiaz, A.; Yen, D.T.; Nor, S.A.M.; Naim, D.M. Molecular identification of commercially important species of Nemipterus (Perciformes: Nemipteridae) in surrounding seas of Malaysia. Biodiversitas J. Biol. Divers. 2016, 17. [Google Scholar] [CrossRef]
- Hung, K.W.; Russell, B.C.; Chen, W.J. Molecular systematics of threadfin breams and relatives (Teleostei, Nemipteridae). Zool. Scr. 2017, 46, 536–551. [Google Scholar] [CrossRef]
- Ping, N. Taxonomy, Molecular Phylogeny, and Zoogeography of the Family Nemipteridae (Pisces: Perciformes) in China Seas. Ph.D. Thesis, University of Chinese Academy of Sciences (Institute of Oceanology), Qingdao, China, 2012. [Google Scholar]
- Jin, W. Fisheries Biology and Trophic Characteristics of Four Species of Nemipterus in the Beibu Gulf. Ph.D. Thesis, Guangdong Ocean University, Zhanjiang, China, 2023. [Google Scholar]
- Hoffmann, A.A.; Hercus, M.J. Environmental stress as an evolutionary force. Bioscience 2000, 50, 217–226. [Google Scholar] [CrossRef]
- Hoffmann, A.A.; Willi, Y. Detecting genetic responses to environmental change. Nat. Rev. Genet. 2008, 9, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Yi, S.K.; Yao, H.; Chi, W.; Guo, Y.; Ma, X.F.; Wang, H.P. Comparative transcriptome analysis reveals potential evolutionary differences in adaptation of temperature and body shape among four Percidae species. PLoS ONE 2019, 14, e0215933. [Google Scholar] [CrossRef] [PubMed]
- Ohlberger, J.; Mehner, T.; Staaks, G.; Hölker, F. Temperature-related physiological adaptations promote ecological divergence in a sympatric species pair of temperate freshwater fish, Coregonus spp. Funct. Ecol. 2008, 22, 501–508. [Google Scholar] [CrossRef]
- Chen, S.; Xiao, Y.; Xiao, Z.; Li, J.; Herrera-Ulloa, A. Suitable habitat shifts and ecological niche overlay assessments among benthic Oplegnathus species in response to climate change. Environ. Res. 2024, 252, 119129. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Sun, J.; Xu, T.; Chen, C.; Tian, R.; Qiu, J.W.; Qian, P.Y. De novo transcriptome assembly and positive selection analysis of an individual deep-sea fish. BMC Genom. 2018, 19, 394. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M. Biology, age, growth and population dynamics of threadfin bream Nemipterus japonicus. J. Mar. Biol. Assoc. India. 1990, 32, 66–76. [Google Scholar]
- Kimoto, H.; Ibrahim, J. The Stock Status and Management Scheme of Kerisi (Nemipterus peronii) on the East Coast of Peninsular Malaysia; SEAFDEC/MFRDMD: Kuala Terengganu, Malaysia, 1996. [Google Scholar]
- Granada, V.P.; Masuda, Y.; Matsuoka, T. Age and growth of the yellowbelly threadfin bream Nemipterus bathybius in Kagoshima Bay, southern Japan. Fish. Sci. 2004, 70, 497–506. [Google Scholar] [CrossRef]
- Wu, C.C.; Weng, J.S.; Liu, K.M.; Su, W.C. Reproductive biology of the notchedfin threadfin bream, Nemipterus peronii (Nemipteridae), in waters of southwestern Taiwan. Zool. Stud.-TAIPEI 2008, 47, 103. [Google Scholar]
- Gandra, M.; Assis, J.; Martins, M.R.; Abecasis, D. Reduced global genetic differentiation of exploited marine fish species. Mol. Biol. Evol. 2021, 38, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Ravitchandirane, V.; Geetha, V.; Ramya, V.; Janifer, B.; Thangaraj, M.; Subburaj, J.; Ramanadevi, V.; Ganesan, T. Molecular identification and phylogenetic relationships of Threadfin Breams (Family: Nemipteridae) using mtDNA marker. Not. Sci. Biol. 2012, 4, 13–18. [Google Scholar] [CrossRef]
