
Understanding Sex Differences in Autoimmune Diseases: Immunologic Mechanisms

Abstract
1. Introduction
2. Experimental Approaches to Investigating Sex Differences in Autoimmunity
2.1. Cellular and Molecular Studies
2.2. Animal Models with Genetic or Hormonal Manipulations
2.3. Clinical and Epidemiological Studies
2.4. Multi-Omics, Systems Biology, and Computational Modeling
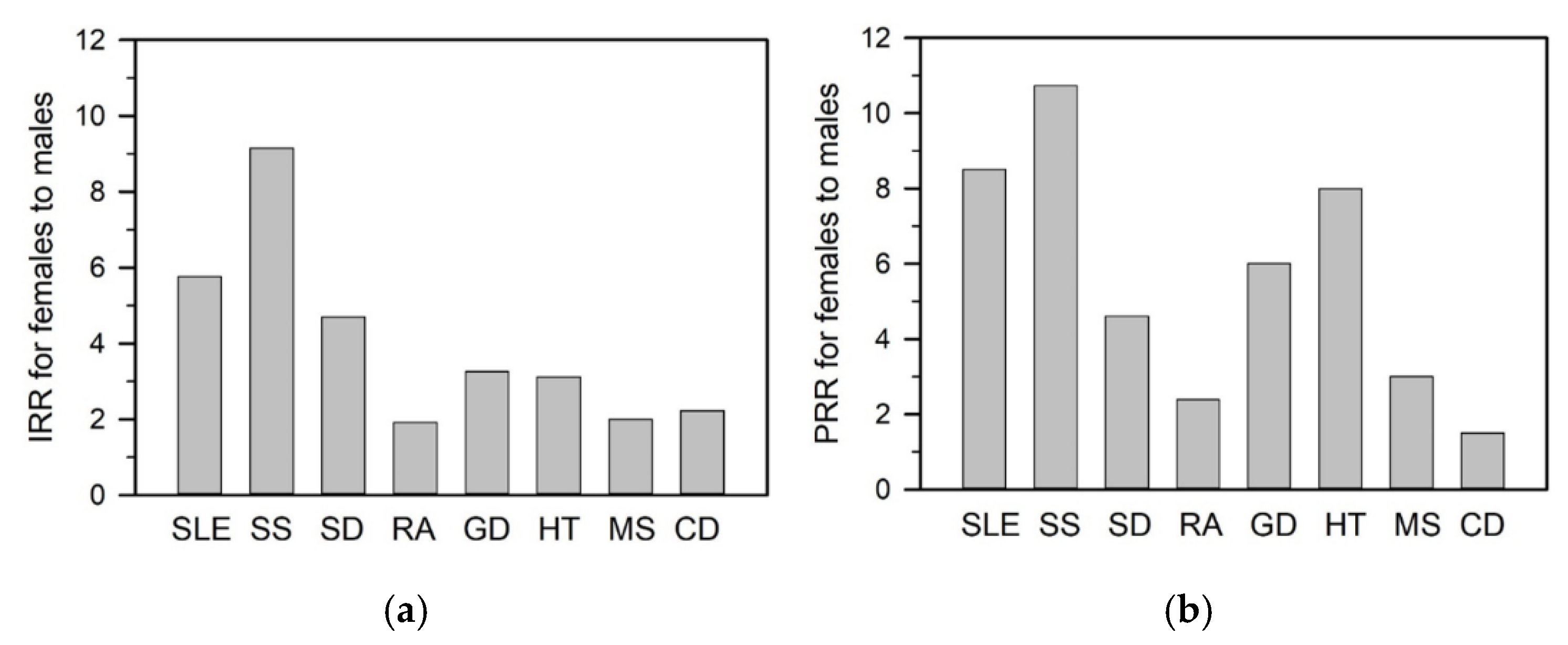
3. Sex-Specific Disparities in Immune Responses
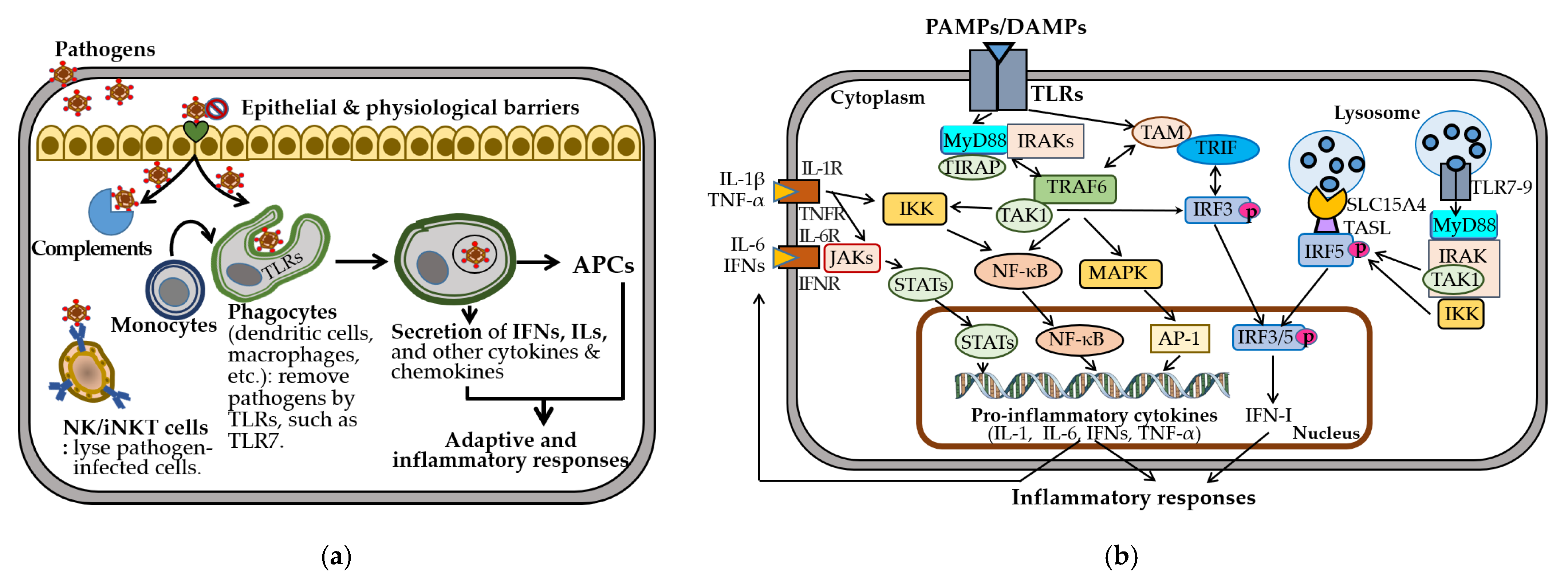
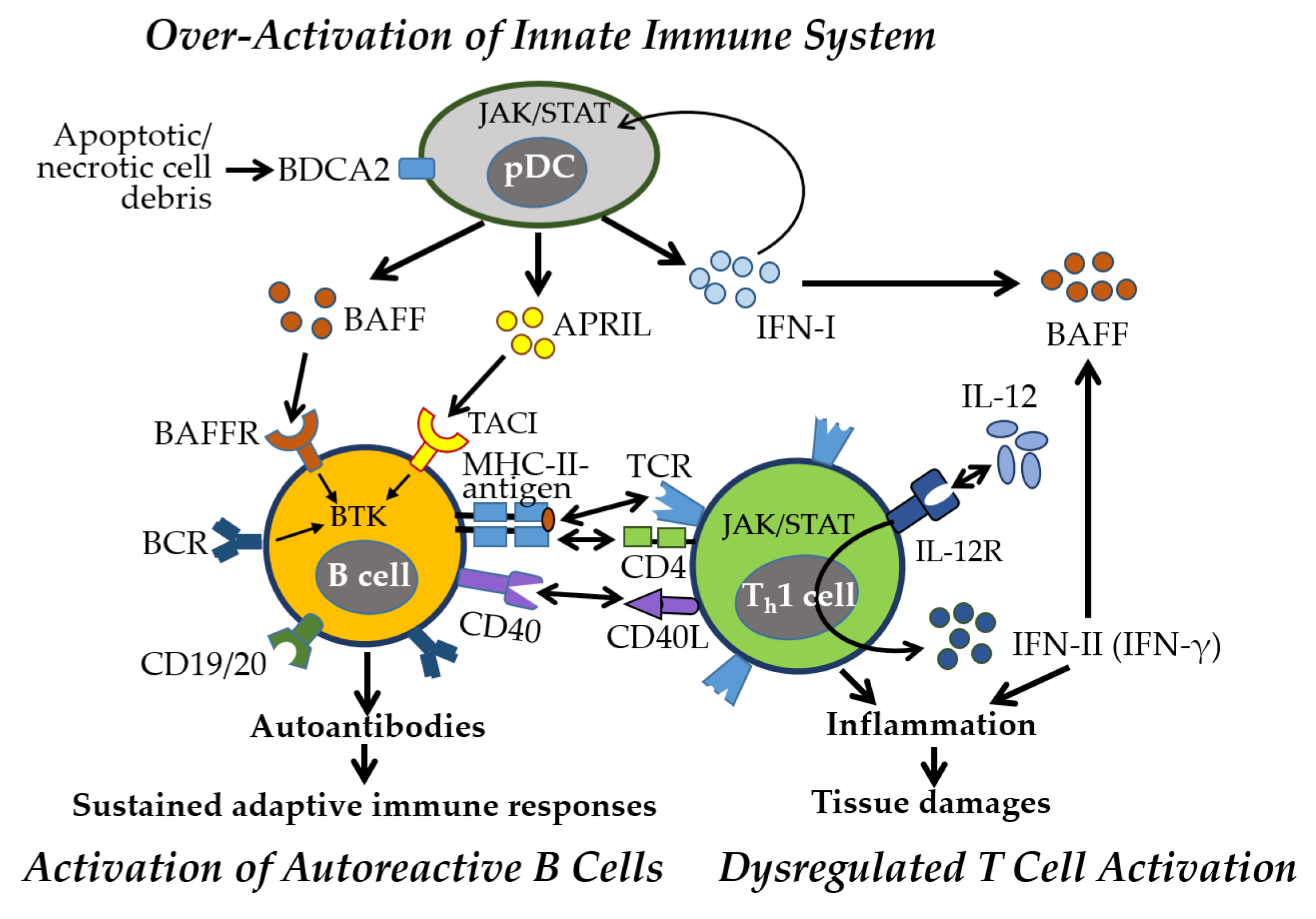
3.1. Innate Immune Responses
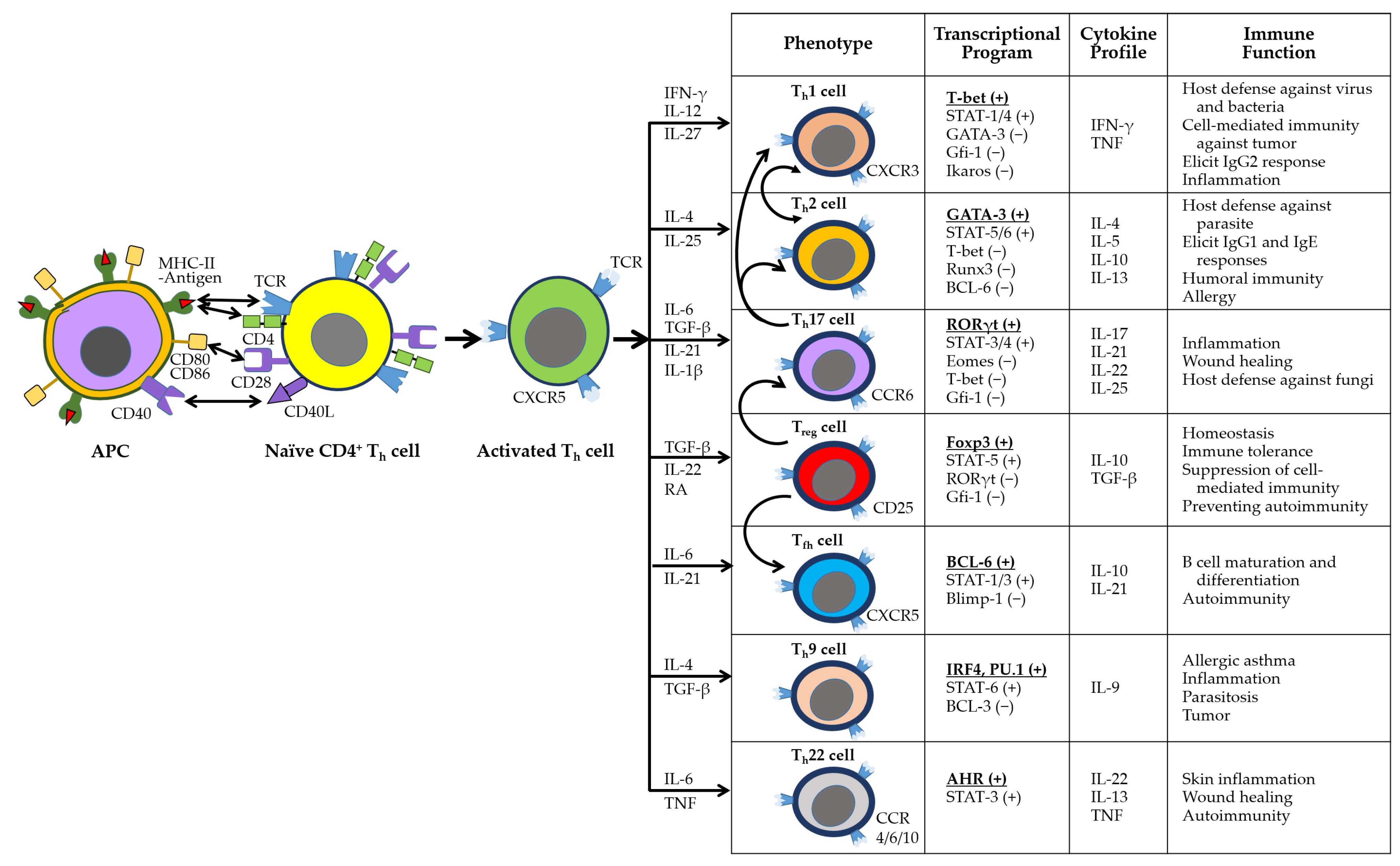
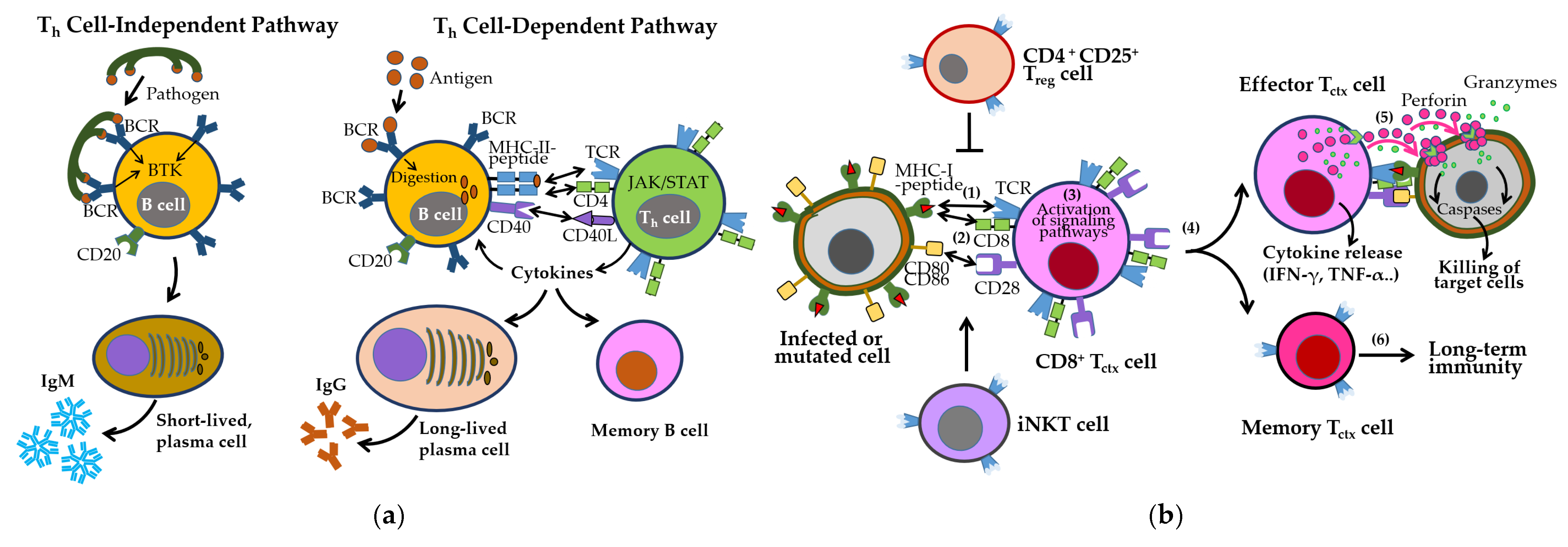
3.2. Adaptive Immune Responses
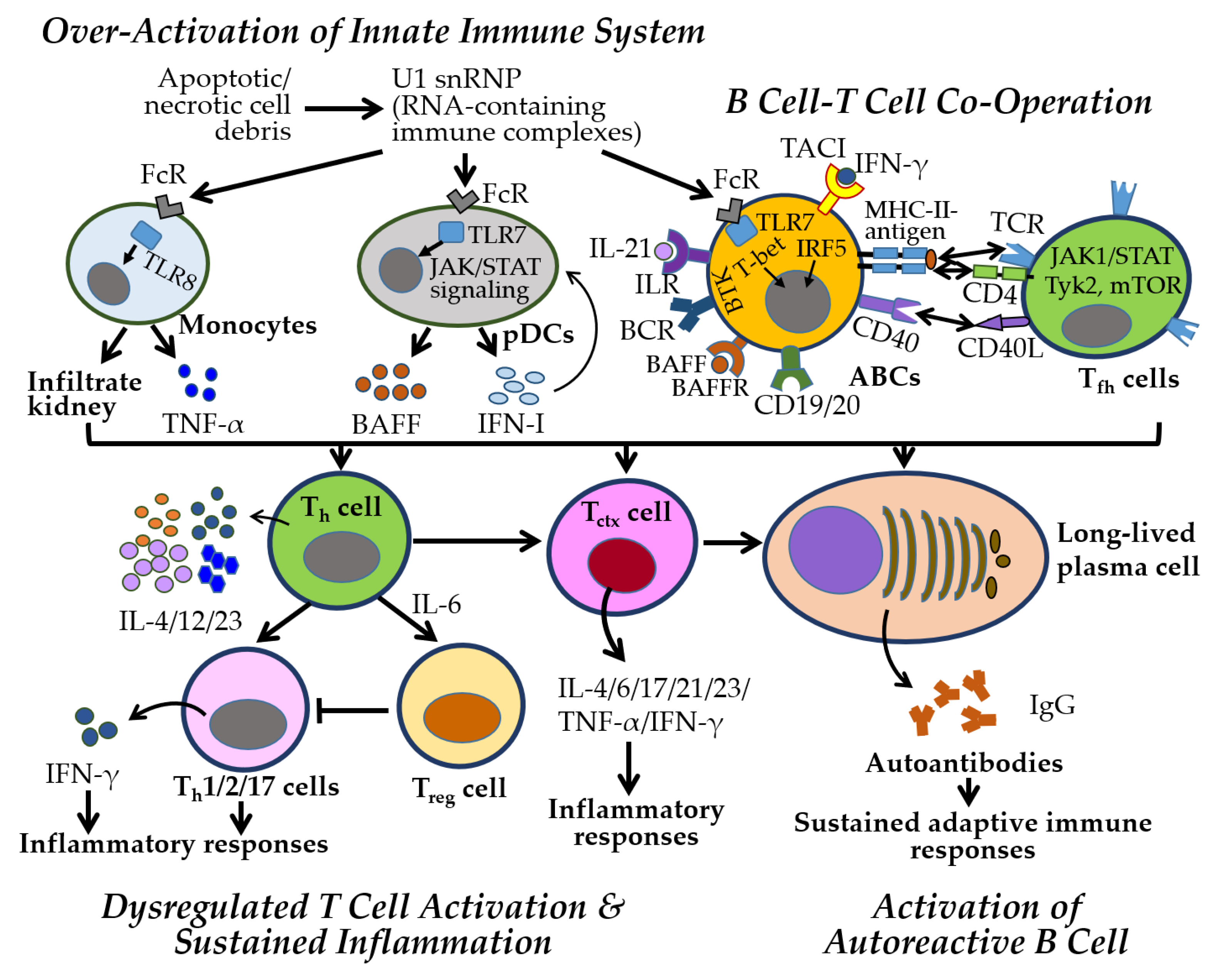
4. Alterations of Immune and Inflammatory Responses in SLE and SS
4.1. Immune and Inflammatory Responses Manifested in SLE
4.2. Immune and Inflammatory Responses Manifested in SS
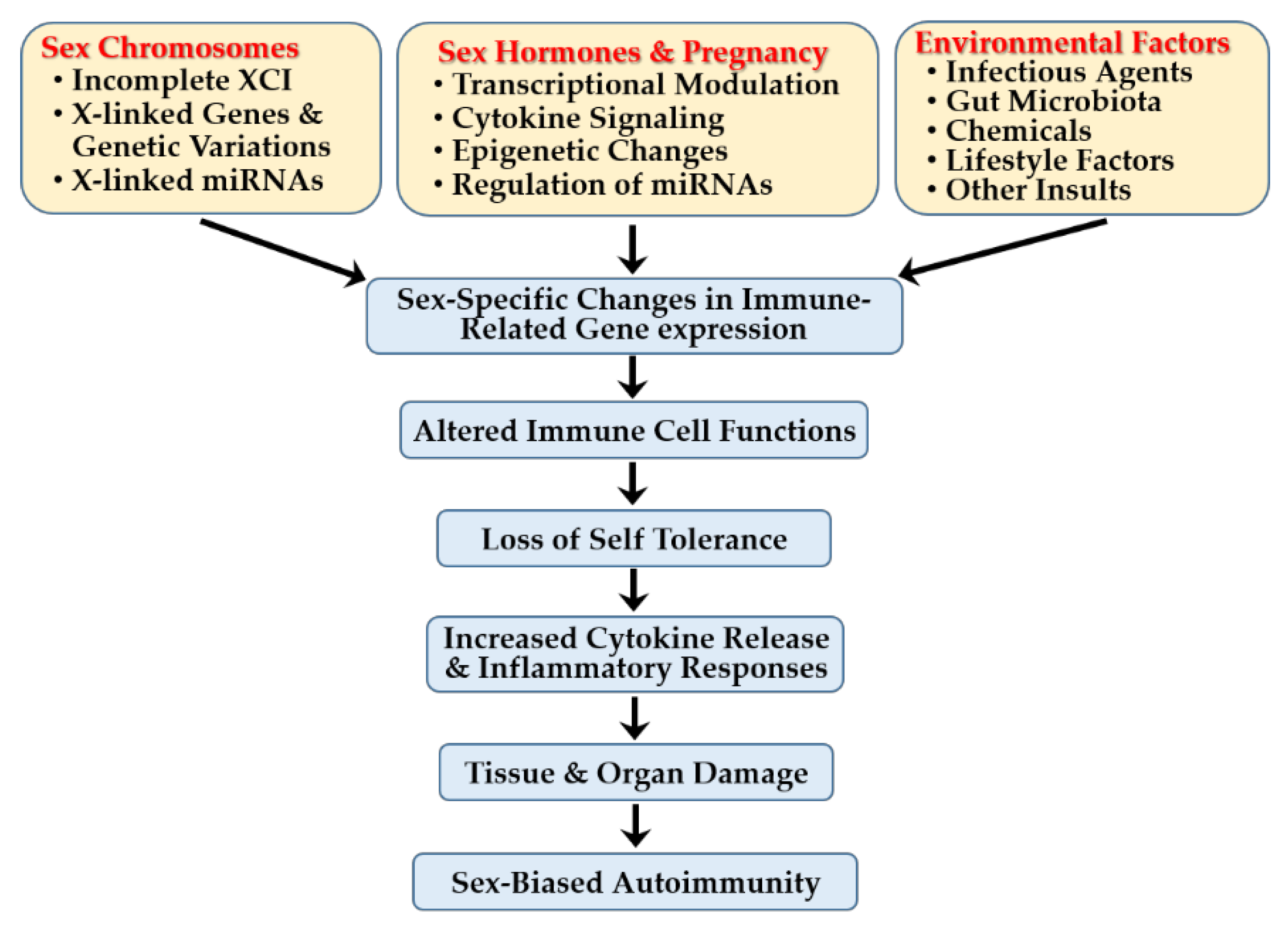
5. Sex-Specific Immune Mechanisms in Autoimmune Diseases
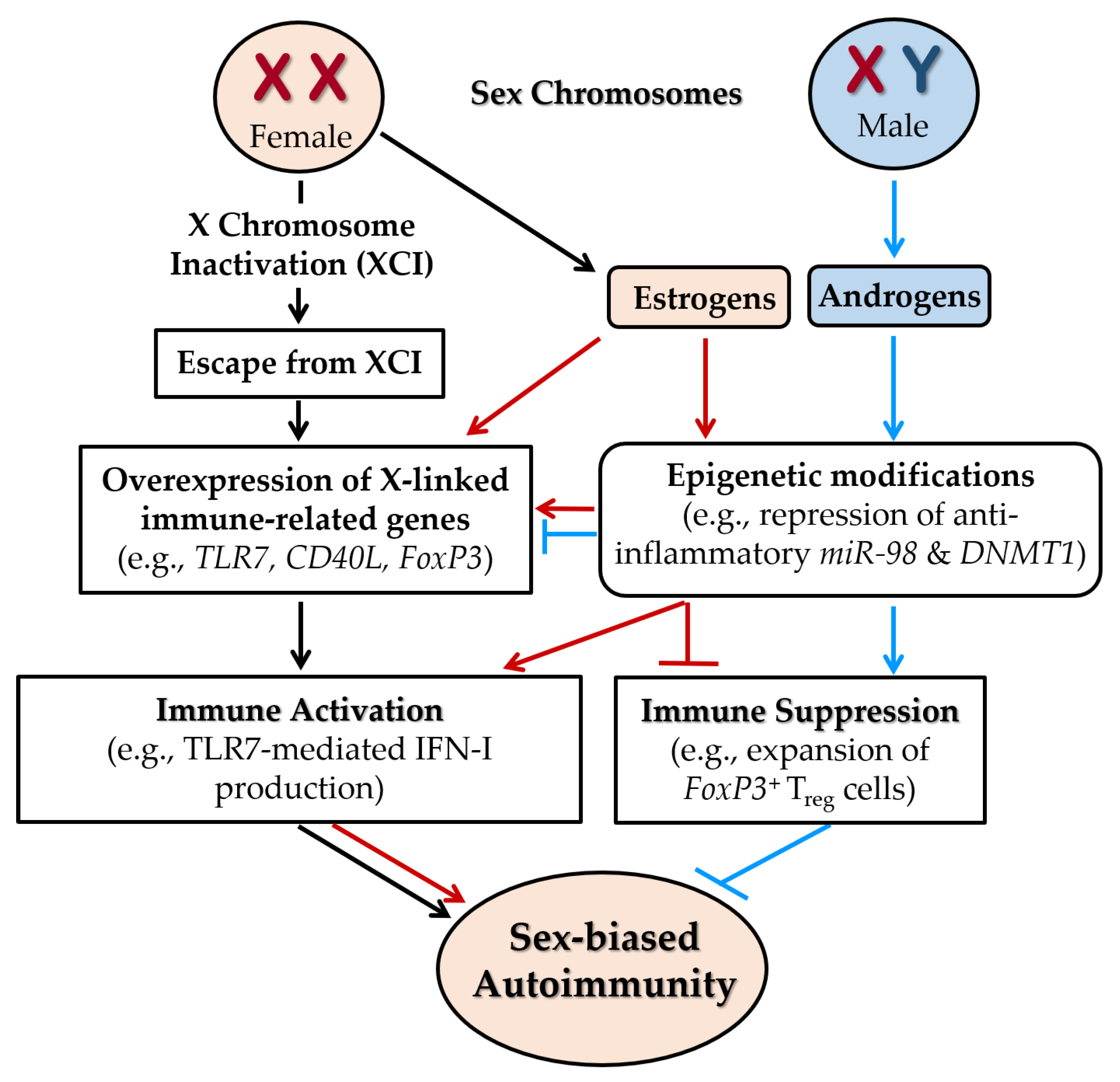
5.1. Sex-Linked Genetic Factors
5.1.1. Escape from XCI
5.1.2. Immune-Associated Genes Escaping XCI
5.1.3. Genetic Variations Across the Genome
5.1.4. Sex-Biased miRs and Gene Expression
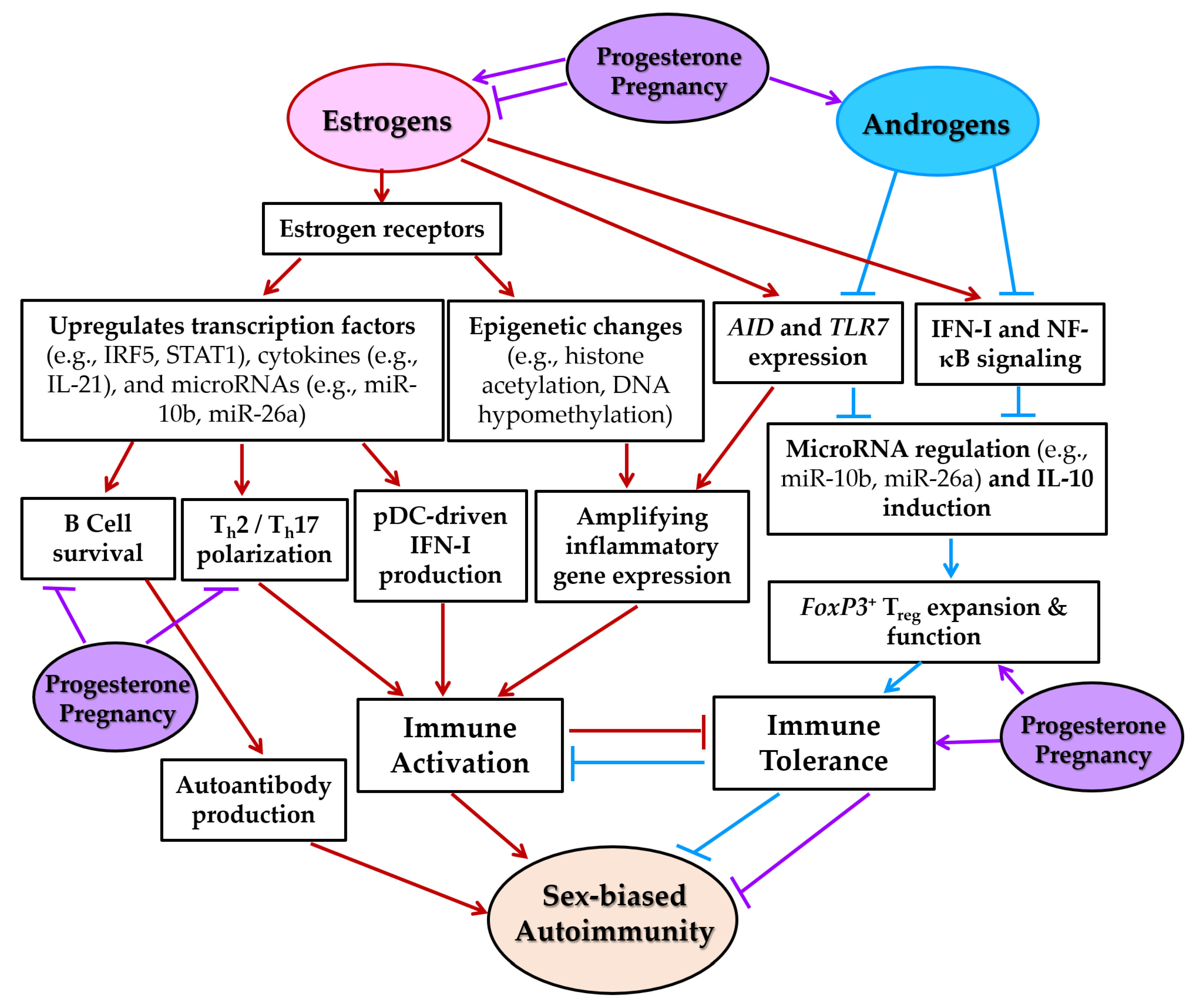
5.2. Sex Hormones, Pregnancy, and Autoimmunity
5.3. Sex Hormone-Dependent Mechanisms of Immune Regulation
5.3.1. Modulation of Transcription Factors
5.3.2. Amplification of Cytokine Signaling
5.3.3. Induction of Epigenetic Changes
5.3.4. Regulation of miR Expression
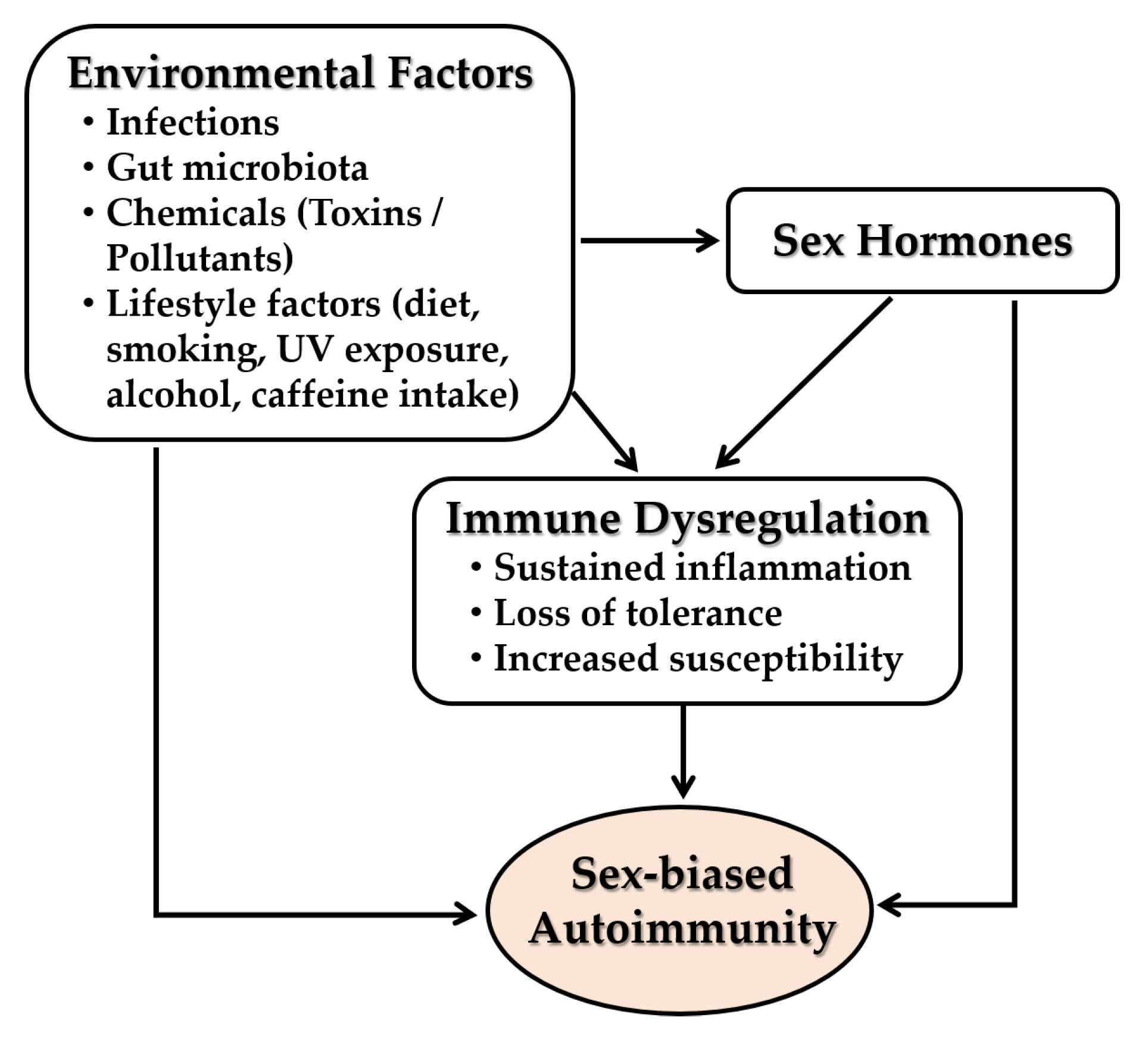
5.4. Interplay with Environmental Factors
5.4.1. Infections
5.4.2. Gut Microbiota
5.4.3. Environmental Chemicals and Pollutants
5.4.4. Lifestyle Behaviors
5.4.5. Psychological and Physical Stressors
6. Conclusions
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dai, X.; Fan, Y.; Zhao, X. Systemic lupus erythematosus: Updated insights on the pathogenesis, diagnosis, prevention and therapeutics. Signal Transduct. Target. Ther. 2025, 10, 102. [Google Scholar] [CrossRef]
- Peckham, H.; Webb, K.; Rosser, E.C.; Butler, G.; Ciurtin, C. Gender-diverse inclusion in immunological research: Benefits to science and health. Front. Med. 2022, 9, 909789. [Google Scholar] [CrossRef]
- Conrad, N.; Misra, S.; Verbakel, J.Y.; Verbeke, G.; Molenberghs, G.; Taylor, P.N.; Mason, J.; Sattar, N.; McMurray, J.J.V.; McInnes, I.B.; et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: A population-based cohort study of 22 million individuals in the uk. Lancet 2023, 401, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.K.; Brinton, R.D. Autoimmune disease in women: Endocrine transition and risk across the lifespan. Front. Endocrinol. 2019, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The prevalence of autoimmune disorders in women: A narrative review. Cureus. 2020, 12, e8094. [Google Scholar] [CrossRef] [PubMed]
- Izmirly, P.M.; Buyon, J.P.; Wan, I.; Belmont, H.M.; Sahl, S.; Salmon, J.E.; Askanase, A.; Bathon, J.M.; Geraldino-Pardilla, L.; Ali, Y.; et al. The incidence and prevalence of adult primary sjogren’s syndrome in new york county. Arthritis Care Res. 2019, 71, 949–960. [Google Scholar] [CrossRef]
- Izmirly, P.M.; Parton, H.; Wang, L.; McCune, W.J.; Lim, S.S.; Drenkard, C.; Ferucci, E.D.; Dall’ERa, M.; Gordon, C.; Helmick, C.G.; et al. Prevalence of systemic lupus erythematosus in the united states: Estimates from a meta-analysis of the centers for disease control and prevention national lupus registries. Arthritis Rheumatol. 2021, 73, 991–996. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, D.; Yao, X.; Huang, Y.; Lu, Q. Global epidemiology of systemic lupus erythematosus: A comprehensive systematic analysis and modelling study. Ann. Rheum. Dis. 2023, 82, 351–356. [Google Scholar] [CrossRef]
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A.; Mak, A.; Kow, N.Y.; Svenungsson, E.; Peterson, J.; Clarke, A.E.; Ramsey-Goldman, R. Global epidemiology of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 515–532. [Google Scholar] [CrossRef]
- Seror, R.; Chiche, L.; Beydon, M.; Desjeux, G.; Zhuo, J.; Vannier-Moreau, V.; Devauchelle-Pensec, V. Estimated prevalence, incidence and healthcare costs of sjögren’s syndrome in france: A national claims-based study. RMD Open 2024, 10, e003591. [Google Scholar] [CrossRef]
- Royle, J.G.; Lanyon, P.C.; Grainge, M.J.; Abhishek, A.; Pearce, F.A. The incidence, prevalence, and survival of systemic sclerosis in the uk clinical practice research datalink. Clin. Rheumatol. 2018, 37, 2103–2111. [Google Scholar] [CrossRef]
- Bairkdar, M.; Rossides, M.; Westerlind, H.; Hesselstrand, R.; Arkema, E.V.; Holmqvist, M. Incidence and prevalence of systemic sclerosis globally: A comprehensive systematic review and meta-analysis. Rheumatology 2021, 60, 3121–3133. [Google Scholar] [CrossRef]
- Myasoedova, E.; Davis, J.; Matteson, E.L.; Crowson, C.S. Is the epidemiology of rheumatoid arthritis changing? Results from a population-based incidence study, 1985–2014. Ann. Rheum. Dis. 2020, 79, 440–444. [Google Scholar] [CrossRef]
- Venetsanopoulou, A.I.; Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Epidemiology and risk factors for rheumatoid arthritis development. Mediterr. J. Rheumatol. 2023, 34, 404–413. [Google Scholar] [CrossRef]
- Black, R.J.; Cross, M.; Haile, L.M.; Culbreth, G.T.; Steinmetz, J.D.; Hagins, H.; A Kopec, J.; Brooks, P.M.; Woolf, A.D.; Ong, K.L.; et al. Global, regional, and national burden of rheumatoid arthritis, 1990–2020, and projections to 2050: A systematic analysis of the global burden of disease study 2021. Lancet Rheumatol 2023, 5, e594–e610. [Google Scholar] [CrossRef] [PubMed]
- Calissendorff, J.; Cramon, P.K.; Hallengren, B.; Khamisi, S.; Lantz, M.; Planck, T.; Sjölin, G.; Wallin, G.; Holmberg, M. Long-term outcome of graves’ disease: A gender perspective. Women’s Health Rep. 2023, 4, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, Y.; Shen, Y.; Tian, R.; Sheng, Y.; Que, H. Global prevalence and epidemiological trends of hashimoto’s thyroiditis in adults: A systematic review and meta-analysis. Front. Public Health 2022, 10, 1020709. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the atlas of ms, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Coyle, P.K. What can we learn from sex differences in ms? J. Pers. Med. 2021, 11, 1006. [Google Scholar] [CrossRef]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Fasano, A. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef]
- King, J.A.; Jeong, J.; Underwood, F.E.; Quan, J.; Panaccione, N.; Windsor, J.W.; Coward, S.; Debruyn, J.; Ronksley, P.E.; Shaheen, A.-A.; et al. Incidence of celiac disease is increasing over time: A systematic review and meta-analysis. Am. J. Gastroenterol. 2020, 115, 507–525. [Google Scholar] [CrossRef]
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun. 2019, 96, 1–13. [Google Scholar] [CrossRef]
- Roveta, A.; Parodi, E.L.; Brezzi, B.; Tunesi, F.; Zanetti, V.; Merlotti, G.; Francese, A.; Maconi, A.G.; Quaglia, M. Lupus nephritis from pathogenesis to new therapies: An update. Int. J. Mol. Sci. 2024, 25, 8981. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Zeeshan, B.; Dissanayake, G.; Elgendy, M.; Billey, A.; Zergaw, M.F. Anti-smith antibodies as a predictive factor for developing lupus nephritis in systemic lupus erythematosus patients: A systematic review. Cureus 2024, 16, e66270. [Google Scholar] [CrossRef] [PubMed]
- Siegel, C.H.; Sammaritano, L.R. Systemic lupus erythematosus: A review. JAMA 2024, 331, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Pryor, K.P.; Barbhaiya, M.; Costenbader, K.H.; Feldman, C.H. Disparities in lupus and lupus nephritis care and outcomes among us medicaid beneficiaries. Rheum. Dis. Clin. N. Am. 2021, 47, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ren, Y.L.; Chang, J.; Gu, L.; Sun, L.-Y. A systematic review and meta-analysis of prevalence of biopsy-proven lupus nephritis. Arch. Rheumatol. 2018, 33, 17–25. [Google Scholar] [CrossRef]
- Morthen, M.K.; Tellefsen, S.; Richards, S.M.; Lieberman, S.M.; Darabad, R.R.; Kam, W.R.; Sullivan, D.A. Testosterone influence on gene expression in lacrimal glands of mouse models of sjogren syndrome. Investig. Opthalmol. Vis. Sci. 2019, 60, 2181–2197. [Google Scholar] [CrossRef]
- Zhan, Q.; Zhang, J.; Lin, Y.; Chen, W.; Fan, X.; Zhang, D. Pathogenesis and treatment of sjogren’s syndrome: Review and update. Front. Immunol. 2023, 14, 1127417. [Google Scholar] [CrossRef]
- Sciarra, F.; Campolo, F.; Franceschini, E.; Carlomagno, F.; Venneri, M.A. Gender-specific impact of sex hormones on the immune system. Int. J. Mol. Sci. 2023, 24, 6302. [Google Scholar] [CrossRef]
- Forsyth, K.S.; Jiwrajka, N.; Lovell, C.D.; Toothacre, N.E.; Anguera, M.C. The connexion between sex and immune responses. Nat. Rev. Immunol. 2024, 24, 487–502. [Google Scholar] [CrossRef]
- Bose, M.; Jefferies, C. Sex bias in systemic lupus erythematosus: A molecular insight. Immunometabolism 2022, 4, e00004. [Google Scholar] [CrossRef]
- Fairweather, D.; Beetler, D.J.; McCabe, E.J.; Lieberman, S.M. Mechanisms underlying sex differences in autoimmunity. J. Clin. Investig. 2024, 134, e180076. [Google Scholar] [CrossRef] [PubMed]
- BerghöfEr, B.; Frommer, T.; Haley, G.; Fink, L.; Bein, G.; Hackstein, H. Tlr7 ligands induce higher ifn-alpha production in females. J. Immunol. 2006, 177, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, M.; Ziegler, S.; Laffont, S.; Smith, N.; Chauveau, L.; Tomezsko, P.; Sharei, A.; Kourjian, G.; Porichis, F.; Hart, M.; et al. Sex differences in plasmacytoid dendritic cell levels of irf5 drive higher ifn-alpha production in women. J. Immunol. 2015, 195, 5327–5336. [Google Scholar] [CrossRef] [PubMed]
