

Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Inflammatory Bowel Disease: Clinical Features and Pathogenesis
1.1. Etiological Factors of IBD
1.2. Immune Dysregulation and Genetic Susceptibility
2. Neutrophils in IBD Pathophysiology
2.1. Neutrophil Recruitment and Functional Diversity in IBD
2.2. Inflammatory Mediators Derived from Neutrophils
3. Neutrophil Extracellular Traps (NETs)
4. Pathological Effects of NETs in IBD
4.1. Increased NET Formation in the Inflamed Intestine
4.2. Defective NET Clearance in IBD
4.3. NETs in Promoting the Recruitment of Immune Cells
4.4. ANCA, Autoimmunity, and NET Amplification
4.5. Neutrophil and NET-Mediated Epithelial Barrier Disruption
5. Neutrophils and NETs Markers of IBD Progression
6. Contribution of NETs to ECM Remodeling and Fibrosis in IBD
7. Prothrombotic Effects of NETs in IBD
8. Microbiota and NET Interactions
9. Protective Roles of Neutrophils and NETs in IBD
10. Therapeutic Modulation of Neutrophils and NETs in IBD
10.1. PAD4 Inhibition Strategies
10.2. Targeting NADPH Oxidase and ROS Pathways
10.3. Targeting DNase I
10.4. Antibodies and Immune-Modulatory Agents
11. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aGMAbs | Anti–granulocyte–macrophage colony-stimulating factor antibodies |
| ANCA | Anti-neutrophil cytoplasmic antibody |
| CAPS | Cryopyrin-associated periodic syndromes |
| CAR | Coxsackie-adenovirus receptor |
| CCL | CC-motif chemokine ligand |
| CD | Crohn’s disease |
| cfDNA | Cell-free DNA |
| cGAS | Cyclic GMP-AMP synthase |
| Cit-H3 | Citrullinated histone H3 |
| CR3 | Complement receptor 3 |
| CRP | C-reactive protein |
| CsA | Cyclosporine A |
| CXCL | C-X-C motif chemokine ligand |
| DDIT4/REDD1 | DNA damage-inducible transcript 4/regulated in development and DNA damage response 1 |
| DAMP | Damage-associated molecular pattern |
| DNase | Deoxyribonuclease |
| DSS | Dextran sulfate sodium |
| ECM | Extracellular matrix |
| FcRn | Neonatal Fc receptor |
| FMF | Familial Mediterranean fever |
| GWAS | Genome-wide association studies |
| GSDMD | Gasdermin D |
| HIF-1α | Hypoxia-inducible factor-1α |
| HMGB1 | High-mobility group box 1 |
| HXA3 | Hepoxilin A3 |
| IBD | Inflammatory Bowel Disease |
| IEC | Intestinal epithelial cell |
| IL | Interleukin |
| ILC | Innate lymphoid cells |
| INFα | Interferon-alpha |
| JAML | Junctional adhesion molecule-like protein |
| LPS | Lipopolysaccharide |
| LTB4 | Leukotriene B4 |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MERTK | MER tyrosine kinase |
| MPO | Myeloperoxidase |
| MMP | Matrix metalloproteinase |
| mtDNA | Mitochondrial DNA |
| NE | Neutrophil elastase |
| NET | Neutrophil extracellular trap |
| NF-κB | Nuclear factor-Κb |
| NGAL | Neutrophil gelatinase-associated lipocalin |
| NOD2 | Nucleotide-binding oligomerization domain 2 |
| NOX | NADPH oxidase |
| PAD4 | Peptidylarginine deiminase 4 |
| PAMP | Pathogen-associated molecular pattern |
| PKC | Protein kinase C |
| PMA | Phorbol 12-myristate 13-acetate |
| PR3 | Proteinase 3 |
| PMP | Platelet-derived microparticle |
| ROS | Reactive oxygen species |
| SCFA | Short-chain fatty acid |
| SIRT6 | Sirtuin 6 |
| SLE | Systemic lupus erythematosus |
| STING | Stimulator of interferon genes |
| TGFβ | Transforming growth factor-β |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-alpha |
| UC | Ulcerative colitis |
References
- Gros, B.; Kaplan, G.G. Ulcerative Colitis in Adults: A Review. JAMA 2023, 330, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Gargallo-Puyuelo, C.J.; Ricart, E.; Iglesias, E.; de Francisco, R.; Gisbert, J.P.; Taxonera, C.; Manosa, M.; Aguas Peris, M.; Navarrete-Munoz, E.M.; Sanahuja, A.; et al. Sex-Related Differences in the Phenotype and Course of Inflammatory Bowel Disease: SEXEII Study of ENEIDA. Clin. Gastroenterol. Hepatol. 2024, 22, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Windsor, J.W. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Ingravalle, F.; Valvano, M.; Barbara, A.; Bardhi, D.; Latella, G.; Viscido, A.; Campanale, M.; Vinci, A.; Viora, C.; Bulfone, G.; et al. Inflammatory Bowel Disease in the Post-STRIDE II Era: Epidemiology and Long-Term Clinical Outcomes from a Population-Based Study. Med. Sci. 2025, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Halloran, J.; McDermott, B.; Ewais, T.; Begun, J.; Karatela, S.; d’Emden, H.; Corias, C.; Denny, S. Psychosocial burden of inflammatory bowel disease in adolescents and young adults. Intern. Med. J. 2021, 51, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Hibi, T. Improving IBD outcomes in the era of many treatment options. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68 (Suppl. S3), s1–s106. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, E.A.; Suskind, D.L. Current Nutritional Therapies in Inflammatory Bowel Disease: Improving Clinical Remission Rates and Sustainability of Long-Term Dietary Therapies. Nutrients 2023, 15, 668. [Google Scholar] [CrossRef] [PubMed]
- M’Koma, A.E. Inflammatory Bowel Disease: Clinical Diagnosis and Surgical Treatment-Overview. Medicina 2022, 58, 567. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Bernstein, C.N.; Burke, K.E.; Lochhead, P.J.; Sasson, A.N.; Agrawal, M.; Tiong, J.H.T.; Steinberg, J.; Kruis, W.; et al. International Organization for Study of Inflammatory Bowel, D. Lifestyle, behaviour, and environmental modification for the management of patients with inflammatory bowel diseases: An International Organization for Study of Inflammatory Bowel Diseases consensus. Lancet Gastroenterol. Hepatol. 2022, 7, 666–678. [Google Scholar] [PubMed]
- Roy, S.; Dhaneshwar, S. Role of prebiotics, probiotics, and synbiotics in management of inflammatory bowel disease: Current perspectives. World J. Gastroenterol. 2023, 29, 2078–2100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13, 835005. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, J.; Hang, R.; Chen, Q.; Wang, D. Neutrophils: From Inflammatory Bowel Disease to Colitis-Associated Colorectal Cancer. J. Inflamm. Res. 2025, 18, 925–947. [Google Scholar] [CrossRef] [PubMed]
- Kiilerich, K.F.; Andresen, T.; Darbani, B.; Gregersen, L.H.K.; Liljensoe, A.; Bennike, T.B.; Holm, R.; Moeller, J.B.; Andersen, V. Advancing Inflammatory Bowel Disease Treatment by Targeting the Innate Immune System and Precision Drug Delivery. Int. J. Mol. Sci. 2025, 26, 575. [Google Scholar] [CrossRef] [PubMed]
- Villanacci, V.; Del Sordo, R.; Mino, S.; Locci, G.; Bassotti, G. Histological healing in IBD: Ready for prime time? Dig. Liver Dis. 2025, 57, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [PubMed]
- Le Berre, C.; Honap, S.; Peyrin-Biroulet, L. Ulcerative colitis. Lancet 2023, 402, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Kappenberger, A.S.; Schardey, J.; Wirth, U.; Kuhn, F.; Werner, J.; Zimmermann, P. Clinical outcomes and perioperative morbidity and mortality following segmental resections of the colon for Crohn’s colitis. Int. J. Color. Dis. 2024, 39, 36. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, R.; Gao, H.; Jung, S.; Gao, X.; Sun, R.; Liu, X.; Kim, Y.; Lee, H.S.; Kawai, Y.; et al. Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries. Nat. Genet. 2023, 55, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Plichta, D.R.; Graham, D.B.; Subramanian, S.; Xavier, R.J. Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell 2019, 178, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Kuffa, P.; Pickard, J.M.; Campbell, A.; Yamashita, M.; Schaus, S.R.; Martens, E.C.; Schmidt, T.M.; Inohara, N.; Nunez, G.; Caruso, R. Fiber-deficient diet inhibits colitis through the regulation of the niche and metabolism of a gut pathobiont. Cell Host Microbe 2023, 31, 2007–2022.e12. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Shukla, S.; Acharya, R.; Birda, C.L.; Sah, P.K.; Singh, S.; Jearth, V.; Shah, J.; Agarwal, A.; Sharma, A.K.; et al. Impact of Clostridioides difficile Infection on the Outcome of Severe Flare of Ulcerative Colitis. J. Clin. Gastroenterol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chen, J.; Ruan, X.; Sun, Y.; Zhang, K.; Wang, X.; Li, X.; Gill, D.; Burgess, S.; Giovannucci, E.; et al. Smoking, alcohol consumption, and 24 gastrointestinal diseases: Mendelian randomization analysis. eLife 2023, 12, e84051. [Google Scholar] [CrossRef] [PubMed]