- Ogwang, J.; Bariche, M.; Bos, A.R. Genetic diversity and phylogenetic relationships of threadfin breams (Nemipterus spp.) from the Red Sea and eastern Mediterranean Sea. Genome 2021, 64, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Neenu, R.; Sandhya, S.; Anjaly, J.; Nisha, K.; Subal, K.R.; Shikha, R.; Shoba, J.K.; Gopalakrishnan, A. Population genetic structure of Randall’s threadfin bream Nemipterus randalli in Indian waters based on mitochondrial and nuclear gene sequences. Sci. Rep. 2024, 14, 7556. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Huang, Z.; Li, Y.; Cheng, G. Sequence analysis of mitochondrial COI and Cytb gene of Nemipterus species in South China Sea. J. Fish. Sci. China 2012, 19, 355–363. [Google Scholar]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced applications of RNA sequencing and challenges. Bioinform. Biol. Insights 2015, 9, BBI.S28991. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole transcriptome analysis with sequencing: Methods, challenges and potential solutions. Cell. Mol. Life Sci. 2015, 72, 3425–3439. [Google Scholar] [CrossRef] [PubMed]
- Mutz, K.O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, 2015, pdb.top084970. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Chatchaiphan, S.; Srisapoome, P.; Kim, J.H.; Devlin, R.H.; Na-Nakorn, U. De novo transcriptome characterization and growth-related gene expression profiling of diploid and triploid bighead catfish (Clarias macrocephalus Günther, 1864). Mar. Biotechnol. 2017, 19, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Weinberger, A.; Carmi, M.; Barkai, N. A genetic signature of interspecies variations in gene expression. Nat. Genet. 2006, 38, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.B.W. Principles of transcriptome analysis and gene expression quantification: An RNA-seq tutorial. Mol. Ecol. Resour. 2013, 13, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, P.; Jones, D.B.; Thompson, E.L.; Parker, L.M.; Ross, P.M.; Raftos, D.A. Transcriptomic profiling of adaptive responses to ocean acidification. Mol. Ecol. 2017, 26, 5974–5988. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Sukumaran, S.; Mukundan, L.; Jose, A.; Mary, S.; Nisha, K.; Gopalakrishnan, A. Complete mitochondrial genome profile of Randall’s threadfin bream, Nemipterus randalli and its phylogenetic analysis. Curr. Sci. 2024, 127, 322–332. [Google Scholar] [CrossRef]
- Wu, Z.; Li, X. Complete mitochondrial genome of the Nemipterus virgatus (Perciformes: Nemipteridae). Mitochondrial DNA Part A 2016, 27, 3485–3486. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Yan, B.; Meng, X. The complete mitochondrial genome of the Japanese threadfin bream, Nemipterus japonicus (Teleostei, Nemipteridae). Mitochondrial DNA Part A 2016, 27, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Jung, Y.H.; Choi, Y.J.; Kim, B.; Nguyen, T.V.; Lee, D.S. Characterization of the complete mitochondrial genome of the fork-tailed threadfin bream, Nemipterus furcosus (Spariformes, Nemipteridae) and phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 3130–3131. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Evolution of genome architecture. Int. J. Biochem. Cell Biol. 2009, 41, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Chen, D.; Su, G.; Zhou, M.; Wu, Z.; Zou, J. Molecular phylogenetic relationships of 15 species of the family Nemipteridae based on COI and RAG2 gene sequences. J. Ocean. Univ. China (Nat. Sci. Ed.) 2017, 47, 74–81. [Google Scholar]