- Bynoe, M.S.; Grimaldi, C.M.; Diamond, B. Estrogen up-regulates bcl-2 and blocks tolerance induction of naive b cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2703–2708. [Google Scholar] [CrossRef]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Väänänen, K.; Watford, W.T.; Chen, Z. Estrogen receptor alpha contributes to t cell—mediated autoimmune inflammation by promoting t cell activation and proliferation. Sci. Signal. 2018, 11, e9415. [Google Scholar] [CrossRef]
- Cunningham, M.A.; Wirth, J.R.; Scott, J.L.; Eudaly, J.; Collins, E.L.; Gilkeson, G.S. Early ovariectomy results in reduced numbers of cd11c+/cd11b+ spleen cells and impacts disease expression in murine lupus. Front. Immunol. 2016, 7, 31. [Google Scholar] [CrossRef]
- Brooks, W.H.; Renaudineau, Y. Epigenetics and autoimmune diseases: The x chromosome-nucleolus nexus. Front. Genet. 2015, 6, 22. [Google Scholar] [CrossRef]
- Billi, A.C.; Kahlenberg, J.M.; Gudjonsson, J.E. Sex bias in autoimmunity. Curr. Opin. Rheumatol. 2019, 31, 53–61. [Google Scholar] [CrossRef]
- Richard, M.L.L.; Wirth, J.R.; Khatiwada, A.; Chung, D.; Gilkeson, G.S.; Cunningham, M.A. Conditional knockout of oestrogen receptor alpha in cd11c(+) cells impacts female survival and inflammatory cytokine profile in murine lupus. Immunology 2022, 167, 354–367. [Google Scholar] [CrossRef]
- Stohl, W.; Yu, N.; Wu, Y. Preferential expansion of foxp3(+) t regulatory cells in ctla-4-deficient and ctla-4-haploinsufficient c57bl/6 mice. Immunohorizons 2022, 6, 507–514. [Google Scholar] [CrossRef]
- Chodisetti, S.B.; Fike, A.J.; Domeier, P.P.; Singh, H.; Choi, N.M.; Corradetti, C.; Kalwasawa, Y.I.; Cooper, T.K.; Caricchio, R.; Rahman, Z.S.M. Type ii but not type i ifn signaling is indispensable for tlr7-promoted development of autoreactive b cells and systemic autoimmunity. J. Immunol. 2020, 204, 796–809. [Google Scholar] [CrossRef]
- Gilbert, E.L.; Ryan, M.J. Impact of early life ovariectomy on blood pressure and body composition in a female mouse model of systemic lupus erythematosus. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R990–R997. [Google Scholar] [CrossRef]
- Laffont, S.; Rouquié, N.; Azar, P.; Seillet, C.; Plumas, J.; Aspord, C.; Guéry, J.-C. X-chromosome complement and estrogen receptor signaling independently contribute to the enhanced tlr7-mediated ifn-alpha production of plasmacytoid dendritic cells from women. J. Immunol. 2014, 193, 5444–5452. [Google Scholar] [CrossRef]
- Arnold, A.P.; Chen, X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front. Neuroendocrinol. 2009, 30, 1–9. [Google Scholar] [CrossRef]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. Tlr7 escapes x chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, e8855. [Google Scholar] [CrossRef]
- Punnanitinont, A.; Kramer, J.M. Sex-specific differences in primary sjogren’s disease. Front. Dent. Med. 2023, 4, 1168645. [Google Scholar] [CrossRef] [PubMed]
- Basu, K.; Sengupta, M.; Mukherjee, S.; Karmakar, S.; Roychowdhury, A.; Bandopadhyay, M. Role of light and immunofluorescence microscopy to differentiate primary and secondary membranous nephropathy. Indian J. Pathol. Microbiol. 2022, 65, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Huang, M.; Luo, A.; Liu, S.; Cai, M.; Li, W.; Yuan, S.; Zheng, Z.; Liu, X.; et al. Mettl3 facilitates kidney injury through promoting irf4-mediated plasma cell infiltration via an m6a-dependent manner in systemic lupus erythematosus. BMC Med. 2024, 22, 511. [Google Scholar] [CrossRef] [PubMed]
- Tengstrand, B.; Carlström, K.; Hafström, I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology 2002, 41, 285–289. [Google Scholar] [CrossRef]
- Ali, F.H.M.; Smatti, M.K.; Elrayess, M.A.; Al Thani, A.A.; Yassine, H.M. Role of genetics in eleven of the most common autoimmune diseases in the post genome-wide association studies era. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 8463–8485. [Google Scholar] [CrossRef]
- Fazel-Najafabadi, M.; Looger, L.L.; Rallabandi, H.R.; Nath, S.K. A multilayered post-genome-wide association study analysis pipeline defines functional variants and target genes for systemic lupus erythematosus. Arthritis Rheumatol. 2024, 76, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Fugmann, C.; Reid, S.; Pucholt, P.; Kvarnström, M.; Björk, A.; Mofors, J.; Sjöwall, C.; Eriksson, P.; Olsson, P.; Mandl, T.; et al. A high polygenic risk score is associated with ssa/ssb antibody positivity and early onset in primary sjogren’s disease. Rheumatology 2025, 64, 4341–4346. [Google Scholar] [CrossRef] [PubMed]
- Wallach, J.D.; Sullivan, P.G.; Trepanowski, J.F.; Steyerberg, E.W.; A Ioannidis, J.P. Sex based subgroup differences in randomized controlled trials: Empirical evidence from cochrane meta-analyses. BMJ 2016, 355, i5826. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, R.; Nicola, S.; Corradi, F.; Badiu, I.; Sardo, L.L.; Rashidy, N.; Quinternetto, A.; Mazzola, M.; Meli, F.; Saracco, E.; et al. Impact of sex on infection risk in patients with systemic lupus erythematosus. Bioengineering 2025, 12, 59. [Google Scholar] [CrossRef]
- Arnaud, L.; Furie, R.; Morand, E.F.; Aringer, M.; Peschken, C.; Desta, B.; Rapsomaniki, E.; Hedberg, J.; Knagenhjelm, J.; Seo, C.; et al. Burden of systemic lupus erythematosus in clinical practice: Baseline data from the sle prospective observational cohort study (spocs) by interferon gene signature. Lupus Sci. Med. 2023, 10, e001032. [Google Scholar] [CrossRef]
- Guthridge, J.M.; Lu, R.; Tran, L.T.-H.; Arriens, C.; Aberle, T.; Kamp, S.; Munroe, M.E.; Dominguez, N.; Gross, T.; DeJager, W.; et al. Adults with systemic lupus exhibit distinct molecular phenotypes in a cross-sectional study. EClinicalMedicine 2020, 20, 100291. [Google Scholar] [CrossRef]
- Crawford, J.D.; Wang, H.; Trejo-Zambrano, D.; Cimbro, R.; Talbot, C.C.; Thomas, M.A.; Curran, A.M.; Girgis, A.A.; Schroeder, J.T.; Fava, A.; et al. The xist lncrna is a sex-specific reservoir of tlr7 ligands in sle. J. Clin. Investig. 2023, 8, e169344. [Google Scholar] [CrossRef]
- Pinheiro, I.; Dejager, L.; Libert, C. X-chromosome-located micrornas in immunity: Might they explain male/female differences? The x chromosome-genomic context may affect x-located mirnas and downstream signaling, thereby contributing to the enhanced immune response of females. Bioessays 2011, 33, 791–802. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.-F. Toll-like receptor signaling and its role in cell-mediated immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef] [PubMed]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing toll-like receptors. Nat. Rev. Immunol. 2022, 22, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.; De Nardo, D. Understanding early tlr signaling through the myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Heinz, L.X.; Lee, J.; Kapoor, U.; Kartnig, F.; Sedlyarov, V.; Papakostas, K.; César-Razquin, A.; Essletzbichler, P.; Goldmann, U.; Stefanovic, A.; et al. Tasl is the slc15a4-associated adaptor for irf5 activation by tlr7–9. Nature 2020, 581, 316–322. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nguyen-Tien, D.; Sorimachi, Y.; Sugiura, Y.; Suzuki, T.; Karyu, H.; Shimabukuro-Demoto, S.; Uemura, T.; Okamura, T.; Taguchi, T.; et al. Slc15a4 mediates m1-prone metabolic shifts in macrophages and guards immune cells from metabolic stress. Proc. Natl. Acad. Sci. USA 2021, 118, e2100295118. [Google Scholar] [CrossRef]
- Arleevskaya, M.I.; Larionova, R.V.; Brooks, W.H.; Bettacchioli, E.; Renaludineau, Y. Toll-like receptors, infections, and rheumatoid arthritis. Clin. Rev. Allergy Immunol. 2020, 58, 172–181. [Google Scholar] [CrossRef]
- Frasca, L.; Lande, R. Toll-like receptors in mediating pathogenesis in systemic sclerosis. Clin. Exp. Immunol. 2020, 201, 14–24. [Google Scholar] [CrossRef]
- Caielli, S.; Wan, Z.; Pascual, V. Systemic lupus erythematosus pathogenesis: Interferon and beyond. Annu. Rev. Immunol. 2023, 41, 533–560. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, B.; Liu, X.; Li, H.; Xie, L.; Gao, Y.; Duan, R.; Li, Z.; Zhang, J.; Zheng, Y.; et al. Effects of sex and aging on the immune cell landscape as assessed by single-cell transcriptomic analysis. Proc. Natl. Acad. Sci. USA 2021, 118, e2023216118. [Google Scholar] [CrossRef]
- Taniuchi, I. Cd4 helper and cd8 cytotoxic t cell differentiation. Annu. Rev. Immunol. 2018, 36, 579–601. [Google Scholar] [CrossRef]
- Khantakova, J.N.; Sennikov, S.V. T-helper cells flexibility: The possibility of reprogramming t cells fate. Front. Immunol. 2023, 14, 1284178. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D. B cell responses: Cell interaction dynamics and decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef]
- Curley, S.M.; Putnam, D. Biological nanoparticles in vaccine development. Front. Bioeng. Biotechnol. 2022, 10, 867119. [Google Scholar] [CrossRef]
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C.; I Johnston, G.; Otoul, C.; Stach, C.; Zamacona, M.; Dörner, T.; Urowitz, M.; et al. Repeated administration of dapirolizumab pegol in a randomised phase i study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2017, 76, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond pd-1 and ctla-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, D.; Kurzrock, R.; Miyashita, H.; Adashek, J.; Lee, S.; Nikanjam, M.; Eskander, R.N.; Patel, H.; Botta, G.; Nesline, M.; et al. Viewing the immune checkpoint vista: Landscape and outcomes across cancers. ESMO Open 2024, 9, 102942. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Bao, X.; Zheng, M. Cd8(+) t-cell immunity orchestrated by inkt cells. Front. Immunol. 2022, 13, 1109347. [Google Scholar] [CrossRef]
