
PriorCCI: Interpretable Deep Learning Framework for Identifying Key Ligand–Receptor Interactions Between Specific Cell Types from Single-Cell Transcriptomes

Abstract
1. Introduction
2. Results
2.1. Input Data Preparation for PriorCCI and Presentation
2.2. Performance Evaluation of the CNN Model
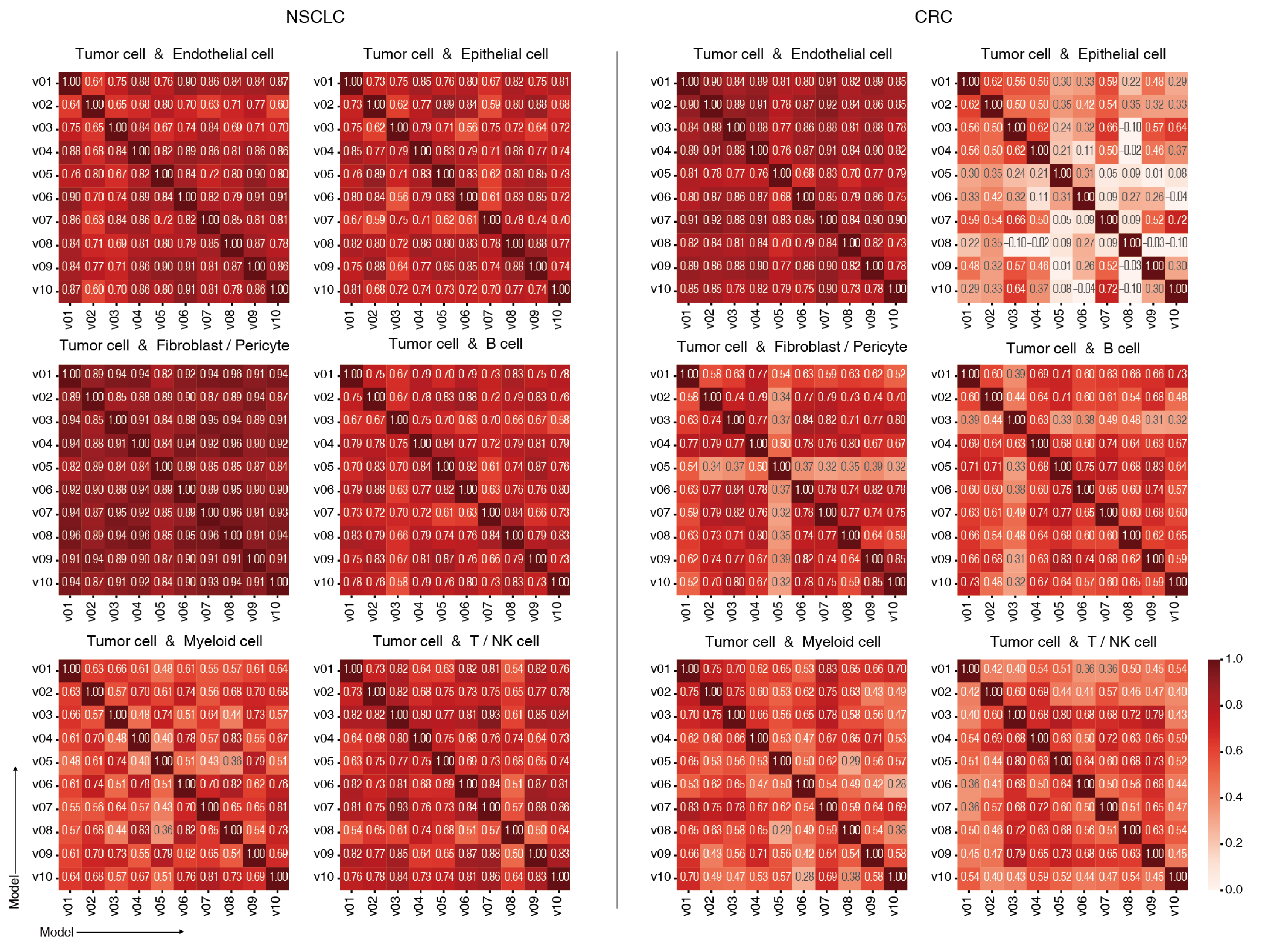
2.3. Model Consistency Across Training Runs
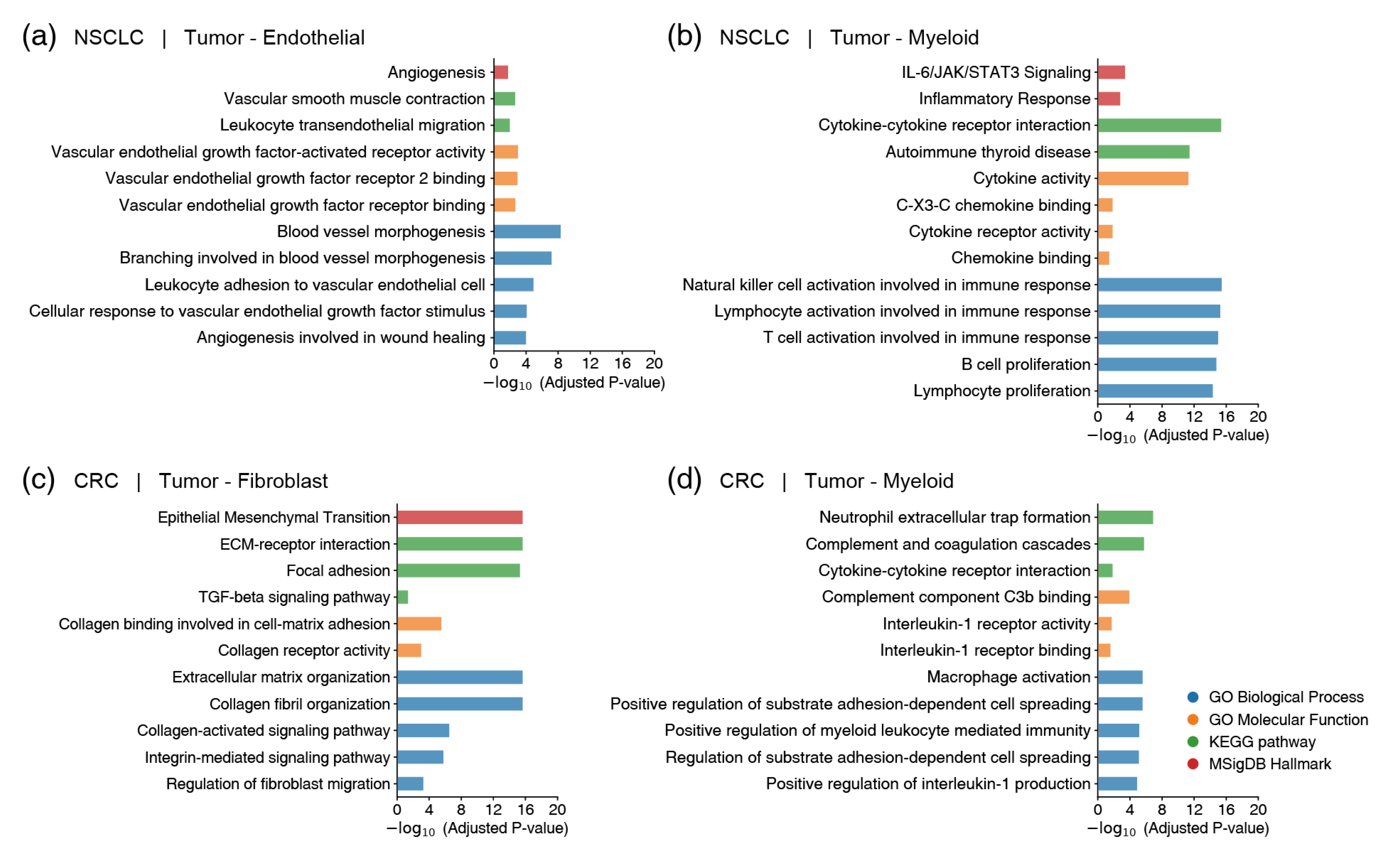
2.4. Functional Validation of Prioritized Gene Pairs Using GSEA
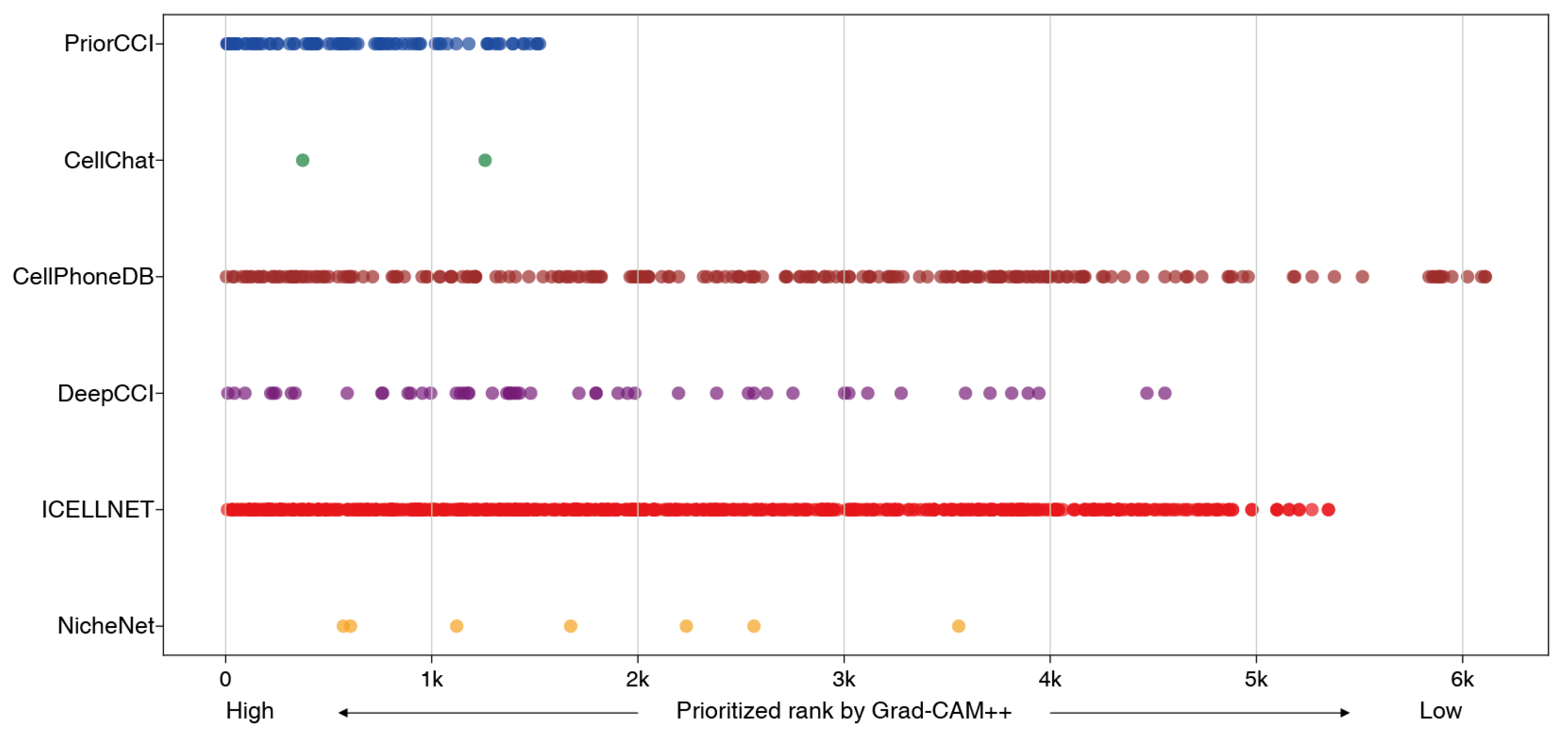
2.5. Comparison with Existing CCI Analysis Tools on Gene Priorities