- Philip, D.; Hodgkiss, R.; Radhakrishnan, S.K.; Sinha, A.; Acharjee, A. Deciphering microbial and metabolic influences in gastrointestinal diseases-unveiling their roles in gastric cancer, colorectal cancer, and inflammatory bowel disease. J. Transl. Med. 2025, 23, 549. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Guan, X.; Ding, W.; Luo, Y.; Wang, W.; Bu, W.; Song, J.; Tan, X.; Sun, E.; Ning, Q.; et al. Scutellaria baicalensis Georgi polysaccharide ameliorates DSS-induced ulcerative colitis by improving intestinal barrier function and modulating gut microbiota. Int. J. Biol. Macromol. 2021, 166, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bris, R.; Saez, A.; Herrero-Fernandez, B.; Rius, C.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2696. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Annibaldi, A.; Kastner, D.L.; Aksentijevich, I. Genetic Regulation of Cell Death: Insights from Autoinflammatory Diseases. Annu. Rev. Immunol. 2025, 43, 313–342. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Ainciburu, M.; Wynne, K.; Bhat, S.A.; Blanco, A.; Tzani, I.; Akiba, Y.; Lalor, S.J.; Kaunitz, J.; Bourke, B.; et al. De novo DUOX2 expression in neutrophil subsets shapes the pathogenesis of intestinal disease. Proc. Natl. Acad. Sci. USA 2025, 122, e2421747122. [Google Scholar] [CrossRef] [PubMed]
- Danne, C.; Skerniskyte, J.; Marteyn, B.; Sokol, H. Neutrophils: From IBD to the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, S.; Kuwaki, K.; Mitsuyama, K.; Takedatsu, H.; Yoshioka, S.; Yamasaki, H.; Yamauchi, R.; Mori, A.; Kakuma, T.; Tsuruta, O.; et al. Detection of calprotectin in inflammatory bowel disease: Fecal and serum levels and immunohistochemical localization. Int. J. Mol. Med. 2018, 41, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, J.H.; Sinkeviciute, D.; Manon-Jensen, T.; Domislovic, V.; McCall, K.; Thudium, C.S.; Brinar, M.; Onnerfjord, P.; Goodyear, C.S.; Krznaric, Z.; et al. A Specific Calprotectin Neo-epitope [CPa9-HNE] in Serum from Inflammatory Bowel Disease Patients Is Associated with Neutrophil Activity and Endoscopic Severity. J. Crohns Colitis 2022, 16, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, A.; Borichevsky, G.M.; Edwards, T.S.; Hirschfeld, E.; Mules, T.C.; Frampton, C.M.A.; Day, A.S.; Hampton, M.B.; Kettle, A.J.; Gearry, R.B. Faecal Myeloperoxidase as a Biomarker of Endoscopic Activity in Inflammatory Bowel Disease. J. Crohns Colitis 2022, 16, 1862–1873. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, B.; Cagan Appak, Y.; Aksit, M.; Cetinoglu, S.; Kahveci, S.; Onbasi Karabag, S.; Guler, S.; Demir, I.; Karakoyun, I.; Baran, M. Leucine-Rich Alpha-2 Glycoprotein 1 as a Biomarker for Evaluation of Inflammatory Bowel Disease Activity in Children. J. Clin. Med. 2025, 14, 2803. [Google Scholar] [CrossRef]
- Tobi, M.; Antaki, F.; Rambus, M.; Hellman, J.; Hatfield, J.; Fligiel, S.; McVicker, B. Inflammatory Bowel Disease from the Perspective of Newer Innate Immune System Biomarkers. Gastrointest. Disord. 2025, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Lange, J.; Benndorf, D.; Welz, L.; Nikolaus, S.; Siever, L.K.; Tran, F.; Schallert, K.; Hellwig, P.; Schreiber, S.; et al. Fecal metaproteomics enables functional characterization of remission in patients with inflammatory bowel disease. J. Proteom. 2025, 318, 105455. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, Y.; Shi, Y.; Zhang, J.; Liu, X.; Liu, Z.; Lv, J.; Leng, Y. The emerging role of neutrophilic extracellular traps in intestinal disease. Gut Pathog. 2022, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2023, 23, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Natsui, M.; Kawasaki, K.; Takizawa, H.; Hayashi, S.I.; Matsuda, Y.; Sugimura, K.; Seki, K.; Narisawa, R.; Sendo, F.; Asakura, H. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J. Gastroenterol. Hepatol. 1997, 12, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, A.A.; Kakirman, H.; Janotta, M.; Dreher, S.; Cremer, P.; Pawlowski, N.N.; Loddenkemper, C.; Heimesaat, M.M.; Grollich, K.; Zeitz, M.; et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology 2007, 133, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhu, F.; Zhang, H.; Wang, Y.; Yang, Y.; Jin, G.; Wang, Y.; Dong, G.; Xiong, H. PTK2B regulates immune responses of neutrophils and protects mucosal inflammation in ulcerative colitis. FASEB J. 2023, 37, e22967. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Yu, L.; Fang, L.; Yang, W.; Yu, T.; Miao, Y.; Chen, M.; Wu, K.; Chen, F.; Cong, Y.; et al. CD177+ neutrophils as functionally activated neutrophils negatively regulate IBD. Gut 2018, 67, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.M.; Allen, L.A. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Fang, X.; Song, Y.H.; He, Z.X.; Wang, Z.J.; Wang, S.L.; Li, Z.S.; Bai, Y. Neutrophil-Epithelial Crosstalk During Intestinal Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Hardisty, G.R.; Llanwarne, F.; Minns, D.; Gillan, J.L.; Davidson, D.J.; Gwyer Findlay, E.; Gray, R.D. High Purity Isolation of Low Density Neutrophils Casts Doubt on Their Exceptionality in Health and Disease. Front. Immunol. 2021, 12, 625922. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, A.J.; Pourfarzad, F.; Aarts, C.E.M.; Tool, A.T.J.; Hiemstra, I.H.; Grassi, L.; Frontini, M.; Meijer, A.B.; van den Biggelaar, M.; Kuijpers, T.W. Dynamic Transcriptome-Proteome Correlation Networks Reveal Human Myeloid Differentiation and Neutrophil-Specific Programming. Cell Rep. 2019, 29, 2505–2519.e4. [Google Scholar] [CrossRef] [PubMed]
- Marini, O.; Costa, S.; Bevilacqua, D.; Calzetti, F.; Tamassia, N.; Spina, C.; De Sabata, D.; Tinazzi, E.; Lunardi, C.; Scupoli, M.T.; et al. Mature CD10+ and immature CD10− neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood 2017, 129, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.X. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease: Role of neutrophils in IBD. J. Dig. Dis. 2017, 18, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, J.; Chen, H.; Ma, X.; Si, T.; Zhu, W. Dual role of CD177 + neutrophils in inflammatory bowel disease: A review. J. Transl. Med. 2024, 22, 813. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Ross, E.A.; McGettrick, H.M.; Osborne, C.E.; Haworth, O.; Schmutz, C.; Stone, P.C.; Salmon, M.; Matharu, N.M.; Vohra, R.K.; et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 2006, 79, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Shi, Q.; Wu, P.; Zhang, X.; Kambara, H.; Su, J.; Yu, H.; Park, S.Y.; Guo, R.; Ren, Q.; et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 2020, 21, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Cao, Q.; Brooks, S.; Naz, F.; Gadkari, M.; Jiang, K.; Gupta, S.; O’Neil, L.; Dell’Orso, S.; Kaplan, M.J.; et al. Single-Cell Analysis Reveals the Range of Transcriptional States of Circulating Human Neutrophils. J. Immunol. 2022, 209, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Montaldo, E.; Lusito, E.; Bianchessi, V.; Caronni, N.; Scala, S.; Basso-Ricci, L.; Cantaffa, C.; Masserdotti, A.; Barilaro, M.; Barresi, S.; et al. Cellular and transcriptional dynamics of human neutrophils at steady state and upon stress. Nat. Immunol. 2022, 23, 1470–1483. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Trigo, A.; Corraliza, A.M.; Veny, M.; Dotti, I.; Melon-Ardanaz, E.; Rill, A.; Crowell, H.L.; Corbi, A.; Gudino, V.; Esteller, M.; et al. Macrophage and neutrophil heterogeneity at single-cell spatial resolution in human inflammatory bowel disease. Nat. Commun. 2023, 14, 4506. [Google Scholar] [CrossRef] [PubMed]