- Villafaña, J.A.; Rivadeneira, M.M. Rise and fall in diversity of Neogene marine vertebrates on the temperate Pacific coast of South America. Paleobiology 2014, 40, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Obura, D.O. An Indian Ocean centre of origin revisited: Palaeogene and Neogene influences defining a biogeographic realm. J. Biogeogr. 2016, 43, 229–242. [Google Scholar] [CrossRef]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.W. Neogene marine climate trends: Implications for biogeography and evolution of the shallow-sea biota. Geology 1984, 12, 647–650. [Google Scholar] [CrossRef]
- Lo, P.C.; Liu, S.H.; Chao, N.L.; Nunoo, F.K.; Mok, H.K.; Chen, W.J. A multi-gene dataset reveals a tropical New World origin and Early Miocene diversification of croakers (Perciformes: Sciaenidae). Mol. Phylogenetics Evol. 2015, 88, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Floeter, S.R.; Bender, M.G.; Siqueira, A.C.; Cowman, P.F. Phylogenetic perspectives on reef fish functional traits. Biol. Rev. 2018, 93, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Feng, J.; Liu, X.; Yan, C.; Ye, Y.; Li, J.; Xu, K.; Guo, B.; Lü, Z. Sequence comparison of the mitochondrial genomes of five brackish water species of the family Neritidae: Phylogenetic implications and divergence time estimation. Ecol. Evol. 2022, 12, e8984. [Google Scholar] [CrossRef] [PubMed]
- Gaither, M.R.; Violi, B.; Gray, H.W.I.; Neat, F.; Drazen, J.C.; Grubbs, R.D.; Roa-Varón, A.; Sutton, T.; Hoelzel, A.R. Depth as a driver of evolution in the deep sea: Insights from grenadiers (Gadiformes: Macrouridae) of the genus Coryphaenoides. Mol. Phylogenet. Evol. 2016, 104, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Thatje, S. Global bottlenecks in the distribution of marine Crustacea: Temperature constraints in the family Lithodidae. J. Biogeogr. 2009, 36, 2125–2135. [Google Scholar] [CrossRef]
- Brown, A.; Thatje, S. Explaining bathymetric diversity patterns in marine benthic invertebrates and demersal fishes: Physiological contributions to adaptation of life at depth. Biol. Rev. 2014, 89, 406–426. [Google Scholar] [CrossRef] [PubMed]
- Gracey, A.Y.; Fraser, E.J.; Li, W.; Fang, Y.; Taylor, R.R.; Rogers, J.; Brass, A.; Cossins, A.R. Coping with cold: An integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proc. Natl. Acad. Sci. USA 2004, 101, 16970–16975. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.A. Stress in fishes: A diversity of responses with particular reference to changes in circulating corticosteroids. Integr. Comp. Biol. 2002, 42, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; You, Q.; Chi, C.; Luo, S.; Song, H.; Lou, B.; Takeuchi, Y. Transcriptional response to low temperature in the yellow drum (Nibea albiflora) and identification of genes related to cold stress. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 28, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Cellular responses in marine animals to hydrostatic pressure. J. Exp. Zool. Part A Ecol. Integr. Physiol. 2020, 333, 398–420. [Google Scholar] [CrossRef] [PubMed]
- Robison, B.H. Deep pelagic biology. J. Exp. Mar. Biol. Ecol. 2004, 300, 253–272. [Google Scholar] [CrossRef]
- Martinez, C.M.; Friedman, S.T.; Corn, K.A.; Larouche, O.; Price, S.A.; Wainwright, P.C. The deep sea is a hot spot of fish body shape evolution. Ecol. Lett. 2021, 24, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.H. Teleost fish osmoregulation: What have we learned since August Krogh, Homer Smith, and Ancel Keys. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 295, R704–R713. [Google Scholar] [CrossRef] [PubMed]