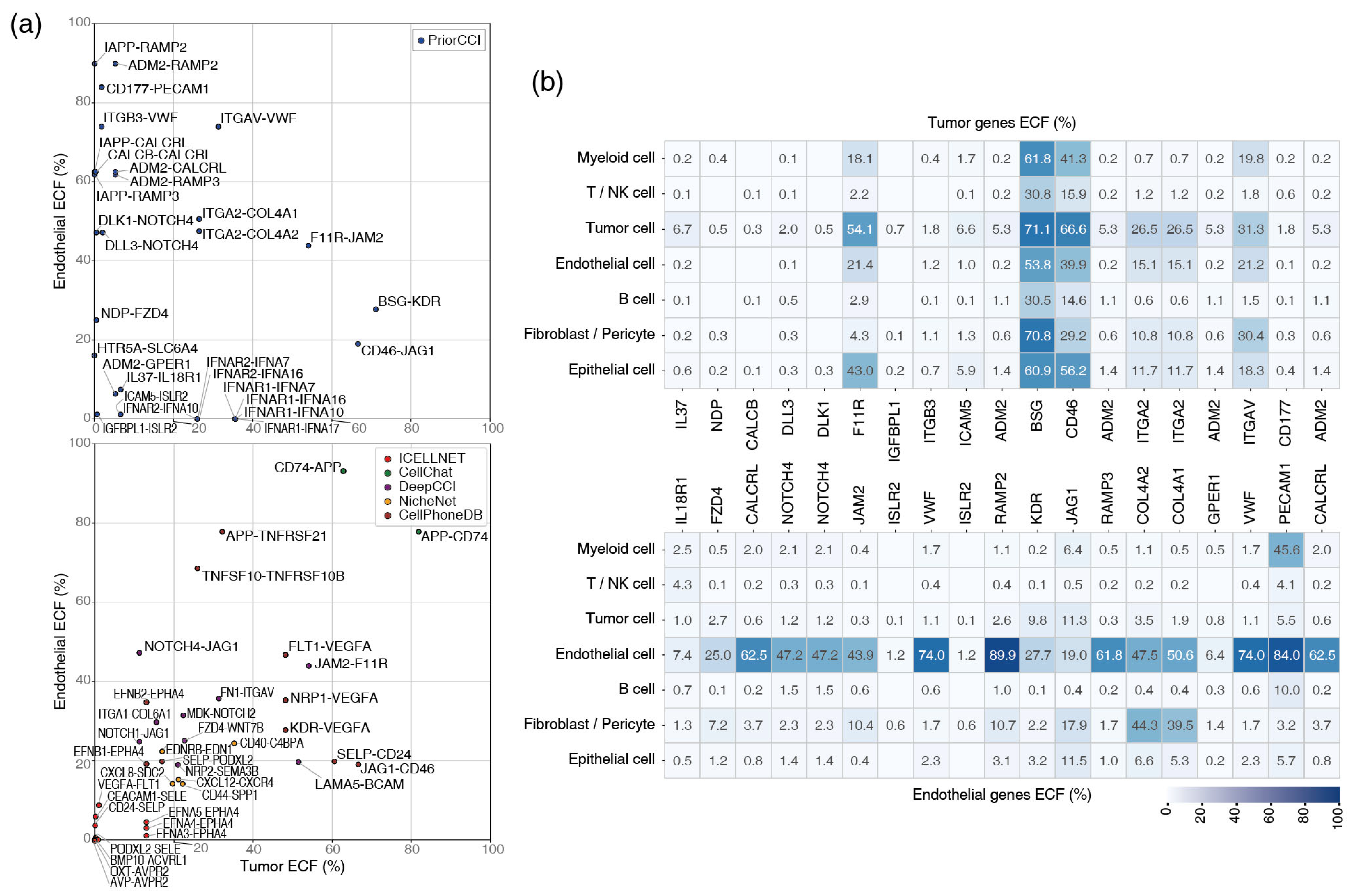
2.6. Single-Cell Expression of Tumor-Endothelial Gene Pairs
3. Discussion
4. Materials and Methods
4.1. Data Preprocessing and Sampling of Representative Cells for Each Cell Type
4.2. CNN in PriorCCI
4.3. Similarity Calculation Within Models
4.4. Visual Interpretation with Grad-CAM++ in PriorCCI
- Model and class definition: The first step was to define the model and class. Let f:X→RC be the trained CNN model, where X∈ℝH×W×D is the input (e.g., ligand–receptor pixel image), C is the number of output classes. We denote the output logit (before softmax) for class c as yc = fc(X).