- Yalom, L.K.; Herrnreiter, C.J.; Bui, T.M.; Lockhart, J.; Piccolo, E.B.; Ren, X.; Wei, C.; Serdiukova, A.; Thorp, E.B.; Dulai, P.S.; et al. Spatially separated epithelium-associated and lamina propria neutrophils present distinct functional identities in the inflamed colon mucosa. Mucosal Immunol. 2025, 18, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.T. Neutrophil Extracellular Traps in Inflammatory Bowel Disease: Pathogenic Mechanisms and Clinical Translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, X.; Xu, C.; Lin, J.; Liu, Z. Dichotomous roles of neutrophils in modulating pathogenic and repair processes of inflammatory bowel diseases. Precis. Clin. Med. 2021, 4, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Jurickova, I.; Karns, R.; Shaw, K.A.; Cutler, D.J.; Okou, D.T.; Dodd, A.; Quinn, K.; Mondal, K.; Aronow, B.J.; et al. Clinical and Genomic Correlates of Neutrophil Reactive Oxygen Species Production in Pediatric Patients with Crohn’s Disease. Gastroenterology 2018, 154, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.A.; Sumagin, R.; McCall, I.C.; Leoni, G.; Neumann, P.A.; Andargachew, R.; Brazil, J.C.; Medina-Contreras, O.; Denning, T.L.; Nusrat, A.; et al. Neutrophil-derived JAML inhibits repair of intestinal epithelial injury during acute inflammation. Mucosal Immunol. 2014, 7, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Slater, T.W.; Finkielsztein, A.; Mascarenhas, L.A.; Mehl, L.C.; Butin-Israeli, V.; Sumagin, R. Neutrophil Microparticles Deliver Active Myeloperoxidase to Injured Mucosa To Inhibit Epithelial Wound Healing. J. Immunol. 2017, 198, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-regulation of MIR200C-3p, Which Degrades Occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [PubMed]
- Angelidou, I.; Chrysanthopoulou, A.; Mitsios, A.; Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Ritis, D.; Tsironidou, V.; Moschos, I.; Dalla, V.; et al. REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1beta Inflammatory Response in Active Ulcerative Colitis. J. Immunol. 2018, 200, 3950–3961. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Si, Y.; Jiang, T.; Ma, R.; Zhang, Y.; Cao, M.; Li, T.; Yao, Z.; Zhao, L.; Fang, S.; et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb. Haemost. 2016, 115, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Cook, W.D.; Okamoto, T.; Murphy, J.; Lawlor, K.E.; Vince, J.E.; Vaux, D.L. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013, 4, e465. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A road to inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [PubMed]
- Wallaeys, C.; Garcia-Gonzalez, N.; Timmermans, S.; Vandewalle, J.; Vanderhaeghen, T.; De Beul, S.; Dufoor, H.; Eggermont, M.; Moens, E.; Bosteels, V.; et al. Paneth cell TNF signaling induces gut bacterial translocation and sepsis. Cell Host Microbe 2024, 32, 1725–1743.e7. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction. Cells 2022, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Cui, H.; Xie, R.; Wu, J.; Shi, H.; Bi, X.; Feng, L.; Shou, J. Neutrophil extracellular traps primed intercellular communication in cancer progression as a promising therapeutic target. Biomark. Res. 2023, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Wang, X.; Yang, C.; Chen, F.; Shi, L.; Xu, W.; Wang, K.; Liu, B.; Wang, C.; Sun, D.; et al. Neutrophil extracellular traps drive intestinal microvascular endothelial ferroptosis by impairing Fundc1-dependent mitophagy. Redox Biol. 2023, 67, 102906. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.A.; Boskovic, D.S. Neutrophil Extracellular DNA Traps in Response to Infection or Inflammation, and the Roles of Platelet Interactions. Int. J. Mol. Sci. 2024, 25, 3025. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Aziz, M.; Wang, P. The vitals of NETs. J. Leukoc. Biol. 2021, 110, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, S.Y.; Kim, J.D.; Park, M.; Kim, Y.H.; Kim, K.W.; Sohn, M.H. Release of sputum neutrophil granules is associated with pulmonary function and disease severity in childhood asthma. BMC Pulm. Med. 2024, 24, 532. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.N.; Leach, S.T.; Lemberg, D.A.; Duvoisin, G.; Gearry, R.B.; Day, A.S. Fecal biomarkers in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2017, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence From Vascular Pathology in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Baz, A.A.; Hao, H.; Lan, S.; Li, Z.; Liu, S.; Chen, S.; Chu, Y. Neutrophil extracellular traps in bacterial infections and evasion strategies. Front. Immunol. 2024, 15, 1357967. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Pivetta, E.; Danussi, C.; Wassermann, B.; Modica, T.M.; Del Bel Belluz, L.; Canzonieri, V.; Colombatti, A.; Spessotto, P. Neutrophil elastase-dependent cleavage compromises the tumor suppressor role of EMILIN1. Matrix Biol. 2014, 34, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.M.; Voorberg, J.; Nicolaes, G.A.F. PAD4 takes charge during neutrophil activation: Impact of PAD4 mediated NET formation on immune-mediated disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Chamardani, T.M.; Amiritavassoli, S. Inhibition of NETosis for treatment purposes: Friend or foe? Mol. Cell. Biochem. 2022, 477, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wu, X.; Zhang, S.; Deng, C.; Zhao, L.; Wang, M.; Wu, Q.; Yang, H.; Zhou, J.; Peng, L.; et al. The potential roles of type I interferon activated neutrophils and neutrophil extracellular traps (NETs) in the pathogenesis of primary Sjogren’s syndrome. Arthritis Res. Ther. 2022, 24, 170. [Google Scholar] [CrossRef] [PubMed]
- Massicotte-Azarniouch, D.; Herrera, C.A.; Jennette, J.C.; Falk, R.J.; Free, M.E. Mechanisms of vascular damage in ANCA vasculitis. Semin. Immunopathol. 2022, 44, 325–345. [Google Scholar] [CrossRef] [PubMed]
- Radic, M.; Muller, S. LL-37, a Multi-Faceted Amphipathic Peptide Involved in NETosis. Cells 2022, 11, 2463. [Google Scholar] [CrossRef] [PubMed]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Zanoni, I. Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease. Trends Microbiol. 2023, 31, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12964. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Kearney, P.L.; Bhatia, M.; Jones, N.G.; Yuan, L.; Glascock, M.C.; Catchings, K.L.; Yamada, M.; Thompson, P.R. Kinetic characterization of protein arginine deiminase 4: A transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005, 44, 10570–10582. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Biol. 2017, 216, 4073–4090. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hong, W.; Wan, M.; Zheng, L. Molecular mechanisms and therapeutic target of NETosis in diseases. MedComm 2022, 3, e162. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Simon, D.; Stojkov, D.; Karsonova, A.; Karaulov, A.; Simon, H.U. In Vivo evidence for extracellular DNA trap formation. Cell Death Dis. 2020, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking In Vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Herre, M.; Cedervall, J.; Mackman, N.; Olsson, A.K. Neutrophil extracellular traps in the pathology of cancer and other inflammatory diseases. Physiol. Rev. 2023, 103, 277–312. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.; Galkin, I.; Pletjushkina, O.; Golyshev, S.; Zinovkin, R.; Prikhodko, A.; Pinegin, V.; Kondratenko, I.; Pinegin, B.; Chernyak, B. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165664. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, O.; Biesiekierska, M.; Panthu, B.; Soszynski, M.; Pirola, L.; Balcerczyk, A. Citrullination in the pathology of inflammatory and autoimmune disorders: Recent advances and future perspectives. Cell. Mol. Life Sci. 2022, 79, 94. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zheng, X.; Chen, M.; Huang, L.; Chen, L.; Huo, R.; Li, X.; Huang, Y.; Sun, M.; Mai, S.; et al. Deficient DNASE1L3 facilitates neutrophil extracellular traps-induced invasion via cyclic GMP-AMP synthase and the non-canonical NF-kappaB pathway in diabetic hepatocellular carcinoma. Clin. Transl. Immunol. 2022, 11, e1386. [Google Scholar] [CrossRef] [PubMed]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [PubMed]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Haider, P.; Kral-Pointner, J.B.; Mayer, J.; Richter, M.; Kaun, C.; Brostjan, C.; Eilenberg, W.; Fischer, M.B.; Speidl, W.S.; Hengstenberg, C.; et al. Neutrophil Extracellular Trap Degradation by Differently Polarized Macrophage Subsets. Arter. Thromb. Vasc. Biol. 2020, 40, 2265–2278. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Miao, C.; Zhang, H. Targeting neutrophil extracellular traps in cancer progression and metastasis. Theranostics 2025, 15, 5846–5869. [Google Scholar] [CrossRef] [PubMed]