- Thao, H.N.; Thu, T.T.; Ly, N.T.; Hong, D.T. Distribution of Nemipterus marginatus (Valenciennes, 1830) (Nemipteridae, Perciformes) in the estuary and coastal areas of North Central, Vietnam. Check List 2022, 18, 1127–1130. [Google Scholar] [CrossRef]
- van Os, C.H.; Kamsteeg, E.J.; Marr, N.; Deen, P.M. Physiological relevance of aquaporins: Luxury or necessity? Pflüg. Arch. 2000, 440, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Benga, G. The first discovered water channel protein, later called aquaporin 1: Molecular characteristics, functions and medical implications. Mol. Asp. Med. 2012, 33, 518–534. [Google Scholar] [CrossRef] [PubMed]
- Umenishi, F.; Schrier, R.W. Hypertonicity-induced aquaporin-1 (AQP1) expression is mediated by the activation of MAPK pathways and hypertonicity-responsive element in the AQP1 gene. J. Biol. Chem. 2003, 278, 15765–15770. [Google Scholar] [CrossRef] [PubMed]
- Cutler, C.P.; Cramb, G. Molecular physiology of osmoregulation in eels and other teleosts: The role of transporter isoforms and gene duplication. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 130, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Iandiev, I.; Pannicke, T.; Reichel, M.B.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Expression of aquaporin-1 immunoreactivity by photoreceptor cells in the mouse retina. Neurosci. Lett. 2005, 388, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Stamer, W.D.; Bok, D.; Hu, J.; Jaffe, G.J.; McKay, B.S. Aquaporin-1 channels in human retinal pigment epithelium: Role in transepithelial water movement. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Lee, E.J.; Martínez-Ruiz, N.; García-Alcázar, A.; Grzywacz, N.M.; De Juan, J. Aquaporin-4 immunoreactivity in Müller and amacrine cells of marine teleost fish retina. Brain Res. 2012, 1432, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.E. On the origins of arrestin and rhodopsin. BMC Evol. Biol. 2008, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Emilsson, V.; Schadt, E.E.; Chutkow, W.A.; Lee, S.; Marsili, A.; Zhang, Y.; Dobrin, R.; Cohen, D.E.; Larsen, P.R.; et al. The arrestin domain-containing 3 protein regulates body mass and energy expenditure. Cell Metab. 2011, 14, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Zhang, N.; Liu, B.S.; Guo, L.; Guo, H.Y.; Jiang, S.G.; Zhang, D.C. A chromosome-level genome assembly of the yellowfin seabream (Acanthopagrus latus; Hottuyn, 1782) provides insights into its osmoregulation and sex reversal. Genomics 2021, 113, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ma, A.; Huang, Z.; Wang, X.; Liu, Z.; Xia, D.; Yang, S.; Zhao, T. Comparative transcriptomic analysis reveals mechanisms of divergence in osmotic regulation of the turbot Scophthalmus maximus. Fish Physiol. Biochem. 2020, 46, 1519–1536. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, J.; Zheng, S. What we know about ST13, a co-factor of heat shock protein, or a tumor suppressor? J. Zhejiang Univ. Sci. B 2007, 8, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xu, X.; Chen, Y.; Wang, J.; Zhang, T.; E, Z.; Chen, S.; Liu, Y. Genome-wide identification, molecular characterization, and involvement in response to abiotic and biotic stresses of the hsp70 gene family in turbot (Scophthalmus maximus). Int. J. Mol. Sci. 2023, 24, 6025. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Liu, H.; He, C.; Shou, C.; Han, Z. Heat shock protein 70 (Hsp70) and heat shock transcription factor (Hsf) gene families in Cynoglossus semilaevis: Genome-wide identification and correlation analysis in response to low salinity stress. Mar. Freshw. Res. 2021, 72, 1132–1141. [Google Scholar] [CrossRef]