- Grad-CAM++ computation: In the second step, an importance map calculation based on Grad-CAM++ is performed. Let Ak∈ℝH′×W′ be the k-th feature map at the last convolutional layer. The importance weight for class c is computed via Grad-CAM++ as:
- 3.
- Classwise average of CAMs: The third step is the calculation of the class-specific average of the CAM. Given N samples from class c, the classwise mean of the CAM is:
- 4.
- Extraction of ligand–receptor importance: In the fourth step, the importance of each ligand–receptor pair must be extracted. Given the predefined ligand–receptor index , the CAM weight for pair i is:
- 5.
- Statistical analysis: The fifth step was the statistical analysis of gene pairs with the top 5% importance values. Let be the weight of the gene pair i in model run j (total M runs). Filtering the top 5% per model, we definewhere Mi is the number of models where pair i is in the top 5%. And then we define the coefficient of variation (CV) and median values.
- 6.
- Final ranking: The final step in the process entails the acquisition and organization of the information set, denoted by , and its subsequent arrangement in descending order of Mi or μi.
4.5. Gene Filtering with ECF
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almet, A.A.; Cang, Z.; Jin, S.; Nie, Q. The Landscape of Cell–Cell Communication through Single-Cell Transcriptomics. Curr. Opin. Syst. Biol. 2021, 26, 12–23. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The Technology and Biology of Single-Cell RNA Sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Natri, H.M.; Del Azodi, C.B.; Peter, L.; Taylor, C.J.; Chugh, S.; Kendle, R.; Chung, M.I.I.; Flaherty, D.K.; Matlock, B.K.; Calvi, C.L.; et al. Cell-Type-Specific and Disease-Associated Expression Quantitative Trait Loci in the Human Lung. Nat. Genet. 2024, 56, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering Cell–Cell Interactions and Communication from Gene Expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Eidi, Z.; Khorasani, N.; Sadeghi, M. Correspondence between Signaling and Developmental Patterns by Competing Cells: A Computational Perspective. bioRxiv 2023. [Google Scholar] [CrossRef]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of Methods and Resources for Cell-Cell Communication Inference from Single-Cell RNA-Seq Data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for Systematic Analysis of Cell–Cell Communication from Single-Cell Transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef]
- Wilk, A.J.; Shalek, A.K.; Holmes, S.; Blish, C.A. Comparative Analysis of Cell–Cell Communication at Single-Cell Resolution. Nat. Biotechnol. 2024, 42, 470–483. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, D.; Wang, C. Evaluation of Cell-Cell Interaction Methods by Integrating Single-Cell RNA Sequencing Data with Spatial Information. Genome Biol. 2022, 23, 218. [Google Scholar] [CrossRef]