- Sogkas, G.; Atschekzei, F.; Adriawan, I.R.; Dubrowinskaja, N.; Witte, T.; Schmidt, R.E. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell Mol. Immunol. 2021, 18, 1122–1140. [Google Scholar] [CrossRef]
- Mohr, A.; Atif, M.; Balderas, R.; Gorochov, G.; Miyara, M. The role of foxp3(+) regulatory t cells in human autoimmune and inflammatory diseases. Clin. Exp. Immunol. 2019, 197, 24–35. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Iadarola, M.J.; Keller, J.M.; Warner, B.M. Autoantibodies targeting intracellular and extracellular proteins in autoimmunity. Front. Immunol. 2021, 12, 548469. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory t cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Marozoff, S.; Li, L.; Sayre, E.C.; Zubieta, J.A.A. All-cause and cause-specific mortality in systemic lupus erythematosus: A population-based study. Rheumatology 2021, 61, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Jiwrajka, N.; Anguera, M.C. The x in sex-biased immunity and autoimmune rheumatic disease. J. Exp. Med. 2022, 219, e20211487. [Google Scholar] [CrossRef]
- Dodd, K.C.; Menon, M. Sex bias in lymphocytes: Implications for autoimmune diseases. Front. Immunol. 2022, 13, 945762. [Google Scholar] [CrossRef]
- Schneider-Hohendorf, T.; Görlich, D.; Savola, P.; Kelkka, T.; Mustjoki, S.; Gross, C.C.; Owens, G.C.; Klotz, L.; Dornmair, K.; Wiendl, H.; et al. Sex bias in mhc i-associated shaping of the adaptive immune system. Proc. Natl. Acad. Sci. USA 2018, 115, 2168–2173. [Google Scholar] [CrossRef]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate immune cells’ contribution to systemic lupus erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kubo, S.; Iwata, S.; Yoshikawa, M.; Nakayamada, S. B cell phenotypes, signaling and their roles in secretion of antibodies in systemic lupus erythematosus. Clin. Immunol. 2018, 186, 21–25. [Google Scholar] [CrossRef]
- Chen, K.; Wu, T.; Wang, D.; Li, R.; Shen, X.; Zhao, T.; Ozato, K.; Li, R. Transcriptomics and quantitative proteomics reveal changes after second stimulation of bone marrow-derived macrophages from lupus-prone mrl/lpr mice. Front. Immunol. 2022, 13, 1004232. [Google Scholar] [CrossRef]
- Xu, J.-W.; Wang, M.-Y.; Mao, Y.; Hu, Z.-Y.; Miao, X.-L.; Jiang, F.; Zhou, G.-P. Inhibition of stat3 alleviates lps-induced apoptosis and inflammation in renal tubular epithelial cells by transcriptionally down-regulating tasl. Eur. J. Med Res. 2024, 29, 34. [Google Scholar] [CrossRef]
- Brown, G.J.; Cañete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Qin, Y.; Cappello, J.; Ellyard, J.I.; et al. Tlr7 gain-of-function genetic variation causes human lupus. Nature 2022, 605, 349–356. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Basdra, E.K.; Papavassiliou, A.G. Targeting tlr signaling cascades in systemic lupus erythematosus and rheumatoid arthritis: An update. Biomedicines 2024, 12, 138. [Google Scholar] [CrossRef]
- Crow, M.K. Pathogenesis of systemic lupus erythematosus: Risks, mechanisms and therapeutic targets. Ann. Rheum. Dis. 2023, 82, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Bruce, I.N.; Dörner, T.; Leon, M.G.; Leszczyński, P.; Urowitz, M.; Haier, B.; Jimenez, T.; Brittain, C.; Liu, J.; et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology 2021, 60, 5397–5407. [Google Scholar] [CrossRef] [PubMed]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjäll, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High levels of circulating interferons type i, type ii and type iii associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Pan, H.; Lu, H.; Song, S.; Jin, C.; Pu, Y. Diagnostic significance of combined anti-extractable nuclear antigens antibody, anti-cardiolipin antibody and anti-beta2-glycoprotein 1 in systemic lupus erythematosus patients. Heliyon 2024, 10, e29230. [Google Scholar] [CrossRef]
- Goel, R.R.; Kotenko, S.V.; Kaplan, M.J. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2021, 17, 349–362. [Google Scholar] [CrossRef]
- Park, J.S.; Perl, A. Endosome traffic modulates pro-inflammatory signal transduction in cd4(+) t cells-implications for the pathogenesis of systemic lupus erythematosus. Int. J. Mol. Sci. 2023, 24, 10749. [Google Scholar] [CrossRef]
- Yuan, S.; Zeng, Y.; Li, J.; Wang, C.; Li, W.; He, Z.; Ye, J.; Li, F.; Chen, Y.; Lin, X.; et al. Phenotypical changes and clinical significance of cd4(+)/cd8(+) t cells in sle. Lupus Sci. Med. 2022, 9, e000660. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, X.; Zhao, Y.; Xu, X.; Liu, Y.; Wu, X. Excessive il-15 promotes cytotoxic cd4 + cd28- t cell-mediated renal injury in lupus nephritis. Immun. Ageing 2022, 19, 50. [Google Scholar] [CrossRef]
- Katsuyama, T.; Tsokos, G.C.; Moulton, V.R. Aberrant t cell signaling and subsets in systemic lupus erythematosus. Front. Immunol. 2018, 9, 1088. [Google Scholar] [CrossRef]
- Morel, L. Immunometabolism in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2017, 13, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Clarke, A. A cellular overview of immunometabolism in systemic lupus erythematosus. Oxf. Open Immunol. 2023, 4, iqad005. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Sumikawa, M.H.; Tanaka, Y. B cell activation via immunometabolism in systemic lupus erythematosus. Front. Immunol. 2023, 14, 1155421. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Hsu, H.C.; Mountz, J.D. Autoreactive b cells in sle, villains or innocent bystanders? Immunol. Rev. 2019, 292, 120–138. [Google Scholar] [CrossRef]
- Odani, T.; Chiorini, J.A. Targeting primary sjogren’s syndrome. Mod. Rheumatol. 2019, 29, 70–86. [Google Scholar] [CrossRef]
- Seror, R.; Nocturne, G.; Mariette, X. Current and future therapies for primary sjogren syndrome. Nat. Rev. Rheumatol. 2021, 17, 475–486. [Google Scholar] [CrossRef]
- Finotti, G.; Tamassia, N.; Cassatella, M.A. Interferon-lambdas and plasmacytoid dendritic cells: A close relationship. Front. Immunol. 2017, 8, 1015. [Google Scholar] [CrossRef]
- Jiang, J.; Zhao, M.; Chang, C.; Wu, H.; Lu, Q. Type i interferons in the pathogenesis and treatment of autoimmune diseases. Clin. Rev. Allergy Immunol. 2020, 59, 248–272. [Google Scholar] [CrossRef]
- Apostolou, E.; Tzioufas, A.G. Type-iii interferons in sjogren’s syndrome. Clin. Exp. Rheumatol. 2020, 38 (Suppl. S126), 245–252. [Google Scholar]
- Jonsson, R.; Brokstad, K.A.; Jonsson, M.V.; Delaleu, N.; Skarstein, K. Current concepts on sjogren’s syndrome—Classification criteria and biomarkers. Eur. J. Oral. Sci. 2018, 126 (Suppl. S1), 37–48. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and distinct functions of type i and type iii interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Cattalini, M.; Soliani, M.; Caparello, M.C.; Cimaz, R. Sex differences in pediatric rheumatology. Clin. Rev. Allergy Immunol. 2019, 56, 293–307. [Google Scholar] [CrossRef]
- Smith, E.M.D.; Lythgoe, H.; Midgley, A.; Beresford, M.W.; Hedrich, C.M. Juvenile-onset systemic lupus erythematosus: Update on clinical presentation, pathophysiology and treatment options. Clin. Immunol. 2019, 209, 108274. [Google Scholar] [CrossRef]
- Miquel, C.-H.; Faz-Lopez, B.; Guéry, J.-C. Influence of x chromosome in sex-biased autoimmune diseases. J. Autoimmun. 2023, 137, 102992. [Google Scholar] [CrossRef]
- Dossin, F.; Heard, E. The molecular and nuclear dynamics of x-chromosome inactivation. Cold Spring Harb. Perspect. Biol. 2022, 14, a040196. [Google Scholar] [CrossRef]
- Fang, H.; Disteche, C.M.; Berletch, J.B. X inactivation and escape: Epigenetic and structural features. Front. Cell Dev. Biol. 2019, 7, 219. [Google Scholar] [CrossRef]
- Loda, A.; Collombet, S.; Heard, E. Gene regulation in time and space during x-chromosome inactivation. Nat. Rev. Mol. Cell Biol. 2022, 23, 231–249. [Google Scholar] [CrossRef]
- Oertelt-Prigione, S.; Mariman, E. The impact of sex differences on genomic research. Int. J. Biochem. Cell Biol. 2020, 124, 105774. [Google Scholar] [CrossRef]
- Gay, L.; Melenotte, C.; Lakbar, I.; Mezouar, S.; Devaux, C.; Raoult, D.; Bendiane, M.-K.; Leone, M.; Mège, J.-L. Sexual dimorphism and gender in infectious diseases. Front. Immunol. 2021, 12, 698121. [Google Scholar] [CrossRef]
- Liu, K.; Kurien, B.T.; Zimmerman, S.L.; Kaufman, K.M.; Taft, D.H.; Kottyan, L.C.; Lazaro, S.; Weaver, C.A.; Ice, J.A.; Adler, A.J.; et al. X chromosome dose and sex bias in autoimmune diseases: Increased prevalence of 47,xxx in systemic lupus erythematosus and sjogren’s syndrome. Arthritis Rheumatol. 2016, 68, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.C.; Pandya-Jones, A.; Plath, K. A lifelong duty: How xist maintains the inactive x chromosome. Curr. Opin. Genet. Dev. 2022, 75, 101927. [Google Scholar] [CrossRef]
- Syrett, C.M.; Paneru, B.; Sandoval-Heglund, D.; Wang, J.; Banerjee, S.; Sindhava, V.; Behrens, E.M.; Atchison, M.; Anguera, M.C. Altered x-chromosome inactivation in t cells may promote sex-biased autoimmune diseases. JCI Insight. 2019, 4. [Google Scholar] [CrossRef]
- Nusbaum, J.S.; Mirza, I.; Shum, J.; Freilich, R.W.; Cohen, R.E.; Pillinger, M.H.; Izmirly, P.M.; Buyon, J.P. Sex differences in systemic lupus erythematosus: Epidemiology, clinical considerations, and disease pathogenesis. Mayo Clin. Proc. 2020, 95, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.R.; Zhao, Y.; Belk, J.A.; Zhao, Y.; Casey, K.M.; Chen, D.C.; Li, R.; Yu, B.; Srinivasan, S.; Abe, B.T.; et al. Xist ribonucleoproteins promote female sex-biased autoimmunity. Cell 2024, 187, 733–749 e716. [Google Scholar] [CrossRef] [PubMed]