- Wahnou, H.; El Kebbaj, R.; Hba, S.; Ouadghiri, Z.; El Faqer, O.; Pinon, A.; Liagre, B.; Limami, Y.; Duval, R.E. Neutrophils and Neutrophil-Based Drug Delivery Systems in Anti-Cancer Therapy. Cancers 2025, 17, 1232. [Google Scholar] [CrossRef] [PubMed]
- Tonello, S.; Vercellino, N.; D’Onghia, D.; Fracchia, A.; Caria, G.; Sola, D.; Tillio, P.A.; Sainaghi, P.P.; Colangelo, D. Extracellular Traps in Inflammation: Pathways and Therapeutic Targets. Life 2025, 15, 627. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.L.; Prasad, R.; Dupont, M.D.; Adu-Rutledge, Y.; Anshumali, S.; Paul, S.; Li Calzi, S.; Qi, X.; Malepati, A.; Johnson, E.; et al. Intestinal neutrophil extracellular traps promote gut barrier damage exacerbating endotoxaemia, systemic inflammation and progression of diabetic retinopathy in type 2 diabetes. Diabetologia 2025, 68, 866–889. [Google Scholar] [CrossRef] [PubMed]
- Filippini, D.F.L.; Jiang, M.; Kramer, L.; van der Poll, T.; Cremer, O.; Hla, T.T.W.; Retter, A.; Bos, L.D.J. MARS Consortium. Plasma H3.1 nucleosomes as biomarkers of infection, inflammation and organ failure. Crit. Care 2025, 29, 198. [Google Scholar] [CrossRef] [PubMed]
- Wan, A.; Chen, D. The Multifaceted Roles of Neutrophil Death in COPD and Lung Cancer. J. Respir. Biol. Transl. Med. 2025, 2, 10022. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guan, L.; Wang, C.; Hu, R.; Ou, L.; Jiang, Q. The role of fibroblast-neutrophil crosstalk in the pathogenesis of inflammatory diseases: A multi-tissue perspective. Front. Immunol. 2025, 16, 1588667. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, R.; Yue, H.; Zhang, X.; Pan, X.; Sun, Y.; Shi, J.; Zhu, G.; Qin, C.; Guo, Y. Interaction between neutrophil extracellular traps and cardiomyocytes contributes to atrial fibrillation progression. Signal Transduct. Target. Ther. 2023, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Lai, X.; Wu, C.; Wang, L.; Shang, J.; Zhang, H.; Jia, S.; Xing, W.; Liu, H. The roles of neutrophils in cardiovascular diseases. Front. Cardiovasc. Med. 2025, 12, 1526170. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Vargas, A.; Gomez-Martin, D.; Carmona-Rivera, C.; Merayo-Chalico, J.; Torres-Ruiz, J.; Manna, Z.; Hasni, S.; Alcocer-Varela, J.; Kaplan, M.J. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13, 838011. [Google Scholar] [CrossRef] [PubMed]
- Therrien, A.; Chapuy, L.; Bsat, M.; Rubio, M.; Bernard, G.; Arslanian, E.; Orlicka, K.; Weber, A.; Panzini, B.P.; Dorais, J.; et al. Recruitment of activated neutrophils correlates with disease severity in adult Crohn’s disease. Clin. Exp. Immunol. 2019, 195, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Minar, P.; Jackson, K.; Tsai, Y.T.; Sucharew, H.; Rosen, M.J.; Denson, L.A. Validation of Neutrophil CD64 Blood Biomarkers to Detect Mucosal Inflammation in Pediatric Crohn’s Disease. Inflamm. Bowel Dis. 2017, 24, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, Y.; Pinar, A.; Hizal, G.; Demir, H.; Saltik Temizel, I.N.; Ozen, H.; Akbiyik, F.; Yuce, A. Neutrophil volume distribution width as a new marker in detecting inflammatory bowel disease activation. Int. J. Lab. Hematol. 2017, 39, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, M.Y.; Ozin, Y.O.; Kaplan, M.; Ates, I.; Kalkan, I.H.; Kilic, Z.M.Y.; Yuksel, M.; Kayacetin, E. Platelet-to-lymphocyte Ratio and Neutrophil-to-lymphocyte Ratio Predict Mucosal Disease Severity in Ulcerative Colitis. J. Med. Biochem. 2018, 37, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Lai, H.J.; Cheng, Y.K.; Leong, K.Q.; Cheng, L.C.; Chou, Y.C.; Peng, Y.C.; Hsu, Y.H.; Chiang, H.S. Neutrophil Extracellular Traps Impair Intestinal Barrier Function during Experimental Colitis. Biomedicines 2020, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. Int. J. Mol. Sci. 2023, 24, 4896. [Google Scholar] [CrossRef] [PubMed]
- Dinallo, V.; Marafini, I.; Di Fusco, D.; Laudisi, F.; Franze, E.; Di Grazia, A.; Figliuzzi, M.M.; Caprioli, F.; Stolfi, C.; Monteleone, I.; et al. Neutrophil Extracellular Traps Sustain Inflammatory Signals in Ulcerative Colitis. J. Crohns Colitis 2019, 13, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, L.; Bsat, M.; Rubio, M.; Sarkizova, S.; Therrien, A.; Bouin, M.; Orlicka, K.; Weber, A.; Soucy, G.; Villani, A.C.; et al. IL-12 and Mucosal CD14+ Monocyte-Like Cells Induce IL-8 in Colonic Memory CD4+ T Cells of Patients with Ulcerative Colitis but not Crohn’s Disease. J. Crohns Colitis 2020, 14, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Li, M.; Zhao, W.; Zhang, K.; Li, M.; Li, W. Hyper alpha2,6-Sialylation Promotes CD4+ T-Cell Activation and Induces the Occurrence of Ulcerative Colitis. Adv. Sci. 2023, 10, e2302607. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Murakami, T.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Tabe, Y.; Nagaoka, I. Neutrophil extracellular traps induce IL-1beta production by macrophages in combination with lipopolysaccharide. Int. J. Mol. Med. 2017, 39, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wittchen, E.S.; Monaghan-Benson, E.; Hahn, C.; Earp, H.S.; Doerschuk, C.M.; Burridge, K. The role of endothelial MERTK during the inflammatory response in lungs. PLoS ONE 2019, 14, e0225051. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Mitselou, A.; Grammeniatis, V.; Varouktsi, A.; Papadatos, S.S.; Katsanos, K.; Galani, V. Proinflammatory cytokines in irritable bowel syndrome: A comparison with inflammatory bowel disease. Intest. Res. 2020, 18, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zheng, B.; Bai, Y.; Zhou, J.; Zhang, X.H.; Yang, Y.Q.; Yu, J.; Zhao, H.Y.; Ma, D.; Wu, H.; et al. Exosomes-transferred LINC00668 Contributes to Thrombosis by Promoting NETs Formation in Inflammatory Bowel Disease. Adv. Sci. 2023, 10, e2300560. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Simonson, T.J.; Jondle, C.N.; Mishra, B.B.; Sharma, J. Mincle-Mediated Neutrophil Extracellular Trap Formation by Regulation of Autophagy. J. Infect. Dis. 2017, 215, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, T.; Cao, M.; Si, Y.; Wu, X.; Zhao, L.; Yao, Z.; Zhang, Y.; Fang, S.; Deng, R.; et al. Extracellular DNA traps released by acute promyelocytic leukemia cells through autophagy. Cell Death Dis. 2016, 7, e2283. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Ramos, A.; Viana, G.C.S.; de Macedo Brigido, M.; Almeida, J.F. Neutrophil extracellular traps in inflammatory bowel diseases: Implications in pathogenesis and therapeutic targets. Pharmacol. Res. 2021, 171, 105779. [Google Scholar] [CrossRef] [PubMed]
- Arias-Loste, M.T.; Bonilla, G.; Moraleja, I.; Mahler, M.; Mieses, M.A.; Castro, B.; Rivero, M.; Crespo, J.; Lopez-Hoyos, M. Presence of anti-proteinase 3 antineutrophil cytoplasmic antibodies (anti-PR3 ANCA) as serologic markers in inflammatory bowel disease. Clin. Rev. Allergy Immunol. 2013, 45, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Malickova, K.; Duricova, D.; Bortlik, M.; Hruskova, Z.; Svobodova, B.; Machkova, N.; Komarek, V.; Fucikova, T.; Janatkova, I.; Zima, T.; et al. Impaired deoxyribonuclease I activity in patients with inflammatory bowel diseases. Autoimmune Dis. 2011, 2011, 945861. [Google Scholar] [CrossRef] [PubMed]
- Vrablicova, Z.; Tomova, K.; Tothova, L.; Babickova, J.; Gromova, B.; Konecna, B.; Liptak, R.; Hlavaty, T.; Gardlik, R. Nuclear and Mitochondrial Circulating Cell-Free DNA Is Increased in Patients with Inflammatory Bowel Disease in Clinical Remission. Front. Med. 2020, 7, 593316. [Google Scholar] [CrossRef] [PubMed]