- Comes, M.C.; Casti, P.; Mencattini, A.; Di Giuseppe, D.; Mermet-Meillon, F.; De Ninno, A.; Parrini, M.C.; Businaro, L.; Di Natale, C.; Martinelli, E. The Influence of Spatial and Temporal Resolutions on the Analysis of Cell-Cell Interaction: A Systematic Study for Time-Lapse Microscopy Applications. Sci. Rep. 2019, 9, 6789. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring Cell–Cell Communication from Combined Expression of Multi-Subunit Ligand–Receptor Complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling Intercellular Communication by Linking Ligands to Target Genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Noël, F.; Massenet-Regad, L.; Carmi-Levy, I.; Cappuccio, A.; Grandclaudon, M.; Trichot, C.; Kieffer, Y.; Mechta-Grigoriou, F.; Soumelis, V. Dissection of Intercellular Communication Using the Transcriptome-Based Framework ICELLNET. Nat. Commun. 2021, 12, 1089. [Google Scholar] [CrossRef] [PubMed]
- Forcato, M.; Romano, O.; Bicciato, S. Computational Methods for the Integrative Analysis of Single-Cell Data. Brief. Bioinform. 2021, 22, bbaa042. [Google Scholar] [CrossRef]
- Yang, W.; Wang, P.; Luo, M.; Cai, Y.; Xu, C.; Xue, G.; Jin, X.; Cheng, R.; Que, J.; Pang, F.; et al. DeepCCI: A Deep Learning Framework for Identifying Cell–Cell Interactions from Single-Cell RNA Sequencing Data. Bioinformatics 2023, 39, btad596. [Google Scholar] [CrossRef]
- Chattopadhay, A.; Sarkar, A.; Howlader, P.; Balasubramanian, V.N. Grad-CAM++: Generalized Gradient-Based Visual Explanations for Deep Convolutional Networks. In Proceedings of the 2018 IEEE Winter Conference on Applications of Computer Vision (WACV), Lake Tahoe, NV, USA, 12–15 March 2018; pp. 839–847. [Google Scholar] [CrossRef]
- Kang, J.; Lee, J.H.; Cha, H.; An, J.; Kwon, J.; Lee, S.; Kim, S.; Baykan, M.Y.; Kim, S.Y.; An, D.; et al. Systematic Dissection of Tumor-Normal Single-Cell Ecosystems across a Thousand Tumors of 30 Cancer Types. Nat. Commun. 2024, 15, 4067. [Google Scholar] [CrossRef]
- Kwon, J.; Kang, J.; Jo, A.; Seo, K.; An, D.; Baykan, M.Y.; Lee, J.H.; Kim, N.; Eum, H.H.; Hwang, S.; et al. Single-Cell Mapping of Combinatorial Target Antigens for CAR Switches Using Logic Gates. Nat. Biotechnol. 2023, 41, 1593–1605. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Somanath, P.R.; Malinin, N.L.; Byzova, T.V. Cooperation between Integrin Aνβ3 and VEGFR2 in Angiogenesis. Angiogenesis 2009, 12, 177–185. [Google Scholar] [CrossRef]
- Ribatti, D. The Crossroad between Tumor and Endothelial Cells. Clin. Exp. Med. 2024, 24, 227. [Google Scholar] [CrossRef]
- Leone, P.; Malerba, E.; Susca, N.; Favoino, E.; Perosa, F.; Brunori, G.; Prete, M.; Racanelli, V. Endothelial Cells in Tumor Microenvironment: Insights and Perspectives. Front. Immunol. 2024, 15, 1367875. [Google Scholar] [CrossRef]
- David, K.; Friedlander, G.; Pellegrino, B.; Radomir, L.; Lewinsky, H.; Leng, L.; Bucala, R.; Becker-Herman, S.; Shachar, I. CD74 as a Regulator of Transcription in Normal B Cells. Cell Rep. 2022, 41, 111572. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Domínguez Conde, C.; Xu, C.; Jarvis, L.B.; Rainbow, D.B.; Wells, S.B.; Gomes, T.; Howlett, S.K.; Suchanek, O.; Polanski, K.; King, H.W.; et al. Cross-Tissue Immune Cell Analysis Reveals Tissue-Specific Features in Humans. Science 2025, 376, eabl5197. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://github.com/broadinstitute/inferCNV/wiki (accessed on 1 August 2023).