- Hagen, S.H.; Henseling, F.; Hennesen, J.; Savel, H.; Delahaye, S.; Richert, L.; Ziegler, S.M.; Altfeld, M. Heterogeneous escape from x chromosome inactivation results in sex differences in type i ifn responses at the single human pdc level. Cell Rep. 2020, 33, 108485. [Google Scholar] [CrossRef]
- Lovell, C.D.; Anguera, M.C. More X’s, more problems: How contributions from the X chromosomes enhance female predisposition for autoimmunity. Curr. Opin. Immunol. 2025, 93, 102543. [Google Scholar] [CrossRef]
- Youness, A.; Miquel, C.-H.; Guéry, J.-C. Escape from x chromosome inactivation and the female predominance in autoimmune diseases. Int. J. Mol. Sci. 2021, 22, 1114. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, M.; E Dorrier, C.; Zhang, R.; Mayer, C.T.; Wagner, D.; McGavern, D.B.; Hodes, R.J. Cd40 drives central nervous system autoimmune disease by inducing complementary effector programs via b cells and dendritic cells. J. Immunol. 2022, 209, 2083–2092. [Google Scholar] [CrossRef]
- Li, X.; Yue, X.; Sepulveda, H.; Burt, R.A.; Scott, D.A.; Carr, S.A.; Myers, S.A.; Rao, A. Ogt controls mammalian cell viability by regulating the proteasome/mtor/mitochondrial axis. Proc. Natl. Acad. Sci. USA 2023, 120, e2218332120. [Google Scholar] [CrossRef]
- Youness, A.; Cenac, C.; Faz-López, B.; Grunenwald, S.; Barrat, F.J.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. Tlr8 escapes x chromosome inactivation in human monocytes and cd4(+) t cells. Biol. Sex Differ. 2023, 14, 60. [Google Scholar] [CrossRef]
- Laffont, S.; Guery, J.C. Deconstructing the sex bias in allergy and autoimmunity: From sex hormones and beyond. Adv. Immunol. 2019, 142, 35–64. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Mejía, J.E.; Chaumeil, J.; Guéry, J.-C. Female predisposition to tlr7-driven autoimmunity: Gene dosage and the escape from x chromosome inactivation. Semin. Immunopathol. 2019, 41, 153–164. [Google Scholar] [CrossRef]
- Satterthwaite, A.B. Tlr7 signaling in lupus b cells: New insights into synergizing factors and downstream signals. Curr. Rheumatol. Rep. 2021, 23, 80. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Golden, L.C.; Itoh, N.; Matsukawa, M.A.; Ren, E.; Tse, V.; Arnold, A.P.; Voskuhl, R.R. The x-linked histone demethylase kdm6a in cd4+ t lymphocytes modulates autoimmunity. J. Clin. Investig. 2019, 129, 3852–3863. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.I.; Li, J.H.; Riggan, L.; Chen, B.; Tafti, R.Y.; Chin, S.; Ma, F.; Pellegrini, M.; Hrncir, H.; Arnold, A.P.; et al. The x-linked epigenetic regulator utx controls nk cell-intrinsic sex differences. Nat. Immunol. 2023, 24, 780–791. [Google Scholar] [CrossRef]
- Oliva, M.; Munoz-Aguirre, M.; Kim-Hellmuth, S.; Wucher, V.; Gewirtz, A.D.H.; Cotter, D.J.; Parsana, P.; Kasela, S.; Balliu, B.; Viñuela, A.; et al. The impact of sex on gene expression across human tissues. Science 2020, 369, 3066. [Google Scholar] [CrossRef]
- Sakkas, L.I.; Chikanza, I.C. Sex bias in immune response: It is time to include the sex variable in studies of autoimmune rheumatic diseases. Rheumatol. Int. 2024, 44, 203–209. [Google Scholar] [CrossRef]
- Aune, T.M.; Crooke, P.S., 3rd; Patrick, A.E.; Tossberg, J.T.; Olsen, N.J.; Spurlock, C.F. Expression of long non-coding rnas in autoimmunity and linkage to enhancer function and autoimmune disease risk genetic variants. J. Autoimmun. 2017, 81, 99–109. [Google Scholar] [CrossRef]
- Boodhoo, K.D.; Liu, S.; Zuo, X. Impact of sex disparities on the clinical manifestations in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Medicine 2016, 95, e4272. [Google Scholar] [CrossRef] [PubMed]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct effector b cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 2018, 49, 725–739 e726. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, S.; Panda, A.K. Association of toll-like receptor 7 (tlr7) polymorphisms with predisposition to systemic lupus erythematosus (sle): A meta and trial sequential analysis. Biochem. Genet. 2024, 62, 3350–3366. [Google Scholar] [CrossRef] [PubMed]
- Leibler, C.; John, S.; Elsner, R.A.; Thomas, K.B.; Smita, S.; Joachim, S.; Levack, R.C.; Callahan, D.J.; Gordon, R.A.; Bastacky, S.; et al. Genetic dissection of tlr9 reveals complex regulatory and cryptic proinflammatory roles in mouse lupus. Nat. Immunol. 2022, 23, 1457–1469. [Google Scholar] [CrossRef]
- Shibata, T.; Sato, R.; Taoka, M.; Saitoh, S.-I.; Komine, M.; Yamaguchi, K.; Goyama, S.; Motoi, Y.; Kitaura, J.; Izawa, K.; et al. Tlr7/8 stress response drives histiocytosis in slc29a3 disorders. J. Exp. Med. 2023, 220, e20230054. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, W.; Tangtanatakul, P.; Zheng, L.; Lei, Y.; Lin, Z.; Qian, C.; Qin, X.; Hou, F.; Zhang, X.; et al. Identification of shared and asian-specific loci for systemic lupus erythematosus and evidence for roles of type iii interferon signaling and lysosomal function in the disease: A multi-ancestral genome-wide association study. Arthritis Rheumatol. 2022, 74, 840–848. [Google Scholar] [CrossRef]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune polyendocrine syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Starokadomskyy, P.; Gluck, N.; Li, H.; Chen, B.; Wallis, M.; Maine, G.N.; Mao, X.; Zaidi, I.W.; Hein, M.Y.; McDonald, F.J.; et al. Ccdc22 deficiency in humans blunts activation of proinflammatory nf-kappab signaling. J. Clin. Investig. 2013, 123, 2244–2256. [Google Scholar] [CrossRef]
- D’amico, F.; Skarmoutsou, E.; Lo, L.J.; Granata, M.; Trovato, C.; Rossi, G.A.; Bellocchi, C.; Marchini, M.; Scorza, R.; Mazzarino, M.C.; et al. Association between rs2294020 in x-linked ccdc22 and susceptibility to autoimmune diseases with focus on systemic lupus erythematosus. Immunol. Lett. 2017, 181, 58–62. [Google Scholar] [CrossRef]
- Postal, M.; Vivaldo, J.F.; Fernandez-Ruiz, R.; Paredes, J.L.; Appenzeller, S.; Niewold, T.B. Type i interferon in the pathogenesis of systemic lupus erythematosus. Curr. Opin. Immunol. 2020, 67, 87–94. [Google Scholar] [CrossRef]
- Wang, Z.; Heid, B.; Dai, R.; Ahmed, S.A. Similar dysregulation of lupus-associated mirnas in peripheral blood mononuclear cells and splenic lymphocytes in mrl/lpr mice. Lupus Sci. Med. 2018, 5, e000290. [Google Scholar] [CrossRef]
- Chen, J.-Q.; Papp, G.; Póliska, S.; Szabó, K.; Tarr, T.; Bálint, B.L.; Szodoray, P.; Zeher, M.; Crispin, J.C. Microrna expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary sjogren’s syndrome. PLoS ONE 2017, 12, e0174585. [Google Scholar] [CrossRef]
- Cui, C.; Yang, W.; Shi, J.; Zhou, Y.; Yang, J.; Cui, Q.; Zhou, Y. Identification and analysis of human sex-biased micrornas. Genom. Proteom. Bioinform. 2018, 16, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V. Sex hormones determine immune response. Front. Immunol. 2018, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Zhou, K.; Liufu, Y.; Huang, X.; Zeng, H.; Zhang, Z. Novel insight into mirna biology and its role in the pathogenesis of systemic lupus erythematosus. Front. Immunol. 2022, 13, 1059887. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, E.N.; Quiroz, R.N.; Lugo, L.P.; Martínez, G.A.; Escorcia, L.G.; Torres, H.G.; Bonfanti, A.C.; Marmolejo, M.d.C.; Sanchez, E.; Camacho, J.L.V.; et al. Integrated analysis of microrna regulation and its interaction with mechanisms of epigenetic regulation in the etiology of systemic lupus erythematosus. PLoS ONE 2019, 14, e0218116. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. Mirbase: From microrna sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, H.; Zhao, M.; Lu, Q. Identifying the differentially expressed micrornas in autoimmunity: A systemic review and meta-analysis. Autoimmunity 2020, 53, 122–136. [Google Scholar] [CrossRef]
- Yuan, S.; Tang, C.; Chen, D.; Li, F.; Huang, M.; Ye, J.; He, Z.; Li, W.; Chen, Y.; Lin, X.; et al. Mir-98 modulates cytokine production from human pbmcs in systemic lupus erythematosus by targeting il-6 mrna. J. Immunol. Res. 2019, 2019, 9827574. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J. Role of mir-98 and its underlying mechanisms in systemic lupus erythematosus. J. Rheumatol. 2018, 45, 1397–1405. [Google Scholar] [CrossRef]
- Yang, X.; Shi, L.; Zheng, X.; Liu, X.; Qian, J. Modulation of mir-548m encoded by x chromosome on the pten pathway in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2022, 40, 56–63. [Google Scholar] [CrossRef]
- So, B.Y.F.; Yap, D.Y.H.; Chan, T.M. Micrornas in lupus nephritis-role in disease pathogenesis and clinical applications. Int. J. Mol. Sci. 2021, 22, 10737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, L.; Lu, W.; Li, J.; Li, Y.; Shao, Y.; Sun, J. Lncrna miat enhances systemic lupus erythematosus by upregulating cfhr5 expression via mir-222 degradation. Central Eur. J. Immunol. 2021, 46, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, B.; He, J.; Hua, H. Labial gland mesenchymal stem cell derived exosomes-mediated mirna-125b attenuates experimental sjogren’s syndrome by targeting prdm1 and suppressing plasma cells. Front. Immunol. 2022, 13, 871096. [Google Scholar] [CrossRef] [PubMed]
- Al-Haidose, A.; Hassan, S.; Elhassan, M.; Ahmed, E.; Al-Riashi, A.; Alharbi, Y.M.; Ghunaim, M.; Alhejaili, T.; Abdallah, A.M. Role of ncrnas in the pathogenesis of sjogren’s syndrome. Biomedicines 2024, 12, 1540. [Google Scholar] [CrossRef]
- Hiramatsu-Asano, S.; Sunahori-Watanabe, K.; Zeggar, S.; Katsuyama, E.; Mukai, T.; Morita, Y.; Wada, J. Deletion of mir223 exacerbates lupus nephritis by targeting s1pr1 in fas(lpr/lpr) mice. Front. Immunol. 2020, 11, 616141. [Google Scholar] [CrossRef]