- Sallai, K.; Nagy, E.; Derfalvy, B.; Muzes, G.; Gergely, P. Antinucleosome antibodies and decreased deoxyribonuclease activity in sera of patients with systemic lupus erythematosus. Clin. Diagn. Lab. Immunol. 2005, 12, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Chertov, O.; Oppenheim, J.J. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J. Leukoc. Biol. 2000, 68, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, S.; Valecha, J.; Hochwald, A.; Berianu, F.; Majithia, V.; Abril, A. Investigating the concomitance of anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitides and inflammatory bowel disease (IBD). Semin. Arthritis Rheum. 2024, 66, 152452. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.C.; Wu, Y.C.; Chen, J.P.; Huang, W.N.; Chen, Y.H.; Chen, Y.M. Association of atypical anti-neutrophil cytoplasmic antibody with comorbidities and outcome in a hospital-based population. Heliyon 2024, 10, e24105. [Google Scholar] [CrossRef] [PubMed]
- Andalucia, C.; Martinez-Prat, L.; Bentow, C.; Aure, M.A.; Horn, M.P.; Mahler, M. Clinical Validity of Anti-Proteinase 3 Antibodies in Patients with Inflammatory Bowel Disease: A Short Meta-Analysis. Diagnostics 2023, 13, 3682. [Google Scholar] [CrossRef] [PubMed]
- Abakarim, O.; Klevor, R.; El Moumou, L.; Hazime, R.; Brahim, I.; Oubaha, S.; Krati, K.; Admou, B. ANCA and ASCA Profiles in a Moroccan Population with Inflammatory Bowel Diseases. J. Community Hosp. Intern. Med. Perspect. 2024, 14, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Hu, H.; Yang, W.; Zhao, Y.; Zheng, L.; Jiang, X.; Wang, L. Targeted inhibition of FcRn reduces NET formation to ameliorate experimental ulcerative colitis by accelerating ANCA clearance. Int. Immunopharmacol. 2022, 113, 109474. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Rath, T.; Lencer, W.I.; Baker, K.; Blumberg, R.S. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J. Immunol. 2015, 194, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- Cadena Castaneda, D.; Brachet, G.; Goupille, C.; Ouldamer, L.; Gouilleux-Gruart, V. The neonatal Fc receptor in cancer FcRn in cancer. Cancer Med. 2020, 9, 4736–4742. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Palaniyandi, S.; Zhu, I.; Tang, J.; Li, W.; Wu, X.; Ochsner, S.P.; Pauza, C.D.; Cohen, J.I.; Zhu, X. Human cytomegalovirus evades antibody-mediated immunity through endoplasmic reticulum-associated degradation of the FcRn receptor. Nat. Commun. 2019, 10, 3020. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, L.J.; Humphries, J.E.; Jones, S.D.; Pearce, L.B.; Holgate, R.; Hearn, A.; Cheung, J.; Mahmood, A.; Del Tito, B.; Graydon, J.S.; et al. Blocking FcRn in humans reduces circulating IgG levels and inhibits IgG immune complex-mediated immune responses. Sci. Adv. 2019, 5, eaax9586. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Lu, F.; Guan, X.; Jiang, X.; Wen, C.; Wang, L. Baicalein Ameliorates Experimental Ulcerative Colitis Recurrency by Downregulating Neonatal Fc Receptor via the NF-kappaB Signaling Pathway. ACS Omega 2025, 10, 10701–10712. [Google Scholar] [CrossRef] [PubMed]
- Jergens, A.E.; Parvinroo, S.; Kopper, J.; Wannemuehler, M.J. Rules of Engagement: Epithelial-Microbe Interactions and Inflammatory Bowel Disease. Front. Med. 2021, 8, 669913. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.J.; Doan, H.T.; Lin, E.Y.; Chiu, Y.L.; Cheng, Y.K.; Lin, Y.H.; Chiang, H.S. Histones of Neutrophil Extracellular Traps Directly Disrupt the Permeability and Integrity of the Intestinal Epithelial Barrier. Inflamm. Bowel Dis. 2023, 29, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.C.; Parkos, C.A. Pathobiology of neutrophil transepithelial migration: Implications in mediating epithelial injury. Annu. Rev. Pathol. 2007, 2, 111–143. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Inca, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Brazil, J.C.; Louis, N.A.; Parkos, C.A. The role of polymorphonuclear leukocyte trafficking in the perpetuation of inflammation during inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.L.; Chami, B.; Liu, Y.; Doyle, C.M.; El Kazzi, M.; Ahlenstiel, G.; Ahmad, G.; Pathma-Nathan, N.; Collins, G.; Toh, J.; et al. Neutrophil Extracellular Trap Density Increases with Increasing Histopathological Severity of Crohn’s Disease. Inflamm. Bowel Dis. 2022, 28, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Wang, Z.; Chang, X.; Wu, H.; Yan, Z.; Wu, J.; He, Z.; Kang, L.; Hu, W.; et al. Neutrophil-derived PAD4 induces citrullination of CKMT1 exacerbates mucosal inflammation in inflammatory bowel disease. Cell. Mol. Immunol. 2024, 21, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Wang, X.; Chen, F.; Yang, C.; Shi, L.; Xu, W.; Wang, K.; Liu, B.; Wang, C.; Sun, D.; et al. Neutrophil extracellular traps aggravate intestinal epithelial necroptosis in ischaemia-reperfusion by regulating TLR4/RIPK3/FUNDC1-required mitophagy. Cell Prolif. 2024, 57, e13538. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, G.; Danese, S.; Peyrin-Biroulet, L.; Sans, M.; Bonelli, F.; Calleri, M.; Zierold, C.; Pollastro, R.; Moretti, F.; Malesci, A. LIAISON((R)) Calprotectin for the prediction of relapse in quiescent ulcerative colitis: The EuReCa study. United Eur. Gastroenterol. J. 2022, 10, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Borichevsky, G.M.; Swaminathan, A.; Smith, B.R.; Edwards, T.S.; Ashby, L.V.; Frampton, C.M.A.; Day, A.S.; Gearry, R.B.; Kettle, A.J. Myeloperoxidase Enzyme Activity in Feces Reflects Endoscopic Severity in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2025, izaf109. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Swaminathan, A.; Borichevsky, G.M.; Frampton, C.M.; Kettle, A.J.; Gearry, R.B. Plasma Calprotectin and Myeloperoxidase as Biomarkers in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2025, izaf110. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.; Ruano-Gallego, D.; Radhakrishnan, S.T.; Lovell, S.; Yu, L.; Kotik, O.; Glegola-Madejska, I.; Tate, E.W.; Choudhary, J.S.; Williams, H.R.T.; et al. Faecal neutrophil elastase-antiprotease balance reflects colitis severity. Mucosal Immunol. 2020, 13, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Jablaoui, A.; Kriaa, A.; Mkaouar, H.; Akermi, N.; Soussou, S.; Wysocka, M.; Woloszyn, D.; Amouri, A.; Gargouri, A.; Maguin, E.; et al. Fecal Serine Protease Profiling in Inflammatory Bowel Diseases. Front. Cell. Infect. Microbiol. 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Gorecka, A.; Komosinska-Vassev, K. Neutrophil Elastase and Elafin in Inflammatory Bowel Diseases: Urinary Biomarkers Reflecting Intestinal Barrier Dysfunction and Proteolytic Activity. J. Clin. Med. 2025, 14, 2466. [Google Scholar] [CrossRef] [PubMed]
- Kardas Yildiz, A.; Urganci, N.; Usta, A.M. Evaluation of fecal neutrophil gelatinase-associated lipocalin levels in childhood inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 2025, 80, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Zollner, A.; Schmiderer, A.; Reider, S.J.; Oberhuber, G.; Pfister, A.; Texler, B.; Watschinger, C.; Koch, R.; Effenberger, M.; Raine, T.; et al. Faecal Biomarkers in Inflammatory Bowel Diseases: Calprotectin Versus Lipocalin-2-a Comparative Study. J. Crohns Colitis 2021, 15, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Thorsvik, S.; Bakke, I.; van Beelen Granlund, A.; Royset, E.S.; Damas, J.K.; Ostvik, A.E.; Sandvik, A.K. Expression of neutrophil gelatinase-associated lipocalin (NGAL) in the gut in Crohn’s disease. Cell Tissue Res. 2018, 374, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Buisson, A.; Vazeille, E.; Minet-Quinard, R.; Goutte, M.; Bouvier, D.; Goutorbe, F.; Pereira, B.; Barnich, N.; Bommelaer, G. Fecal Matrix Metalloprotease-9 and Lipocalin-2 as Biomarkers in Detecting Endoscopic Activity in Patients with Inflammatory Bowel Diseases. J. Clin. Gastroenterol. 2018, 52, e53–e62. [Google Scholar] [CrossRef] [PubMed]
- Thorsvik, S.; Damas, J.K.; Granlund, A.V.; Flo, T.H.; Bergh, K.; Ostvik, A.E.; Sandvik, A.K. Fecal neutrophil gelatinase-associated lipocalin as a biomarker for inflammatory bowel disease. J. Gastroenterol. Hepatol. 2017, 32, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Abd El Hafez, A.; Mohamed, A.S.; Shehta, A.; Sheta, H. Neutrophil extracellular traps-associated protein peptidyl arginine deaminase 4 immunohistochemical expression in ulcerative colitis and its association with the prognostic predictors. Pathol. Res. Pract. 2020, 216, 153102. [Google Scholar] [CrossRef] [PubMed]