- Sun, Y.; Wen, H.; Tian, Y.; Mao, X.; Li, X.; Li, J.; Hu, Y.; Liu, Y.; Li, J.; Li, Y. HSP90 and HSP70 families in Lateolabrax maculatus: Genome-wide identification, molecular characterization, and expression profiles in response to various environmental stressors. Front. Physiol. 2021, 12, 784803. [Google Scholar] [CrossRef] [PubMed]
- Papakostas, S.; Vasemaegi, A.; Vähä, J.P.; Himberg, M.; Peil, L.; Primmer, C.R. A proteomics approach reveals divergent molecular responses to salinity in populations of European whitefish (Coregonus lavaretus). Mol. Ecol. 2012, 21, 3516–3530. [Google Scholar] [CrossRef] [PubMed]
- DeVries, A.L.; Wohlschlag, D.E. Freezing resistance in some Antarctic fishes. Science 1969, 163, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, H.; Dong, C.; Ren, Y.; Pan, N.; Kong, B.; Liu, H.; Xia, X. Effect of ice structuring protein on the microstructure and myofibrillar protein structure of mirror carp (Cyprinus carpio L.) induced by freeze-thaw processes. Lwt 2021, 139, 110570. [Google Scholar] [CrossRef]
- Qian, B. Molecular characterization and mRNA expression of ISP2 and ISP4 in the Large Yellow Croaker (Larimichthys Crocea) under acute cold stress. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Martos-Sitcha, J.A.; Bermejo-Nogales, A.; Calduch-Giner, J.A.; Pérez-Sánchez, J. Gene expression profiling of whole blood cells supports a more efficient mitochondrial respiration in hypoxia-challenged gilthead sea bream (Sparus aurata). Front. Zool. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Mai, K.; Ai, Q. Regulation of hepatic lipid deposition by phospholipid in large yellow croaker. Br. J. Nutr. 2017, 118, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.H.; Pavalko, F.M. Mechanotransduction and functional response of the skeleton to physical stress: The mechanisms and mechanics of bone adaptation. J. Orthop. Sci. 1998, 3, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, Y.T.; Ma, J.; Ruuttilal, P.; Hukkanen, M.; Santavirta, S. Chondrocyte-mediated collagenolysis correlates with cartilage destruction grades in osteoarthritis. Clin. Exp. Rheumatol. 2005, 23, 19–26. [Google Scholar] [PubMed]
- Pradillon, F.; Gaill, F. Pressure and life: Some biological strategies. Life Extrem. Environ. 2006, 341–355. [Google Scholar] [CrossRef]
- Myllyharju, J.; Kivirikko, K.I. Collagens and collagen-related diseases. Ann. Med. 2001, 33, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, L.G.; Youn, I.; Bonaldo, P.; Guilak, F. Developmental and osteoarthritic changes in Col6a1-knockout mice: Biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; Bloemberg, D.; Quadrilatero, J. Caspase-2 is required for skeletal muscle differentiation and myogenesis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.T.; Zheng, B.S. Systematic Synopsis of Chinese Fishes; Science Press: Beijing, China, 1987; pp. 339–341. [Google Scholar]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. BioRxiv 2018, 34, 274100. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNAseq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular evolutionary genetics analysis software for microcomputers. Bioinformatics 1994, 10, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Reads Ratio (%) | Clean Bases (bp) | Clean Reads Q20 (%) | Clean Reads Q30 (%) |