- Qi, X.; Wang, R.; Jin, L.; Tian, Y.; Jin, H.; Han, Y.; Sun, C.; Ding, M.; Guo, H. Mir-223-3p aggravates ocular inflammation in sjogren’s syndrome. Endocrine, Metab. Immune Disord.—Drug Targets 2023, 23, 1087–1095. [Google Scholar] [CrossRef]
- Young, N.A.; Valiente, G.R.; Hampton, J.M.; Wu, L.-C.; Burd, C.J.; Willis, W.L.; Bruss, M.; Steigelman, H.; Gotsatsenko, M.; Amici, S.A.; et al. Estrogen-regulated stat1 activation promotes tlr8 expression to facilitate signaling via microrna-21 in systemic lupus erythematosus. Clin. Immunol. 2017, 176, 12–22. [Google Scholar] [CrossRef]
- Lu, M.-C.; Lai, N.-S.; Chen, H.-C.; Yu, H.-C.; Huang, K.-Y.; Tung, C.-H.; Huang, H.-B.; Yu, C.-L. Decreased microrna(mir)-145 and increased mir-224 expression in t cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin. Exp. Immunol. 2013, 171, 91–99. [Google Scholar] [CrossRef]
- Cornet, A.; Andersen, J.; Myllys, K.; Edwards, A.; Arnaud, L. Living with systemic lupus erythematosus in 2020: A european patient survey. Lupus Sci. Med. 2021, 8, e000469. [Google Scholar] [CrossRef]
- Kim, J.-W.; Kim, H.-A.; Suh, C.-H.; Jung, J.-Y. Sex hormones affect the pathogenesis and clinical characteristics of systemic lupus erythematosus. Front. Med. 2022, 9, 906475. [Google Scholar] [CrossRef]
- Taneja, V. Sexual dimorphism, aging and immunity. Vitam. Horm. 2021, 115, 367–399. [Google Scholar] [CrossRef]
- A Rey, R. The role of androgen signaling in male sexual development at puberty. Endocrinology 2021, 162, bqaa215. [Google Scholar] [CrossRef]
- Santi, M.; Graf, S.; Zeino, M.; Cools, M.; Van De Vijver, K.; Trippel, M.; Aliu, N.; E Flück, C. Approach to the virilizing girl at puberty. J. Clin. Endocrinol. Metab. 2021, 106, 1530–1539. [Google Scholar] [CrossRef]
- Steeg, L.G.V.; Klein, S.L. Sex and sex steroids impact influenza pathogenesis across the life course. Semin. Immunopathol. 2019, 41, 189–194. [Google Scholar] [CrossRef]
- Yang, Q.; Kennicott, K.; Zhu, R.; Kim, J.; Wakefield, H.; Studener, K.; Lialng, Y. Sex hormone influence on female-biased autoimmune diseases hints at puberty as an important factor in pathogenesis. Front. Pediatr. 2023, 11, 1051624. [Google Scholar] [CrossRef] [PubMed]
- Keestra, S.M.; Male, V.; Salali, G.D. Out of balance: The role of evolutionary mismatches in the sex disparity in autoimmune disease. Med. Hypotheses 2021, 151, 110558. [Google Scholar] [CrossRef] [PubMed]
- Merz, W.M.; Fischer-Betz, R.; Hellwig, K.; Lamprecht, G.; Gembruch, U. Pregnancy and autoimmune disease. Dtsch. Arztebl. Int. 2022, 119, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lipoldova, M.; Demant, P. Gene-specific sex effects on susceptibility to infectious diseases. Front. Immunol. 2021, 12, 712688. [Google Scholar] [CrossRef]
- Singh, R.P.; Hahn, B.H.; Bischoff, D.S. Interferon genes are influenced by 17beta-estradiol in sle. Front. Immunol. 2021, 12, 725325. [Google Scholar] [CrossRef]
- Jaiswal, A.; Roy, R.; Tamrakar, A.; Singh, A.K.; Kar, P.; Kodgire, P. Activation-induced cytidine deaminase an antibody diversification enzyme interacts with chromatin modifier ubn1 in b-cells. Sci. Rep. 2023, 13, 19615. [Google Scholar] [CrossRef]
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R.; Novakovic, B. Sexual dimorphism in innate immunity: The role of sex hormones and epigenetics. Front. Immunol. 2020, 11, 604000. [Google Scholar] [CrossRef]
- Moulton, V.R. Sex hormones in acquired immunity and autoimmune disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef]
- Singh, R.P.; Bischoff, D.S. Sex hormones and gender influence the expression of markers of regulatory t cells in sle patients. Front. Immunol. 2021, 12, 619268. [Google Scholar] [CrossRef] [PubMed]
- Fuseini, H.; Cephus, J.-Y.; Wu, P.; Davis, J.B.; Contreras, D.C.; Gandhi, V.D.; Rathmell, J.C.; Newcomb, D.C. Eralpha signaling increased il-17a production in th17 cells by upregulating il-23r expression, mitochondrial respiration, and proliferation. Front. Immunol. 2019, 10, 2740. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.; Shen, T.; Xu, Y.; Qiu, Z.; Chupp, D.P.; Im, J.; Xu, Z.; Zan, H. Estrogen reverses hdac inhibitor-mediated repression of aicda and class-switching in antibody and autoantibody responses by downregulation of mir-26a. Front. Immunol. 2020, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Henze, L.; Schwinge, D.; Schramm, C. The effects of androgens on t cells: Clues to female predominance in autoimmune liver diseases? Front. Immunol. 2020, 11, 1567. [Google Scholar] [CrossRef]
- Harding, A.T.; Heaton, N.S. The impact of estrogens and their receptors on immunity and inflammation during infection. Cancers 2022, 14, 909. [Google Scholar] [CrossRef]
- Ciarambino, T.; Para, O.; Giordano, M. Immune system and COVID-19 by sex differences and age. Women’s Health 2021, 17, 17455065211022262. [Google Scholar] [CrossRef]
- Shepherd, R.; Bretherton, I.; Pang, K.; Mansell, T.; Czajko, A.; Kim, B.; Vlahos, A.; Zajac, J.D.; Saffery, R.; Cheung, A.; et al. Gender-affirming hormone therapy induces specific DNA methylation changes in blood. Clin. Epigenetics 2022, 14, 24. [Google Scholar] [CrossRef]
- Gulati, G.; Brunner, H.I. Environmental triggers in systemic lupus erythematosus. Semin. Arthritis Rheum. 2018, 47, 710–717. [Google Scholar] [CrossRef]
- Brooks, W.H.; Arleevskaya, M.I.; Renaudineau, Y. Editorial: Epigenetic aspects of autoimmune diseases. Front. Cell Dev. Biol. 2022, 10, 991693. [Google Scholar] [CrossRef]
- Golden, L.C.; Itoh, Y.; Itoh, N.; Iyengar, S.; Coit, P.; Salama, Y.; Arnold, A.P.; Sawalha, A.H.; Voskuhl, R.R. Parent-of-origin differences in DNA methylation of x chromosome genes in t lymphocytes. Proc. Natl. Acad. Sci. USA 2019, 116, 26779–26787. [Google Scholar] [CrossRef]
- Márquez, E.J.; Chung, C.-H.; Marches, R.; Rossi, R.J.; Nehar-Belaid, D.; Eroglu, A.; Mellert, D.J.; Kuchel, G.A.; Banchereau, J.; Ucar, D. Sexual-dimorphism in human immune system aging. Nat. Commun. 2020, 11, 751. [Google Scholar] [CrossRef]
- Charras, A.; Garau, J.; Hofmann, S.R.; Carlsson, E.; Cereda, C.; Russ, S.; Abraham, S.; Hedrich, C.M. DNA methylation patterns in cd8(+) t cells discern psoriasis from psoriatic arthritis and correlate with cutaneous disease activity. Front. Cell Dev. Biol. 2021, 9, 746145. [Google Scholar] [CrossRef] [PubMed]
- Vecellio, M.; Paraboschi, E.M.; Ceribelli, A.; Isailovic, N.; Motta, F.; Cardamone, G.; Robusto, M.; Asselta, R.; Brescianini, S.; Sacrini, F.; et al. DNA methylation signature in monozygotic twins discordant for psoriatic disease. Front. Cell Dev. Biol. 2021, 9, 778677. [Google Scholar] [CrossRef] [PubMed]
- Kabeerdoss, J.; Danda, D.; Goel, R.; Mohan, H.; Danda, S.; Scofield, R.H. Genome-wide DNA methylation profiling in cd8 t-cells and gamma delta t-cells of asian indian patients with takayasu arteritis. Front. Cell Dev. Biol. 2022, 10, 843413. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Song, Z.; Wang, B.; Jia, X.; Song, R.; Zhang, J. Identification of lncrna and mrna expression profile in relapsed graves’ disease. Front. Cell Dev. Biol. 2021, 9, 756560. [Google Scholar] [CrossRef]
- Bost, C.; Arleevskaya, M.I.; Brooks, W.H.; Plaza, S.; Guery, J.-C.; Renaudineau, Y. Long non-coding rna xist contribution in systemic lupus erythematosus and rheumatoid arthritis. Clin. Immunol. 2022, 236, 108937. [Google Scholar] [CrossRef]
- Lv, X.; Liu, X.; Zhao, M.; Wu, H.; Zhang, W.; Lu, Q.; Chen, X. Rna methylation in systemic lupus erythematosus. Front. Cell Dev. Biol. 2021, 9, 696559. [Google Scholar] [CrossRef]
- Chao, Y.; Li, H.-B.; Zhou, J. Multiple functions of rna methylation in t cells: A review. Front. Immunol. 2021, 12, 627455. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Gong, S.; Avellino, G.; Barbagallo, R.; Gaccioli, F.; Jenkins, B.J.; Koulman, A.; Murray, A.J.; Charnock-Jones, D.S.; Smith, G.C.S. Placental sex-dependent spermine synthesis regulates trophoblast gene expression through acetyl-coa metabolism and histone acetylation. Commun. Biol. 2022, 5, 586. [Google Scholar] [CrossRef]
- Jiang, Y.; Peng, Z.; Man, Q.; Wang, S.; Huang, X.; Meng, L.; Wang, H.; Zhu, G. H3k27ac chromatin acetylation and gene expression analysis reveal sex- and situs-related differences in developing chicken gonads. Biol. Sex Differ. 2022, 13, 6. [Google Scholar] [CrossRef]
- Bacalao, M.A.; Satterthwaite, A.B. Recent advances in lupus b cell biology: Pi3k, ifngamma, and chromatin. Front. Immunol. 2020, 11, 615673. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Yang, Y.; Li, T.; Xu, Y.; Zhang, W.; Li, M.; Xiao, Y.; Hu, J.; Liu, K.; et al. The tgf-beta/mir-31/ceacam1-s axis inhibits cd4(+) cd25(+) treg differentiation in systemic lupus erythematosus. Immunol. Cell Biol. 2021, 99, 697–710. [Google Scholar] [CrossRef]
- Pashangzadeh, S.; Motallebnezhad, M.; Vafashoar, F.; Khalvandi, A.; Mojtabavi, N. Implications the role of mir-155 in the pathogenesis of autoimmune diseases. Front. Immunol. 2021, 12, 669382. [Google Scholar] [CrossRef]
- Li, S.; Wu, Q.; Jiang, Z.; Wu, Y.; Li, Y.; Ni, B.; Xiao, J.; Zhai, Z. Mir-31-5p regulates type i interferon by targeting slc15a4 in plasmacytoid dendritic cells of systemic lupus erythematosus. J. Inflamm. Res. 2022, 15, 6607–6616. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Woo, J.M.; Parks, C.G.; Costenbader, K.H.; Jacobsen, S.; Bernatsky, S. Systemic lupus erythematosus risk: The role of environmental factors. Rheum. Dis. Clin. N. Am. 2022, 48, 827–843. [Google Scholar] [CrossRef] [PubMed]