- Neuenfeldt, F.; Schumacher, J.C.; Grieshaber-Bouyer, R.; Habicht, J.; Schroder-Braunstein, J.; Gauss, A.; Merle, U.; Niesler, B.; Heineken, N.; Dalpke, A.; et al. Inflammation induces pro-NETotic neutrophils via TNFR2 signaling. Cell Rep. 2022, 39, 110710. [Google Scholar] [CrossRef] [PubMed]
- Bennike, T.B.; Carlsen, T.G.; Ellingsen, T.; Bonderup, O.K.; Glerup, H.; Bogsted, M.; Christiansen, G.; Birkelund, S.; Stensballe, A.; Andersen, V. Neutrophil Extracellular Traps in Ulcerative Colitis: A Proteome Analysis of Intestinal Biopsies. Inflamm. Bowel Dis. 2015, 21, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ye, Z.; Jiang, W.; Wang, S.; Zhang, H. Cyclosporine A alleviates colitis by inhibiting the formation of neutrophil extracellular traps via the regulating pentose phosphate pathway. Mol. Med. 2023, 29, 169. [Google Scholar] [CrossRef] [PubMed]
- Di Vincenzo, F.; Yadid, Y.; Petito, V.; Emoli, V.; Masi, L.; Gerovska, D.; Arauzo-Bravo, M.J.; Gasbarrini, A.; Regenberg, B.; Scaldaferri, F. Circular and Circulating DNA in Inflammatory Bowel Disease: From Pathogenesis to Potential Molecular Therapies. Cells 2023, 12, 1953. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Maronek, M.; Gromova, B.; Liptak, R.; Konecna, B.; Pastorek, M.; Cechova, B.; Harsanyova, M.; Budis, J.; Smolak, D.; Radvanszky, J.; et al. Extracellular DNA Correlates with Intestinal Inflammation in Chemically Induced Colitis in Mice. Cells 2021, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Ngo, A.T.; Skidmore, A.; Oberg, J.; Yarovoi, I.; Sarkar, A.; Levine, N.; Bochenek, V.; Zhao, G.; Rauova, L.; Kowalska, M.A.; et al. Platelet factor 4 limits neutrophil extracellular trap- and cell-free DNA-induced thrombogenicity and endothelial injury. JCI Insight 2023, 8, e171054. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Potter, J.E.; Brownlie, R.; Ficzycz, A.D.; Griebel, P.J.; Mookherjee, N.; Mutwiri, G.K.; Babiuk, L.A.; Napper, S. Nucleic acids exert a sequence-independent cooperative effect on sequence-dependent activation of Toll-like receptor 9. J. Biol. Chem. 2007, 282, 13944–13953. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Verma, P.; Varga, J.; Bhattacharyya, S. Extracellular Matrix-Derived Damage-Associated Molecular Patterns (DAMP): Implications in Systemic Sclerosis and Fibrosis. J. Investig. Dermatol. 2023, 143, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 2015, 62, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Nanini, H.F.; Bernardazzi, C.; Castro, F.; de Souza, H.S.P. Damage-associated molecular patterns in inflammatory bowel disease: From biomarkers to therapeutic targets. World J. Gastroenterol. 2018, 24, 4622–4634. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, M.; Wang, X.; Xu, H.; Jiang, L.; Wu, F.; Wei, L.; Qi, G.; Zhang, D. Dihydromyricetin ameliorates experimental ulcerative colitis by inhibiting neutrophil extracellular traps formation via the HIF-1alpha/VEGFA signaling pathway. Int. Immunopharmacol. 2024, 138, 112572. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Portier, I.; Rustad, J.L.; Cody, M.J.; de Araujo, C.V.; Hoki, C.; Alexander, M.D.; Grandhi, R.; Dyer, M.R.; Neal, M.D.; et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J. Clin. Investig. 2022, 132, e154225. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Ling, Y.; Deng, Q.; Qiu, Y.; Shen, J.; Lai, H.; Chen, Z.; Huang, C.; Liang, L.; Li, X.; et al. HMGB1-Mediated Neutrophil Extracellular Trap Formation Exacerbates Intestinal Ischemia/Reperfusion-Induced Acute Lung Injury. J. Immunol. 2022, 208, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bao, S.; Liu, M.; Han, Z.; Tan, J.; Zhu, Q.; Huang, X.; Tian, X. Inhibition of HMGB1 improves experimental mice colitis by mediating NETs and macrophage polarization. Cytokine 2024, 176, 156537. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.; Vandergheynst, F.; Ilzkovitz, M. Exploring ANCA Pathogenicity in Ulcerative Colitis: A Case Report Highlighting the Risk of Progression to ANCA-Associated Vasculitis. Eur. J. Case Rep. Intern. Med. 2025, 12, 005061. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Choi, Y.M.; Jung, S.A.; Yang, H.R. Diagnostic utility, disease activity, and disease phenotype correlation of serum ASCA, pANCA, and PR3-ANCA in pediatric inflammatory bowel disease. J. Pediatr. 2024, 100, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P.R. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Lindemann, A.; Gosswein, S.; Paulus, S.; Roth, D.; Hartung, A.; Liebing, E.; Zundler, S.; Gonzalez-Acera, M.; Patankar, J.V.; et al. Neutrophils prevent rectal bleeding in ulcerative colitis by peptidyl-arginine deiminase-4-dependent immunothrombosis. Gut 2022, 71, 2414–2429. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, D.; Zhou, Z.; Liu, F.; Shen, Y.; You, Q.; Lu, S.; Wu, J. The role of protein arginine deiminase 4-dependent neutrophil extracellular traps formation in ulcerative colitis. Front. Immunol. 2023, 14, 1144976. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, P.; Zhai, X.; Wang, Z.; Miao, X.; Yang, Y.; Wu, J. Neutrophil extracellular traps induce barrier dysfunction in DSS-induced ulcerative colitis via the cGAS-STING pathway. Int. Immunopharmacol. 2024, 143, 113358. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Ko, G.R.; Lee, Y.Y.; Park, J.; Park, W.; Park, T.E.; Jin, Y.; Kim, S.N.; Lee, J.S.; Park, C.G. Polymeric DNase-I nanozymes targeting neutrophil extracellular traps for the treatment of bowel inflammation. Nano Converg. 2024, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Qian, K.; Zhao, Y.; Hong, J.; Chen, H.; Wang, X.; Yang, N.; Zhang, C.; Cao, J.; Jia, K.; et al. Association of Neutrophil Extracellular Traps with Fistula Healing in Patients with Complex Perianal Fistulizing Crohn’s Disease. J. Crohns Colitis 2023, 17, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Hu, M.; Yang, N.; Qian, K.; Hong, J.; Tang, J.; Bian, Y.; Zhang, C.; Wang, X.; Wu, G.; et al. Microbial and Transcriptomic Landscape Associated with Neutrophil Extracellular Traps in Perianal Fistulizing Crohn’s Disease. Inflamm. Bowel Dis. 2025, 31, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ai, F.; Wu, X.; Yu, W.; Jin, X.; Ma, J.; Xiang, B.; Shen, S.; Li, X. Analysis of neutrophil extracellular trap-related genes in Crohn’s disease based on bioinformatics. J. Cell. Mol. Med. 2024, 28, e70013. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, L.; Wei, L.; Yu, J.; Zhu, S.; Li, X.; Liu, J.; Liang, R.; Peng, W.; Ge, F.; et al. Da-yuan-yin decoction alleviates ulcerative colitis by inhibiting complement activation, LPS-TLR4/NF-kappaB signaling pathway and NET formation. J. Ethnopharmacol. 2024, 332, 118392. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.N.; Wang, J.; Mukherjee, P.K.; Veisman, I.; Massey, W.J.; Mao, R.; Chandra, J.; Fiocchi, C.; Rieder, F. The functional role of the extracellular matrix in inflammatory bowel disease associated gut fibrosis. Matrix Biol. 2025, 139, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 129–169. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, M.; Di Sabatino, A.; Manon-Jensen, T.; Mazza, G.; Madsen, G.I.; Giuffrida, P.; Pinzani, M.; Krag, A.; Karsdal, M.A.; Kjeldsen, J.; et al. A Serological Biomarker of Laminin Gamma 1 Chain Degradation Reflects Altered Basement Membrane Remodeling in Crohn’s Disease and DSS Colitis. Dig. Dis. Sci. 2022, 67, 3662–3671. [Google Scholar] [CrossRef] [PubMed]
- Latella, G.; Sferra, R.; Speca, S.; Vetuschi, A.; Gaudio, E. Can we prevent, reduce or reverse intestinal fibrosis in IBD? Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1283–1304. [Google Scholar] [PubMed]
- Schniers, A.; Anderssen, E.; Fenton, C.G.; Goll, R.; Pasing, Y.; Paulssen, R.H.; Florholmen, J.; Hansen, T. The Proteome of Ulcerative Colitis in Colon Biopsies from Adults—Optimized Sample Preparation and Comparison with Healthy Controls. Proteom. Clin. Appl. 2017, 11, 1700053. [Google Scholar] [CrossRef] [PubMed]