|---|---|---|---|---|---|---|
| N. bathybius | 48,063,784 | 47,827,824 | 99.51 | 7,144,556,370 | 98.01 | 94.45 |
| N. furcosus | 55,724,924 | 55,422,050 | 99.46 | 8,274,446,963 | 97.75 | 93.92 |
| N. nemurus | 52,998,374 | 52,690,592 | 99.42 | 7,869,665,822 | 97.65 | 93.62 |
| N. virgatus | 51,121,272 | 50,875,276 | 99.52 | 7,607,939,561 | 97.94 | 94.26 |
| N. aurora | 46,469,258 | 46,255,048 | 99.54 | 6,915,888,873 | 97.82 | 94.03 |
| N. japonicus | 49,153,734 | 48,869,976 | 99.42 | 7,311,785,453 | 97.13 | 92.37 |
| N. marginatus | 41,533,576 | 41,302,034 | 99.44 | 6,177,399,999 | 97.16 | 92.40 |
| Sample | Total Number | Max Length | Min Length | Mean Length | N50 | GC (%) |
|---|---|---|---|---|---|---|
| N. bathybius | 32,661 | 33,374 | 201 | 794 | 1205 | 49.21 |
| N. furcosus | 37,904 | 37,710 | 201 | 877 | 1400 | 49.27 |
| N. nemurus | 38,342 | 60,447 | 201 | 971 | 1701 | 48.06 |
| N. virgatus | 41,024 | 43,285 | 201 | 877 | 1433 | 48.55 |
| N. aurora | 34,882 | 70,286 | 201 | 1096 | 1958 | 48.57 |
| N. japonicus | 40,036 | 67,520 | 201 | 1177 | 2165 | 47.88 |
| N. marginatus | 51,540 | 51,855 | 201 | 965 | 1872 | 47.73 |
| N. bathybius | N. furcosus | N. nemurus | N. virgatus | N. aurora | N. japonicus | N. marginatus | |
|---|---|---|---|---|---|---|---|
| NR | 20,641 (63.20%) | 22,907 (60.43%) | 22,637 (59.04%) | 25,751 (62.77%) | 22,016 (63.12%) | 24,115 (60.23%) | 28,636 (55.56%) |
| KEGG | 20,291 (62.13%) | 22,596 (59.61%) | 22,248 (58.03%) | 25,356 (61.81%) | 21,708 (62.23%) | 23,673 (59.13%) | 28,058 (54.44%) |
| KOG | 13,648 (41.79%) | 15,293 (40.35%) | 14,915 (38.90%) | 16,582 (40.42%) | 14,802 (42.43%) | 15,348 (38.34%) | 18,562 (36.01%) |
| Swiss-Prot | 16,878 (51.68%) | 18,962 (50.03%) | 18,567 (48.42%) | 20,761 (50.61%) | 18,397 (52.74%) | 19,404 (48.47%) | 23,225 (45.06%) |
| annotation genes | 20,858 (63.86%) | 23,178 (61.15%) | 22,842 (59.57%) | 25,948 (63.25%) | 22,331 (64.02%) | 24,154 (60.33%) | 28,699 (55.68%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, Z.; Wu, Q.; Zhou, Y.; Wang, L.; Liu, Y.; Yang, C.; Liu, M.; Xie, Q.; Chen, C.; Ma, S.; et al. Comparative Transcriptomic Analysis for Identification of Environmental-Responsive Genes in Seven Species of Threadfin Breams (Nemipterus). Int. J. Mol. Sci. 2025, 26, 7118. https://doi.org/10.3390/ijms26157118
Dang Z, Wu Q, Zhou Y, Wang L, Liu Y, Yang C, Liu M, Xie Q, Chen C, Ma S, et al. Comparative Transcriptomic Analysis for Identification of Environmental-Responsive Genes in Seven Species of Threadfin Breams (Nemipterus). International Journal of Molecular Sciences. 2025; 26(15):7118. https://doi.org/10.3390/ijms26157118
Chicago/Turabian StyleDang, Zhaoke, Qiaer Wu, Yanbo Zhou, Liangming Wang, Yan Liu, Changping Yang, Manting Liu, Qijian Xie, Cheng Chen, Shengwei Ma, and et al. 2025. "Comparative Transcriptomic Analysis for Identification of Environmental-Responsive Genes in Seven Species of Threadfin Breams (Nemipterus)" International Journal of Molecular Sciences 26, no. 15: 7118. https://doi.org/10.3390/ijms26157118
APA StyleDang, Z., Wu, Q., Zhou, Y., Wang, L., Liu, Y., Yang, C., Liu, M., Xie, Q., Chen, C., Ma, S., & Shan, B. (2025). Comparative Transcriptomic Analysis for Identification of Environmental-Responsive Genes in Seven Species of Threadfin Breams (Nemipterus). International Journal of Molecular Sciences, 26(15), 7118. https://doi.org/10.3390/ijms26157118