- Illescas-Montes, R.; Corona-Castro, C.C.; Melguizo-Rodríguez, L.; Ruiz, C.; Costela-Ruiz, V.J. Infectious processes and systemic lupus erythematosus. Immunology 2019, 158, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Chen, Y.; Wu, W.; Guo, L.; Xu, W.; Chen, J.; Sun, S.; Li, J.; Chen, Z.; Gu, L.; et al. Varicella zoster virus infections increase the risk of disease flares in patients with sle: A matched cohort study. Lupus Sci. Med. 2019, 6, e000339. [Google Scholar] [CrossRef]
- Tsai, P.H.; Jang, S.S.; Liou, L.B. Septicaemia is associated with increased disease activity and mortality in systemic lupus erythematosus: A retrospective analysis from taiwan. Lupus 2020, 29, 191–198. [Google Scholar] [CrossRef]
- Bin Joo, Y.; Kim, K.-J.; Park, K.-S.; Park, Y.-J. Influenza infection as a trigger for systemic lupus erythematosus flares resulting in hospitalization. Sci. Rep. 2021, 11, 4630. [Google Scholar] [CrossRef]
- Fu, X.-L.; Qian, Y.; Jin, X.-H.; Yu, H.-R.; Du, L.; Wu, H.; Chen, H.-L.; Shi, Y.-Q. COVID-19 in patients with systemic lupus erythematosus: A systematic review. Lupus 2022, 31, 684–696. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of epstein-barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The role of viral infections in the onset of autoimmune diseases. Viruses 2023, 15, 782. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, S.; Nannini, G.; Cianchi, F.; Coratti, F.; Amedei, A. The impact of microbiota-immunity-hormone interactions on autoimmune diseases and infection. Biomedicines 2024, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Ronnblom, L.; Leonard, D. Interferon pathway in sle: One key to unlocking the mystery of the disease. Lupus Sci. Med. 2019, 6, e000270. [Google Scholar] [CrossRef]
- Huang, J.; Hong, W.; Wan, M.; Zheng, L. Molecular mechanisms and therapeutic target of netosis in diseases. MedComm. 2022, 3, e162. [Google Scholar] [CrossRef]
- el Hadiyen, F.; Tsang-A-Sjoe, M.W.P.; I Lissenberg-Witte, B.; E Voskuyl, A.; Bultink, I.E.M. Intercurrent infection as a risk factor for disease flares in patients with systemic lupus erythematosus. Lupus Sci. Med. 2024, 11, e001131. [Google Scholar] [CrossRef]
- Xuan, J.; Ji, Z.; Wang, B.; Zeng, X.; Chen, R.; He, Y.; Rao, P.; Wu, P.; Shi, G. Serological evidence for the association between epstein-barr virus infection and sjogren’s syndrome. Front. Immunol. 2020, 11, 590444. [Google Scholar] [CrossRef]
- Liu, Z.; Chu, A. Sjogren’s syndrome and viral infections. Rheumatol. Ther. 2021, 8, 1051–1059. [Google Scholar] [CrossRef]
- Lemus, Y.B.; Martínez, G.A.; Lugo, L.P.; Escorcia, L.G.; Peñata, E.Z.; Llanos, N.S.; Bonfanti, A.C.; Acosta-Hoyos, A.J.; Quiroz, E.N. Gene profiling of epstein-barr virus and human endogenous retrovirus in peripheral blood mononuclear cells of sle patients: Immune response implications. Sci. Rep. 2024, 14, 20236. [Google Scholar] [CrossRef]
- Agostini, S.; Mancuso, R.; Guerini, F.R.; D’aLfonso, S.; Agliardi, C.; Hernis, A.; Zanzottera, M.; Barizzone, N.; Leone, M.A.; Caputo, D.; et al. Hla alleles modulate ebv viral load in multiple sclerosis. J. Transl. Med. 2018, 16, 80. [Google Scholar] [CrossRef]
- Barber, M.R.; Clarke, A.E. Systemic lupus erythematosus and risk of infection. Expert. Rev. Clin. Immunol. 2020, 16, 527–538. [Google Scholar] [CrossRef]
- Silva, J.d.M.; Alves, C.E.d.C.; Pontes, G.S. Epstein-barr virus: The mastermind of immune chaos. Front. Immunol. 2024, 15, 1297994. [Google Scholar] [CrossRef]
- Kim, Y.S.; Unno, T.; Kim, B.-Y.; Park, M.-S. Sex differences in gut microbiota. World J. Men’s Health 2020, 38, 48–60. [Google Scholar] [CrossRef]
- Yao, K.; Xie, Y.; Wang, J.; Lin, Y.; Chen, X.; Zhou, T. Gut microbiota: A newly identified environmental factor in systemic lupus erythematosus. Front. Immunol. 2023, 14, 1202850. [Google Scholar] [CrossRef] [PubMed]
- Rio, P.; Caldarelli, M.; Chiantore, M.; Ocarino, F.; Candelli, M.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Immune cells, gut microbiota, and vaccines: A gender perspective. Cells 2024, 13, 526. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Gaudreau, M.-C.; Gudi, R.; Brown, R.; Gilkeson, G.; Vasu, C. Gut microbiota differently contributes to intestinal immune phenotype and systemic autoimmune progression in female and male lupus-prone mice. J. Autoimmun. 2020, 108, 102420. [Google Scholar] [CrossRef] [PubMed]
- Santos-Marcos, J.A.; Mora-Ortiz, M.; Tena-Sempere, M.; Lopez-Miranda, J.; Camargo, A. Interaction between gut microbiota and sex hormones and their relation to sexual dimorphism in metabolic diseases. Biol. Sex Differ. 2023, 14, 4. [Google Scholar] [CrossRef]
- Silverman, G.J.; Deng, J.; Azzouz, D.F. Sex-dependent lupus blautia (ruminococcus) gnavus strain induction of zonulin-mediated intestinal permeability and autoimmunity. Front. Immunol. 2022, 13, 897971. [Google Scholar] [CrossRef]
- Park, H.-J.; Choi, J.-M. Sex-specific regulation of immune responses by ppars. Exp. Mol. Med. 2017, 49, e364. [Google Scholar] [CrossRef]
- Greiling, T.M.; Dehner, C.; Chen, X.; Hughes, K.; Iñiguez, A.J.; Boccitto, M.; Ruiz, D.Z.; Renfroe, S.C.; Vieira, S.M.; Ruff, W.E.; et al. Commensal orthologs of the human autoantigen ro60 as triggers of autoimmunity in lupus. Sci. Transl. Med. 2018, 10, 2306. [Google Scholar] [CrossRef]
- Chen, B.; Jia, X.; Xu, J.; Zhao, L.; Ji, J.; Wu, B.; Ma, Y.; Li, H.; Zuo, X.; Pan, W.; et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol. 2021, 73, 232–243. [Google Scholar] [CrossRef]
- Moon, J.; Choi, S.H.; Yoon, C.H.; Kim, M.K.; Appel, S. Gut dysbiosis is prevailing in sjogren’s syndrome and is related to dry eye severity. PLoS ONE 2020, 15, e0229029. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Trujillo-Vargas, C.M.; Midani, F.S.; Pflugfelder, S.C.; Britton, R.A.; de Paiva, C.S. Gut microbiota from sjogren syndrome patients causes decreased t regulatory cells in the lymphoid organs and desiccation-induced corneal barrier disruption in mice. Front. Med. 2022, 9, 852918. [Google Scholar] [CrossRef] [PubMed]
- Refai, R.H.; Hussein, M.F.; Abdou, M.H.; Abou-Raya, A.N. Environmental risk factors of systemic lupus erythematosus: A case-control study. Sci. Rep. 2023, 13, 10219. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.; Costenbader, K.; Long, S.; Hofmann, J.; Beane, F.L.; Sandler, D. Pesticide use and risk of systemic autoimmune diseases in the agricultural health study. Environ. Res. 2022, 209, 112862. [Google Scholar] [CrossRef]
- Jochmanová, I.; Lazúrová, Z.; Rudnay, M.; Bačová, I.; Mareková, M.; Lazúrová, I. Environmental estrogen bisphenol a and autoimmunity. Lupus 2015, 24, 392–399. [Google Scholar] [CrossRef]
- Pollard, K.M. Silica, silicosis, and autoimmunity. Front. Immunol. 2016, 7, 97. [Google Scholar] [CrossRef]
- Lueschow, S.R.; McElroy, S.J. The paneth cell: The curator and defender of the immature small intestine. Front. Immunol. 2020, 11, 587. [Google Scholar] [CrossRef]
- Fee, L.; Kumar, A.; Tighe, R.M.; Foster, M.H. Autoreactive b cells recruited to lungs by silica exposure contribute to local autoantibody production in autoimmune-prone bxsb and b cell receptor transgenic mice. Front. Immunol. 2022, 13, 933360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qu, W.; Sun, L.; Chen, J.; Kong, W.; Wang, F.; Pan, W.; Liu, L.; Wu, M.; Ding, F.; et al. The relationship of polluted air and drinking water sources with the prevalence of systemic lupus erythematosus: A provincial population-based study. Sci. Rep. 2021, 11, 18591. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Pontalti, M.; Cattani, G.; Rossini, M.; Viapiana, O.; Orsolini, G.; Benini, C.; Bertoldo, E.; Fracassi, E.; Gatti, D.; et al. Association between long-term exposure to air pollution and immune-mediated diseases: A population-based cohort study. RMD Open 2022, 8, e002055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Hu, S.; Wu, Y. Causal relationships between air pollution and common autoimmune diseases: A two-sample mendelian randomization study. Sci. Rep. 2025, 15, 135. [Google Scholar] [CrossRef]
- Rasking, L.; Roelens, C.; Sprangers, B.; Thienpont, B.; Nawrot, T.S.; De Vusser, K. Lupus, DNA methylation, and air pollution: A malicious triad. Int. J. Environ. Res. Public Health 2022, 19, 15050. [Google Scholar] [CrossRef]
- Chen, J.; Liao, S.; Pang, W.; Guo, F.; Yang, L.; Liu, H.-F.; Pan, Q. Life factors acting on systemic lupus erythematosus. Front. Immunol. 2022, 13, 986239. [Google Scholar] [CrossRef]
- Touil, H.; Mounts, K.; De Jager, P.L. Differential impact of environmental factors on systemic and localized autoimmunity. Front. Immunol. 2023, 14, 1147447. [Google Scholar] [CrossRef]
- Jin, L.; Dai, M.; Li, C.; Wang, J.; Wu, B. Risk factors for primary sjogren’s syndrome: A systematic review and meta-analysis. Clin. Rheumatol. 2023, 42, 327–338. [Google Scholar] [CrossRef]
- Cozier, Y.C.; Barbhaiya, M.; Castro-Webb, N.; Conte, C.; Tedeschi, S.K.; Leatherwood, C.; Costenbader, K.H.; Rosenberg, L. Relationship of cigarette smoking and alcohol consumption to incidence of systemic lupus erythematosus in a prospective cohort study of black women. Arthritis Care Res. 2019, 71, 671–677. [Google Scholar] [CrossRef]
- Hahn, J.; Leatherwood, C.; Malspeis, S.; Liu, X.; Lu, B.; Roberts, A.L.; Sparks, J.A.; Karlson, E.W.; Feldman, C.H.; Munroe, M.E.; et al. Associations between smoking and systemic lupus erythematosus-related cytokines and chemokines among us female nurses. Arthritis Care Res. 2021, 73, 1583–1589. [Google Scholar] [CrossRef]