- Hie, B.; Cho, H.; DeMeo, B.; Bryson, B.; Berger, B. Geometric Sketching Compactly Summarizes the Single-Cell Transcriptomic Landscape. Cell Syst. 2019, 8, 483–493.e7. [Google Scholar] [CrossRef]
- Agarap, A.F. Deep Learning Using Rectified Linear Units (Relu). arXiv 2018, arXiv:1803.08375. [Google Scholar]
- Kingma, D.P. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | NSCLC | CRC | ||
|---|---|---|---|---|
| Not Applied | Applied | Not Applied | Applied | |
| T/NK | 198,927 | 18,594 | 252,232 | 43,586 |
| Tumor | 131,662 | 17,856 | 196,589 | 42,991 |
| B | 73,508 | 17,729 | 100,116 | 42,142 |
| Myeloid | 40,660 | 15,502 | 69,275 | 36,787 |
| Epithelial | 15,427 | 12,734 | 34,874 | 32,111 |
| Fibroblast/Pericyte | 12,177 | 11,704 | 25,829 | 25,652 |
| Endothelial | 9990 | 9990 | 23,742 | 23,742 |
| Total | 482,351 | 104,109 | 702,657 | 27,001 |
| Model | NSCLC | CRC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Loss | Accuracy | Precision | Recall | F1 Score | Macro AUC | Loss | Accuracy | Precision | Recall | F1 Score | Macro AUC | |
| v1 | 0.021 | 0.993 | 0.994 | 0.993 | 0.993 | 1.000 | 0.141 | 0.954 | 0.961 | 0.954 | 0.953 | 0.999 |
| v2 | 0.043 | 0.983 | 0.985 | 0.983 | 0.983 | 1.000 | 0.200 | 0.942 | 0.945 | 0.942 | 0.942 | 0.999 |
| v3 | 0.018 | 0.997 | 0.997 | 0.997 | 0.997 | 0.999 | 0.075 | 0.977 | 0.979 | 0.977 | 0.977 | 0.999 |
| v4 | 0.021 | 0.994 | 0.994 | 0.994 | 0.994 | 1.000 | 0.198 | 0.949 | 0.952 | 0.949 | 0.948 | 0.999 |
| v5 | 0.029 | 0.989 | 0.990 | 0.989 | 0.989 | 1.000 | 0.177 | 0.943 | 0.945 | 0.943 | 0.943 | 0.999 |
| v6 | 0.043 | 0.986 | 0.987 | 0.986 | 0.986 | 1.000 | 0.191 | 0.944 | 0.946 | 0.944 | 0.944 | 0.999 |
| v7 | 0.027 | 0.991 | 0.991 | 0.991 | 0.991 | 0.999 | 0.157 | 0.949 | 0.950 | 0.949 | 0.949 | 0.999 |
| v8 | 0.032 | 0.988 | 0.989 | 0.988 | 0.988 | 1.000 | 0.171 | 0.929 | 0.942 | 0.929 | 0.927 | 0.999 |
| v9 | 0.026 | 0.992 | 0.992 | 0.992 | 0.992 | 0.999 | 0.168 | 0.955 | 0.957 | 0.955 | 0.955 | 0.999 |
| v10 | 0.028 | 0.992 | 0.993 | 0.992 | 0.992 | 1.000 | 0.064 | 0.981 | 0.981 | 0.981 | 0.980 | 1.000 |
| Avg. | 0.029 | 0.991 | 0.991 | 0.991 | 0.991 | 1.000 | 0.154 | 0.952 | 0.956 | 0.952 | 0.952 | 0.999 |
| Class No. | NSCLC | CRC | ||||||
|---|---|---|---|---|---|---|---|---|
| Cosine | Spearman | Cosine | Spearman | |||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 0 | 0.867 | 0.049 | 0.652 | 0.090 | 0.884 | 0.047 | 0.632 | 0.136 |
| 1 | 0.902 | 0.030 | 0.672 | 0.100 | 0.854 | 0.035 | 0.603 | 0.113 |
| 2 | 0.899 | 0.058 | 0.830 | 0.062 | 0.904 | 0.027 | 0.822 | 0.043 |
| 3 | 0.970 | 0.011 | 0.902 | 0.038 | 0.907 | 0.022 | 0.731 | 0.088 |
| 4 | 0.883 | 0.042 | 0.700 | 0.104 | 0.930 | 0.025 | 0.848 | 0.060 |
| 5 | 0.934 | 0.024 | 0.756 | 0.080 | 0.841 | 0.053 | 0.474 | 0.172 |