- Koller, F.L.; Dozier, E.A.; Nam, K.T.; Swee, M.; Birkland, T.P.; Parks, W.C.; Fingleton, B. Lack of MMP10 exacerbates experimental colitis and promotes development of inflammation-associated colonic dysplasia. Lab. Investig. 2012, 92, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Kirov, S.; Sasson, A.; Zhang, C.; Chasalow, S.; Dongre, A.; Steen, H.; Stensballe, A.; Andersen, V.; Birkelund, S.; Bennike, T.B. Degradation of the extracellular matrix is part of the pathology of ulcerative colitis. Mol. Omics 2019, 15, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Deraison, C.; Bonnart, C.; Langella, P.; Roget, K.; Vergnolle, N. Elafin and its precursor trappin-2: What is their therapeutic potential for intestinal diseases? Br. J. Pharmacol. 2023, 180, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Jaberi, S.A.; Cohen, A.; D’Souza, C.; Abdulrazzaq, Y.M.; Ojha, S.; Bastaki, S.; Adeghate, E.A. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 2021, 142, 112002. [Google Scholar] [CrossRef] [PubMed]
- Komosinska-Vassev, K.; Kaluzna, A.; Jura-Poltorak, A.; Derkacz, A.; Olczyk, K. Circulating Profile of ECM-Related Proteins as Diagnostic Markers in Inflammatory Bowel Diseases. J. Clin. Med. 2022, 11, 5618. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mao, R.; Kurada, S.; Wang, J.; Lin, S.; Chandra, J.; Rieder, F. Pathogenesis of fibrostenosing Crohn’s disease. Transl. Res. 2019, 209, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.O.; Agrawal, N.; Willis, E.; Goldblum, J.R.; Lopez, R.; Allende, D.; Liu, X.; Patil, D.Y.; Yerian, L.; El-Khider, F.; et al. Fibrosis in ulcerative colitis is directly linked to severity and chronicity of mucosal inflammation. Aliment. Pharmacol. Ther. 2018, 47, 922–939. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.O.; Bettenworth, D.; Bokemeyer, A.; Srivastava, A.; Rosty, C.; de Hertogh, G.; Robert, M.E.; Valasek, M.A.; Mao, R.; Kurada, S.; et al. Histopathology Scoring Systems of Stenosis Associated with Small Bowel Crohn’s Disease: A Systematic Review. Gastroenterology 2020, 158, 137–150.e1. [Google Scholar] [CrossRef] [PubMed]
- Petrey, A.C.; de la Motte, C.A. The extracellular matrix in IBD: A dynamic mediator of inflammation. Curr. Opin. Gastroenterol. 2017, 33, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Koelink, P.J.; Overbeek, S.A.; Braber, S.; Morgan, M.E.; Henricks, P.A.; Abdul Roda, M.; Verspaget, H.W.; Wolfkamp, S.C.; te Velde, A.A.; Jones, C.W.; et al. Collagen degradation and neutrophilic infiltration: A vicious circle in inflammatory bowel disease. Gut 2014, 63, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A.; Alexdottir, M.S.; Riis, L.B.; Ovesen, P.D.; Rasmussen, J.; Wewer, M.D.; Lin, V.; Lagstrom, R.M.B.; Al-Sheikh, M.; Dahl, E.; et al. Serum biomarkers of collagen remodeling are associated with intestinal fibrosis and differentiate stenotic from luminal Crohn’s disease patients: A pre- and post-resection longitudinal study. J. Crohns Colitis 2025, 19, jjaf085. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, J.; Li, M.; Qi, G.; Wei, L.; Zhang, D. Nets in fibrosis: Bridging innate immunity and tissue remodeling. Int. Immunopharmacol. 2024, 137, 112516. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 2021, 21, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Gavriilidis, E.; Divolis, G.; Natsi, A.M.; Kafalis, N.; Kogias, D.; Antoniadou, C.; Synolaki, E.; Pavlos, E.; Koutsi, M.A.; Didaskalou, S.; et al. Neutrophil-fibroblast crosstalk drives immunofibrosis in Crohn’s disease through IFNalpha pathway. Front. Immunol. 2024, 15, 1447608. [Google Scholar] [CrossRef] [PubMed]
- Kerami, Z.; Duijvis, N.W.; Vogels, E.W.; van Dooren, F.H.; Moerland, P.D.; Te Velde, A.A. Effect of interleukin-17 on gene expression profile of fibroblasts from Crohn’s disease patients. J. Crohns Colitis 2014, 8, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, G.; Ke, B.J.; Picariello, L.; Abdurahiman, S.; Ceni, E.; Biscu, F.; Mello, T.; Polvani, S.; Innocenti, T.; Spalart, V.; et al. The Impact of Peptidyl Arginine Deiminase 4-Dependent Neutrophil Extracellular Trap Formation on the Early Development of Intestinal Fibrosis in Crohn’s Disease. J. Crohns Colitis 2025, 19, jjae121. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Jackson, M.A.; Korsunsky, I.; Bullers, S.J.; Rue-Albrecht, K.; Christoforidou, Z.; Sathananthan, D.; Thomas, T.; Ravindran, R.; et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 2021, 27, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Xu, S.; Ji, K.; Tang, C. Myeloid Cell-Derived IL-1 Signaling Damps Neuregulin-1 from Fibroblasts to Suppress Colitis-Induced Early Repair of the Intestinal Epithelium. Int. J. Mol. Sci. 2024, 25, 4469. [Google Scholar] [CrossRef] [PubMed]
- Gala, D.; Newsome, T.; Roberson, N.; Lee, S.M.; Thekkanal, M.; Shah, M.; Kumar, V.; Bandaru, P.; Gayam, V. Thromboembolic Events in Patients with Inflammatory Bowel Disease: A Comprehensive Overview. Diseases 2022, 10, 4469. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, L.; Braun, O.O.; Westman, J.; Madhi, R.; Herwald, H.; Morgelin, M.; Thorlacius, H. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci. Rep. 2018, 8, 4020. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Novacek, G.; Weltermann, A.; Sobala, A.; Tilg, H.; Petritsch, W.; Reinisch, W.; Mayer, A.; Haas, T.; Kaser, A.; Feichtenschlager, T.; et al. Inflammatory bowel disease is a risk factor for recurrent venous thromboembolism. Gastroenterology 2010, 139, 779–787.e1. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Russell, J.; Senchenkova, E.Y.; Almeida Paula, L.D.; Granger, D.N. Interleukin-1beta mediates the extra-intestinal thrombosis associated with experimental colitis. Am. J. Pathol. 2010, 177, 2774–2781. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Yilmaz, C.E.; Granger, D.N. Role of tumor necrosis factor-alpha in the extraintestinal thrombosis associated with colonic inflammation. Inflamm. Bowel Dis. 2011, 17, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Senchenkova, E.Y.; Komoto, S.; Russell, J.; Almeida-Paula, L.D.; Yan, L.S.; Zhang, S.; Granger, D.N. Interleukin-6 mediates the platelet abnormalities and thrombogenesis associated with experimental colitis. Am. J. Pathol. 2013, 183, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Varju, I.; Longstaff, C.; Szabo, L.; Farkas, A.Z.; Varga-Szabo, V.J.; Tanka-Salamon, A.; Machovich, R.; Kolev, K. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb. Haemost. 2015, 113, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A. Physiological Role of Gut Microbiota for Maintaining Human Health. Digestion 2016, 93, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejon, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Tai, W.C.; Liang, C.M.; Wu, C.K.; Tsai, M.C.; Hu, W.H.; Huang, P.Y.; Chen, C.H.; Kuo, Y.H.; Yao, C.C.; et al. Alternations of the gut microbiota and the Firmicutes/Bacteroidetes ratio after biologic treatment in inflammatory bowel disease. J. Microbiol. Immunol. Infect. 2025, 58, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Loubinoux, J.; Bronowicki, J.P.; Pereira, I.A.; Mougenel, J.L.; Faou, A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002, 40, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Zinkevich, V.V.; Beech, I.B. Screening of sulfate-reducing bacteria in colonoscopy samples from healthy and colitic human gut mucosa. FEMS Microbiol. Ecol. 2000, 34, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rowan, F.; Docherty, N.G.; Murphy, M.; Murphy, B.; Calvin Coffey, J.; O’Connell, P.R. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis. Colon Rectum 2010, 53, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [PubMed]