- Speyer, C.B.; Costenbader, K.H. Cigarette smoking and the pathogenesis of systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2018, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.J.; Estadt, S.N.; Theros, J.; Moore, T.; Ellis, J.; Liu, J.; Reed, T.J.; Jacob, C.O.; Gudjonsson, J.E.; Kahlenberg, J.M. Ultraviolet light induces increased t cell activation in lupus-prone mice via type i ifn-dependent inhibition of t regulatory cells. J. Autoimmun. 2019, 103, 102291. [Google Scholar] [CrossRef] [PubMed]
- Coss, S.L.; Zhou, D.; Chua, G.T.; Aziz, R.A.; Hoffman, R.P.; Wu, Y.L.; Ardoin, S.P.; Atkinson, J.P.; Yu, C.-Y. The complement system and human autoimmune diseases. J. Autoimmun. 2023, 137, 102979. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Ultraviolet b radiation: The vitamin d connection. Adv. Exp. Med. Biol. 2017, 996, 137–154. [Google Scholar] [CrossRef]
- Awuah, W.A.M.; Huang, H.M.; Kalmanovich, J.B.; Mehta, A.; Mikhailova, T.; Ng, J.C.; Abdul-Rahman, T.M.; Adebusoye, F.T.M.; Tan, J.K.M.; Kamanousa, K.B.; et al. Circadian rhythm in systemic autoimmune conditions: Potential of chrono-immunology in clinical practice: A narrative review. Medicine 2023, 102, e34614. [Google Scholar] [CrossRef]
- A Young, K.; Munroe, M.E.; Harley, J.B.; Guthridge, J.M.; Kamen, D.L.; Gilkensen, G.S.; Weisman, M.H.; Karp, D.R.; Wallace, D.J.; A James, J.; et al. Less than 7 hours of sleep per night is associated with transitioning to systemic lupus erythematosus. Lupus 2018, 27, 1524–1531. [Google Scholar] [CrossRef]
- Choi, M.Y.; Malspeis, S.; Sparks, J.A.; Cui, J.; Yoshida, K.; Costenbader, K.H. Association of sleep deprivation and the risk of developing systemic lupus erythematosus among women. Arthritis Care Res. 2023, 75, 1206–1212. [Google Scholar] [CrossRef]
- Anaya, J.-M.; Restrepo-Jiménez, P.; Ramírez-Santana, C. The autoimmune ecology: An update. Curr. Opin. Rheumatol. 2018, 30, 350–360. [Google Scholar] [CrossRef]
- Hsu, T.-W.; Bai, Y.-M.; Tsai, S.-J.; Chen, T.-J.; Chen, M.-H.; Liang, C.-S. Risk of autoimmune diseases after post-traumatic stress disorder: A nationwide cohort study. Eur. Arch. Psychiatry Clin. Neurosci. 2024, 274, 487–495. [Google Scholar] [CrossRef]
- Mandagere, K.; Stoy, S.; Hammerle, N.; Zapata, I.; Brooks, B. Systematic review and meta-analysis of post-traumatic stress disorder as a risk factor for multiple autoimmune diseases. Front. Psychiatry 2025, 16, 1523994. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Product | Immunological Function |
|---|---|---|
| BTK | Burton’s tyrosine kinase | A protein tyrosine kinase that mediates pre-BCR signaling for Ig heavy chain rearrangement, crucial for B cell development and IgE-dependent mast cell activation |
| CD40L | CD40 ligand | A costimulatory molecule on T cells that binds CD40 on APCs, priming pathogenic Th cells, driving immune responses, enabling B-T cell communication and B cell class switching |
| CXCR3 | C-X-C motif chemokine receptor 3 | A chemokine receptor involved in immune cell trafficking, recruiting killer T cells to sites of inflammation |
| CXorf21 | Chromosome X open reading frame 21 | A TLR adaptor that interacts with SLC15A4 on the lysosomal membrane |
| CYBB | Cytochrome b-245 beta chain | A component of the NADPH oxidase complex that generates ROS for microbial killing |
| FoxP3 | Forkhead box P3 | A key regulator of Treg cell development that functions to suppress immune responses |
| IL13RA1/2 | Interleukin 13 receptor subunit alpha 1/2 | A component of the IL-13 receptor complex that mediates immune regulatory functions |
| IL2RG | Interleukin 2 receptor subunit gamma | A component of the IL-2 receptor essential for T cell development and function |
| IL9R | Interleukin 9 receptor | A component of the IL-9 signaling pathway that regulates diverse immune responses |
| IRAK1 | Interleukin 1 receptor associated kinase 1 | A protein kinase that mediates IL-1R and TLR signaling to activate NF-κB and MAPK pathways, promoting innate immune responses and inflammation |
| KDM6a (UTX) | Lysine demethylase 6a | An enzyme that demethylates H3K27me3 to regulate gene expression, skewing immunity toward inflammation and enhancing NK cell effector function. |
| OGT | O-linked N-acetylglucosamine transferase | An enzyme involved in protein glycosylation that regulates mechanistic target of rapamycin (mTOR) activity and influences diverse cellular processes |
| SLC15A4 | Solute carrier family 15 member 4 | A proton-coupled amino acid transporter essential for endolysosomal TLR activation and TLR-mediated IFN-I production in innate immune responses |
| TLR7 | Toll like receptor 7 | A receptor protein that enhances viral RNA sensing and IFN-α production, contributing to female-biased antiviral defense, and promotes ABC accumulation, immune activation, and inflammation |
| TLR8 | Toll like receptor 8 | A receptor protein involved in TLR signaling via MyD88, sufficient to drive B cell tolerance loss, class-switched autoantibody production, enhanced granulopoiesis, and increased IFN-I production |
| miR | Target Genes | Changes * | Functional Consequences in Autoimmunity |
|---|---|---|---|
| miR-20b | RELA (NF-κB subunit), STAT3 | ↓ | Its downregulation in SLE T cells lifts repression on RELA and STAT3, amplifying NF-κB signaling and Th17 differentiation, thereby promoting inflammation in lupus pathogenesis. |
| miR-23b | TAB2/3 and IKKα | ↓ | Its downregulation in inflamed tissues of SLE, RA, and MS upregulates NF-κB signaling, promoting proinflammatory cytokine production and autoimmunity, while its ectopic expression suppresses inflammation and reduces disease severity in autoimmune models. |
| miR-92a | KLF2, BCL2L11 (Bim) | ↓ | Its dysregulation in salivary glands and PBMCs of SS patients may disrupt glandular epithelial cell survival and immune cell homeostasis by altering apoptosis and T cell differentiation. |
| miR-98 | IL-6, FAS (CD95), TNF-α | ↓ | Its downregulation in SLE PBMCs lifts suppression of IL-6, increasing proinflammatory cytokines and STAT3 signaling, while FAS upregulation promotes CD4+ T cell apoptosis, worsening immune dysregulation and disease activity. |
| miR-106a | IL-10, SOCS5 | ↑ | Its upregulation in CD4+ T cells and PBMCs from SLE and pSS patients suppresses IL-10, a regulatory cytokine, and SOCS5, enhancing JAK-STAT signaling and promoting T cell activation and inflammation. |
| miR-125b | PRDM1 (Blimp-1) | ↓ | Its downregulation in activated CD4+ T cells of SS patients lifts repression on PRDM1 (Blimp-1: B-lymphocyte-induced maturation protein 1), enhancing plasma cell differentiation and autoantibody production, while its exosomal delivery from salivary gland-derived mesenchymal stem cells suppresses plasma cell formation and restores secretory function. |
| miR-188 | NFATc2, FOXO1, CBL | ↑ | Its upregulation in PBMCs of SLE, RA, and PA patients suppresses FOXO1, impairing Treg differentiation and immune tolerance, while reducing CBL expression and dampening TCR signaling in lupus CD4+ Th cells, collectively enhancing effector T cell activity. |
| miR-221 /222 cluster | CDKN1B (p27kip1), ETS1 | ↑ | Its upregulation in SLE PBMCs downregulates CDKN1B and ETS1, driving lymphocyte proliferation and plasma cell differentiation, which enhances autoreactive B cell activity and autoantibody production. |
| miR-222 | CFHR5 | ↓ | Its downregulation in LN patients increases CFHR5 expression, overactivating the alternative complement pathway and promoting immune complex–mediated tissue injury. |
| miR-223 | S1PR1, CXCL2, CCL3 (in SLE) ITPR3 (in SS) | ↑/↓ | Its upregulation in CD4+ T cells from SLE patients and in epithelial cells of SS patients suppresses S1PR1 and chemokines, limiting T cell egress and inflammatory cell recruitment, while downregulating ITPR3 to impair Ca2+ signaling and activate NF-κB, promoting epithelial inflammation. It is also linked to X chromosome demethylation in female lupus predisposition, whereas its deficiency in LN leads to T cell accumulation and exacerbated renal inflammation. |
| miR-224 | SMAD4, HOXD10, API5 | ↑ | Its upregulation in PBMCs and T cells of SLE, SD/SSc, and RA targets SMAD4, disrupting TGF-β signaling and promoting fibrosis and tissue dysfunction. It also enhances cell proliferation and migration, downregulates apoptosis inhibitor 5 (API5) to facilitate activation-induced cell death in Jurkat and SLE T cells, and upregulates STAT-1, contributing to LN. |
| miR-361-5P | VEGFA, IL-6R | ↑ | Its overexpression in labial salivary glands of SS patients reduces VEGFA and IL-6R expression, potentially compromising vascular integrity and altering cytokine responses in glandular tissues. |
| miR-374a | SOCS1, PTEN, IL-10 | ↑ | Its upregulation in inflamed synovium of SLE and RA downregulates SOCS1 and PTEN, activating JAK-STAT and PI3K pathways, thereby promoting cytokine-driven inflammation and lymphocyte survival, and increased susceptibility to SLE with renal involvement. |
| miR-421 | ATM, E2F1, PDCD4 | ↑ | Its upregulation in LN kidney biopsies and RA synovial tissue impairs the DNA damage response by inhibiting ATM and E2F1 in LN renal tissues, suppresses apoptosis-related genes like PDCD4, promoting fibroblast-like synoviocyte survival and proliferation in RA. |
| miR-424 | CCND1, CDK6 | ↑ | Its upregulation induces cell cycle arrest in SLE PBMCs and SS salivary gland epithelial cells by downregulating CCND1 and CDK6, resulting in tissue atrophy and impaired glandular regeneration. |
| miR-452 | BMI1, RAB11A, CDKN1B | ↓ | Its downregulation derepresses genes that promote T cell proliferation and survival, thereby enhancing autoreactive T cell responses in MS and RA patients. |
| miR-506 | NFATC1 | ↓ | Its underexpression lifts repression on NFATC1, leading to increased CD4+ T cell activation and proliferation in SS patients. |