| 6 | 0.895 | 0.044 | 0.867 | 0.039 | 0.835 | 0.109 | 0.628 | 0.149 |
| 7 | 0.959 | 0.016 | 0.944 | 0.019 | 0.766 | 0.086 | 0.321 | 0.223 |
| 8 | 0.943 | 0.022 | 0.915 | 0.035 | 0.845 | 0.057 | 0.658 | 0.106 |
| 9 | 0.957 | 0.014 | 0.904 | 0.036 | 0.940 | 0.015 | 0.914 | 0.030 |
| 10 | 0.717 | 0.113 | 0.418 | 0.140 | 0.861 | 0.064 | 0.734 | 0.074 |
| 11 | 0.822 | 0.077 | 0.671 | 0.098 | 0.892 | 0.047 | 0.804 | 0.056 |
| 12 | 0.894 | 0.039 | 0.624 | 0.114 | 0.857 | 0.036 | 0.662 | 0.100 |
| 13 | 0.819 | 0.058 | 0.557 | 0.130 | 0.825 | 0.086 | 0.657 | 0.157 |
| 14 | 0.861 | 0.035 | 0.655 | 0.068 | 0.902 | 0.026 | 0.757 | 0.085 |
| 15 | 0.883 | 0.044 | 0.662 | 0.093 | 0.819 | 0.085 | 0.565 | 0.172 |
| 16 | 0.958 | 0.011 | 0.884 | 0.047 | 0.811 | 0.049 | 0.668 | 0.100 |
| 17 | 0.779 | 0.095 | 0.580 | 0.099 | 0.845 | 0.061 | 0.606 | 0.120 |
| 18 | 0.924 | 0.025 | 0.737 | 0.098 | 0.919 | 0.026 | 0.834 | 0.058 |
| 19 | 0.922 | 0.022 | 0.751 | 0.069 | 0.824 | 0.072 | 0.587 | 0.116 |
| 20 | 0.867 | 0.049 | 0.652 | 0.090 | 0.818 | 0.122 | 0.561 | 0.117 |
| Cell Type | Gene Candidates | |||||
|---|---|---|---|---|---|---|
| APP | CD74 | FLT1 | TNFRSF21 | TNFSF10 | TNFRSF10B | |
| T/NK | 29.4 | 95.9 | 7.9 | 10.4 | 24.1 | 14.2 |
| Tumor | 3.6 | 74.1 | 0.4 | 0.3 | 12.2 | 3.1 |
| B | 62.8 | 81.8 | 1.1 | 32.2 | 39.9 | 25.9 |
| Myeloid | 77.8 | 93.1 | 46.6 | 2.7 | 68.6 | 13.7 |
| Epithelial | 6.9 | 99.1 | 0.5 | 0.7 | 9.1 | 3.8 |
| Fibroblast/Pericyte | 57.7 | 60.0 | 1.2 | 12.7 | 20.2 | 9.7 |
| Endothelial | 45.6 | 87.5 | 0.7 | 13.9 | 30.8 | 17.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Choi, E.; Shim, Y.; Kwon, J. PriorCCI: Interpretable Deep Learning Framework for Identifying Key Ligand–Receptor Interactions Between Specific Cell Types from Single-Cell Transcriptomes. Int. J. Mol. Sci. 2025, 26, 7110. https://doi.org/10.3390/ijms26157110
Kim H, Choi E, Shim Y, Kwon J. PriorCCI: Interpretable Deep Learning Framework for Identifying Key Ligand–Receptor Interactions Between Specific Cell Types from Single-Cell Transcriptomes. International Journal of Molecular Sciences. 2025; 26(15):7110. https://doi.org/10.3390/ijms26157110
Chicago/Turabian StyleKim, Hanbyeol, Eunyoung Choi, Yujeong Shim, and Joonha Kwon. 2025. "PriorCCI: Interpretable Deep Learning Framework for Identifying Key Ligand–Receptor Interactions Between Specific Cell Types from Single-Cell Transcriptomes" International Journal of Molecular Sciences 26, no. 15: 7110. https://doi.org/10.3390/ijms26157110
APA StyleKim, H., Choi, E., Shim, Y., & Kwon, J. (2025). PriorCCI: Interpretable Deep Learning Framework for Identifying Key Ligand–Receptor Interactions Between Specific Cell Types from Single-Cell Transcriptomes. International Journal of Molecular Sciences, 26(15), 7110. https://doi.org/10.3390/ijms26157110