| miR-548m | PTEN | ↑ | Its overexpression reduces PTEN expression, resulting in hyperactivation of the PI3K–AKT pathway and promoting immune cell survival and activation in SLE patients. |
| hsa-miR-503 | BCL2, CCND1, FGF2 | ↓ | Its downregulation enhances BCL2-mediated survival and CCND1-driven proliferation, leading to synovial hyperplasia and joint inflammation in RA, while its expression is elevated in demethylated CD4+ T cells from women with lupus following 5-azacytidine treatment. |
| hsa-miR-545 | RIG-I, TP53INP1, ZEB2 | ↑ | Its upregulation, observed in some LN datasets, inhibits RIG-I-mediated antiviral responses and regulates p53-dependent apoptosis, potentially shifting immune balance away from effective antiviral surveillance. |
| hsa-let-7f-2 | STAT3, IL-13, TGFBR1 | ↑ | Its upregulation in the plasma and salivary glands of SLE and SS patients modulates Th2 and Th17 differentiation by suppressing STAT3 and IL-13, potentially disrupting effector T cell balance and promoting proinflammatory cytokine production. |
| Gene Products | Impacts by Sex Hormones |
|---|---|
| TLRs (Toll-like receptors) | Estrogens and ERα signaling differentially regulate TLR family members, enhancing TLR7/9-mediated IRF5 activation and IFN production in female pDCs, while modulating TLR8 expression independently of IFNs through direct ERα binding to an ERE near the TLR8 locus or indirectly via STAT1-mediated transcriptional activation. |
| IRF5 (Interferon regulatory factor 5) | Estrogens and ERα signaling upregulate IRF5, a key transcription factor involved in immune responses and a lupus susceptibility factor, leading to IFN-I overproduction and contributing to autoimmune disease progression in SLE. |
| IFNs (Interferons) | Estrogens enhance IFN-α and IFN-γ production via ERα and TLR7/9-mediated IRF5 activation in pDCs, amplifying cytokine output and innate immunity, thereby contributing to the female bias in autoimmunity. IFN-α upregulates MHC-I, while IFN-γ induces MHC-II and alters proteasome composition, facilitating self-peptide presentation to T cells—processes central to SLE pathogenesis. |
| ILs (Interleukins) | Estrogens and ERα signaling promote IL-6 expression and inflammation in both mice and humans, while elevated estradiol in SLE patients enhances the secretion of IL-8, IL-18, and IL-23. Estrogen-regulated cytokines such as IL-4, IL-5, and IL-10 support B cell activation and antibody production. Notably, increased IL-6 and IL-10 levels correlate with higher SLE disease activity index (SLEDAI) scores, linking estrogen-driven cytokine expression to disease activity in SLE. |
| BAFF (B cell activating factor) | Estrogens enhance BAFF production, which supports B cell survival and maturation, leading to elevated antibody levels and potentially influencing thyroid dysfunction in GD. |
| UNC93B1 (Unc-93 homolog B1) | Estrogens enhance UNC93B1 expression via IFN-α or IFN-γ signaling, with notably higher levels observed in lupus-prone female mice and PBMCs from SLE patients compared to healthy controls. |
| S1PR2 (Sphingosine-1-phosphate receptor 2) | Estrogens regulate S1PR2 expression, a G protein-coupled receptor, potentially contributing to the female-biased severity of CNS-related autoimmune diseases like MS. |
| AIRE (Autoimmune regulator) | Estrogens increase methylation of CpG sites in the AIRE promoter, inducing epigenetic silencing of AIRE—a central tolerance regulator controlling tissue-specific antigen expression—thereby enhancing autoimmune susceptibility. In contrast, androgens upregulate AIRE, contributing to sex bias in CNS autoimmune diseases. |
| AID (Activation-induced cytidine deaminase) | Estrogens promote AID transcription, enhancing somatic hypermutation and class switch recombination in activated B cells—key processes for antibody diversification—likely through AID’s interaction with the chromatin modifier UBN1, a component of the HIRA chaperone complex. |
| SLC15A4 (Solute carrier family 15 member 4) | Estrogens upregulate SLC15A4 expression, enhancing IFN-I and proinflammatory cytokine production in pDCs, thereby contributing to autoimmune disease progression in SLE and colitis models. |
| Cathepsin S | Estrogens promote inflammation by activating cathepsin S—a lysosomal acidic protease involved in immune regulation—elevated in the lacrimal glands and tears of female SS murine models., whereas testosterone reduces inflammation and enhances glandular function in SS, potentially through cathepsin S suppression. |
| miR | Location | Change * | Target Gene | Impacts by Sex Hormones |
|---|---|---|---|---|
| miR-10b | Chr2 | ↑ | SRSF1, MAPK7 (TAK1) | Estrogens upregulate miR-10b-5p, which inhibits SRSF1 and MAPK7, enhancing NF-κB signaling, Th17 differentiation, and proinflammatory cytokine expression in T cells. |
| miR-26a | Chr3/ Chr12 | ↓ | AICDA, HMGA2, COX-2, TLR4, MALT1, HMGA1 | Estrogens suppress miR-26a, enhancing class-switch recombination and autoantibody production, while androgens induce miR-26a to restrain AICDA expression and B cell activation. miR-26a downregulation also enhances proinflammatory cytokines via other target genes. |
| miR-31 | Chr9 | ↓ | RhoA, CEACAM1, IRF5, STAT-1, SLC15A4 | Estrogens downregulate miR-31 via TGF-β and NF-κB in SLE T cells, impairing IL-2 production by disrupting NFAT, NF-κB, and AP-1 activity, while increasing CREM and dysregulating CaMK-IV and PP2A. This leads to defective IL-2 signaling, impaired Treg differentiation, and T cell dysfunction. |
| miR-96 | Chr7 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | Estrogen-induced miR-96 upregulates immune genes such as FoxP3, RHOA, FCGR1, IL-2, CD138, and CEACAM1, influencing SLE susceptibility, onset, clinical heterogeneity, and progression. |
| miR-127 | Chr14 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | Estrogen-induced miR-127 promote immune and inflammatory responses, contributing to SLE susceptibility, onset, clinical heterogeneity, and progression. |
| miR-145 | Chr5 | ↓ | STAT1, OPG | Estrogen-mediated miR-145-5p downregulation elevates osteoprotegerin, reducing osteoclast activity and bone resorption, and contributing to joint damage in RA. |
| miR-145a | Chr5 | ↓ | ADAM17, KLF4, SIRT1 | miR-145a targets inflammation- and stress-related genes to suppress immune activation, but estrogen downregulates miR-145a in B cells, promoting immune activation. |
| miR-146a | Chr5 | ↑↓ | IRAK1, TRAF6, IRF5, STAT-1, SLC15A4 | miR-146a suppresses IRAK1 and TRAF6 translation, serving as a negative regulator of immune activation. Estrogens dysregulate miR-146a in PBMCs and splenocytes of MRL/lpr mice, linking it to epigenetic changes, B cell hyperactivity, and autoantibody production in autoimmunity. |
| miR-148a | Chr7 | ↑ | DNA methytransferase 1 (DNMT1) | Upregulated miR-148a promotes DNA hypomethylation, contributing to autoimmune disease pathogenesis. |
| miR-148b | Chr12 | ↑ | CaMKIIα, Gadd45α, PTEN, Bim | miR-148b suppresses TLR-induced cytokine and IFN-I production, impairing DC-medicated innate responses. It also promotes DNA hypomethylation and survival of autoreactive B cells, contributing systemic autoimmunity. |
| miR-155 | Chr21 | ↑ | MAPK, INS, Wnt, NF-κB, BIC, Pu.1, c-Maf, c-Fos, IFNγRα, c-Rel, c-Fos, Peli1, p27kip1, KPC1, SOCS1 | miR-155 regulates immune cell homeostasis, Th1 differentiation, tolerance, and development. It supports B cell maturation, isotype switching, germinal center formation, high-affinity IgG1 production, DC activation, apoptosis, and IL-12 production, promoting autoimmune susceptibility. |
| miR-183 | Chr7 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | miR-183 modulates immune and inflammatory responses, contributing to susceptibility to SLE and influencing its onset, clinical heterogeneity, and progression. |
| miR-379 | Chr14 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | miR-379 modulates immune and inflammatory responses, influencing SLE susceptibility, onset, heterogeneity, and progression. |
| let-7e | Chr19 | ↓ | SOCS1, TLR6, TLR9 | let-7e targets SOCS1 and TLR pathway components, regulating cytokine signaling and innate immunity. Estrogens suppress let-7e in B cells enhancing immune activation. |
| let-7f | Chr9/ ChrX | ↓ | IL-23R, NLRP3, IL-6, A20 (TNFAIP3) | let-7f-5p targets NLRP3, mitigating inflammation in bone marrow-derived mesenchymal stem cells, and also regulates IL-6 and A20, key modulators of inflammatory and NF-κB signaling pathways. Estradiol and progesterone suppress let-7f and upregulate IL-23R, enhancing IL-17A production in Th17 cells, thereby promoting Th17 differentiation and inflammation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.R.; Jung, Y.; Kang, I.; Yeo, E.-J. Understanding Sex Differences in Autoimmune Diseases: Immunologic Mechanisms. Int. J. Mol. Sci. 2025, 26, 7101. https://doi.org/10.3390/ijms26157101
Kim YR, Jung Y, Kang I, Yeo E-J. Understanding Sex Differences in Autoimmune Diseases: Immunologic Mechanisms. International Journal of Molecular Sciences. 2025; 26(15):7101. https://doi.org/10.3390/ijms26157101
Chicago/Turabian StyleKim, Yu Rin, YunJae Jung, Insug Kang, and Eui-Ju Yeo. 2025. "Understanding Sex Differences in Autoimmune Diseases: Immunologic Mechanisms" International Journal of Molecular Sciences 26, no. 15: 7101. https://doi.org/10.3390/ijms26157101
APA StyleKim, Y. R., Jung, Y., Kang, I., & Yeo, E.-J. (2025). Understanding Sex Differences in Autoimmune Diseases: Immunologic Mechanisms. International Journal of Molecular Sciences, 26(15), 7101. https://doi.org/10.3390/ijms26157101