- Vrakas, S.; Mountzouris, K.C.; Michalopoulos, G.; Karamanolis, G.; Papatheodoridis, G.; Tzathas, C.; Gazouli, M. Intestinal Bacteria Composition and Translocation of Bacteria in Inflammatory Bowel Disease. PLoS ONE 2017, 12, e0170034. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Zapater, P.; Juanola, O.; Sempere, L.; Garcia, M.; Laveda, R.; Martinez, A.; Scharl, M.; Gonzalez-Navajas, J.M.; Such, J.; et al. Gut Bacterial DNA Translocation is an Independent Risk Factor of Flare at Short Term in Patients with Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Frances, R.; Amoros, A.; Zapater, P.; Garmendia, M.; Ndongo, M.; Cano, R.; Jover, R.; Such, J.; Perez-Mateo, M. Cytokine association with bacterial DNA in serum of patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 508–514. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ito, S.; Nishio, N.; Cheng, Z.; Suzuki, H.; Isobe, K. Up-regulation of Gr1+CD11b+ population in spleen of dextran sulfate sodium administered mice works to repair colitis. Inflamm. Allergy Drug Targets 2011, 10, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, Y.; Kanai, T.; Tohda, S.; Totsuka, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; Sakamoto, N.; Fukuda, T.; Miura, O.; et al. Negative feedback regulation of colitogenic CD4+ T cells by increased granulopoiesis. Inflamm. Bowel Dis. 2008, 14, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.H.; Che, X.; Kim, S.; Kim, D.H.; Ma, H.W.; Kim, J.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; Cheon, J.H. Triggering Receptor Expressed on Myeloid Cells-1 Agonist Regulates Intestinal Inflammation via CD177+ Neutrophils. Front. Immunol. 2021, 12, 650864. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, H.; Jia, L.; Lai, D.; Jia, L.; Li, Q.; Guo, E.; Yang, F.; Zhang, B.; Luo, Q. Regulatory role of CD177+ neutrophils in BMP signaling pathway and its implications for inflammatory bowel disease, sepsis, and intestinal tumors. Transl. Oncol. 2025, 55, 102336. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Grieshaber-Bouyer, R.; Wang, J.; Schmider, A.B.; Wilson, Z.S.; Zeng, L.; Halyabar, O.; Godin, M.D.; Nguyen, H.N.; Levescot, A.; et al. CD177 modulates human neutrophil migration through activation-mediated integrin and chemoreceptor regulation. Blood 2017, 130, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Planell, N.; Masamunt, M.C.; Leal, R.F.; Rodriguez, L.; Esteller, M.; Lozano, J.J.; Ramirez, A.; Ayrizono, M.L.S.; Coy, C.S.R.; Alfaro, I.; et al. Usefulness of Transcriptional Blood Biomarkers as a Non-invasive Surrogate Marker of Mucosal Healing and Endoscopic Response in Ulcerative Colitis. J. Crohns Colitis 2017, 11, 1335–1346. [Google Scholar] [CrossRef] [PubMed]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [PubMed]
- Christoffersson, G.; Vagesjo, E.; Vandooren, J.; Liden, M.; Massena, S.; Reinert, R.B.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, G.; Xu, C.; Lu, H.; Zhang, C.; Pang, Z.; Liu, Z. Monocyte Chemotactic Protein 1-Induced Protein 1 Is Highly Expressed in Inflammatory Bowel Disease and Negatively Regulates Neutrophil Activities. Mediat. Inflamm. 2020, 2020, 8812020. [Google Scholar] [CrossRef] [PubMed]
- Kienle, K.; Glaser, K.M.; Eickhoff, S.; Mihlan, M.; Knopper, K.; Reategui, E.; Epple, M.W.; Gunzer, M.; Baumeister, R.; Tarrant, T.K.; et al. Neutrophils self-limit swarming to contain bacterial growth In Vivo. Science 2021, 372, eabe7729. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, S.; Ponzetta, A.; Rigatelli, A.; Carriero, R.; Puccio, S.; Supino, D.; Grieco, G.; Molisso, P.; Di Ceglie, I.; Scavello, F.; et al. Neutrophils Mediate Protection Against Colitis and Carcinogenesis by Controlling Bacterial Invasion and IL22 Production by gammadelta T Cells. Cancer Immunol. Res. 2024, 12, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, J.; Wang, Z.; Han, R.; Zhao, Y.; Lou, Y.; Wang, H. S100a10 deficiency in neutrophils aggravates ulcerative colitis in mice. Int. Immunopharmacol. 2024, 128, 111499. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wu, Y.; Lu, D.; Pang, J.; Hu, J.; Zhang, X.; Wang, Z.; Zhang, G.; Wang, J. Polyphenol-rich diet mediates interplay between macrophage-neutrophil and gut microbiota to alleviate intestinal inflammation. Cell Death Dis. 2023, 14, 656. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Chaaban, H.; Burge, K.; Eckert, J.; Keshari, R.S.; Silasi, R.; Lupu, C.; Warner, B.; Escobedo, M.; Caplan, M.; Lupu, F. Neutrophil extracellular trap inhibition increases inflammation, bacteraemia and mortality in murine necrotizing enterocolitis. J. Cell. Mol. Med. 2021, 25, 10814–10824. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef] [PubMed]
- Padhy, D.S.; Palit, P.; Ikbal, A.M.A.; Das, N.; Roy, D.K.; Banerjee, S. Selective inhibition of peptidyl-arginine deiminase (PAD): Can it control multiple inflammatory disorders as a promising therapeutic strategy? Inflammopharmacology 2023, 31, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, Y.; Masuta, Y.; Minaga, K.; Okai, N.; Hara, A.; Takada, R.; Masaki, S.; Kamata, K.; Honjo, H.; Yamashita, K.; et al. Reciprocal regulation of protein arginine deiminase 2 and 4 expression in the colonic mucosa of ulcerative colitis. J. Clin. Biochem. Nutr. 2024, 75, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Mei, Y.; Dong, W.; Wang, J.; Huang, F.; Wu, J. Evaluation of protein arginine deiminase-4 inhibitor in TNBS- induced colitis in mice. Int. Immunopharmacol. 2020, 84, 106583. [Google Scholar] [CrossRef] [PubMed]
- Witalison, E.E.; Cui, X.; Hofseth, A.B.; Subramanian, V.; Causey, C.P.; Thompson, P.R.; Hofseth, L.J. Inhibiting protein arginine deiminases has antioxidant consequences. J. Pharmacol. Exp. Ther. 2015, 353, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Torok, S.; Almasi, N.; Valkusz, Z.; Posa, A.; Varga, C.; Kupai, K. Investigation of H2S Donor Treatment on Neutrophil Extracellular Traps in Experimental Colitis. Int. J. Mol. Sci. 2021, 22, 12729. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Louttit, C.; Fry, C.; Najafabadi, A.H.; Han, K.; Nemzek, J.; Moon, J.J. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials 2020, 238, 119836. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, T.; Hermann, E.; Moller, S.; Klinger, M.; Solbach, W.; Laskay, T.; Behnen, M. Flavonoids and 5-aminosalicylic acid inhibit the formation of neutrophil extracellular traps. Mediat. Inflamm. 2013, 2013, 710239. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, N.; Gao, X.J.; Li, T.; Yan, Z.X.; Wang, P.P.; Wei, B.; Li, S.; Zhang, Z.J.; Li, S.L.; et al. Dextran sulfate sodium-induced colitis and ginseng intervention altered oral pharmacokinetics of cyclosporine A in rats. J. Ethnopharmacol. 2021, 265, 113251. [Google Scholar] [CrossRef] [PubMed]
- Song, E.M.; Oh, E.H.; Hwang, S.W.; Park, S.H.; Yang, D.H.; Byeon, J.S.; Myung, S.J.; Yang, S.K.; Ye, B.D. Comparison of outcomes of cyclosporine A and infliximab for steroid-refractory acute severe ulcerative colitis. J. Gastroenterol. Hepatol. 2021, 36, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guo, J.; Yang, M.; Han, C.; Zhang, M.; Chen, W.; Liu, Q.; Wang, J.; Cao, X. Cyclosporin A impairs dendritic cell migration by regulating chemokine receptor expression and inhibiting cyclooxygenase-2 expression. Blood 2004, 103, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Giaglis, S.; Hasler, P.; Hahn, S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS ONE 2014, 9, e97088. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.B.; Aliprantis, A.; Hu, B.; Glimcher, L.H. Calcineurin regulates innate antifungal immunity in neutrophils. J. Exp. Med. 2010, 207, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Lin, J.; Xu, C.; Sun, M.; Zuo, K.; Zhang, X.; Li, M.; Huang, H.; Li, Z.; Wu, W.; et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1alpha-glycolysis axis to alleviate severe ulcerative colitis. Clin. Transl. Med. 2021, 11, e334. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.; Khan, M.A.; Palaniyar, N. Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules 2019, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.E.; Sandor, N.; Karpati, E.; Jozsi, M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol. Immunol. 2016, 72, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Bayliss, C.; Bond, S.; Dowling, F.; Galea, J.; Jairath, V.; Lamb, C.; Probert, C.; Timperley-Preece, E.; Watson, A.; et al. Trial summary and protocol for a phase II randomised placebo-controlled double-blinded trial of Interleukin 1 blockade in Acute Severe Colitis: The IASO trial. BMJ Open 2019, 9, e023765. [Google Scholar] [CrossRef] [PubMed]
- Truyens, M.; Hoste, L.; Geldof, J.; Hoorens, A.; Haerynck, F.; Huis In ‘t Veld, D.; Lobaton, T. Successful treatment of ulcerative colitis with anakinra: A case report. Acta Gastroenterol. Belg. 2023, 86, 573–576. [Google Scholar] [CrossRef] [PubMed]
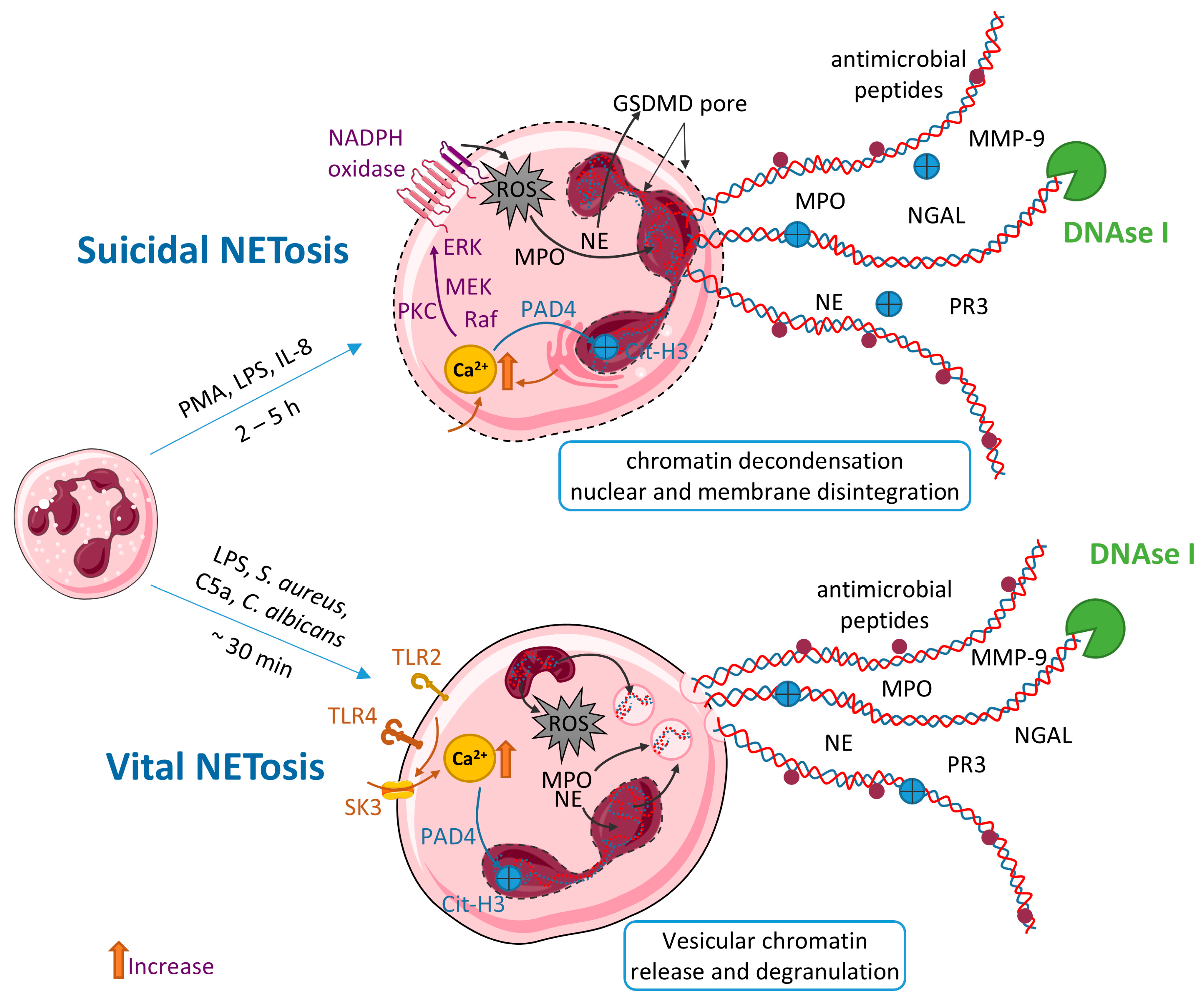
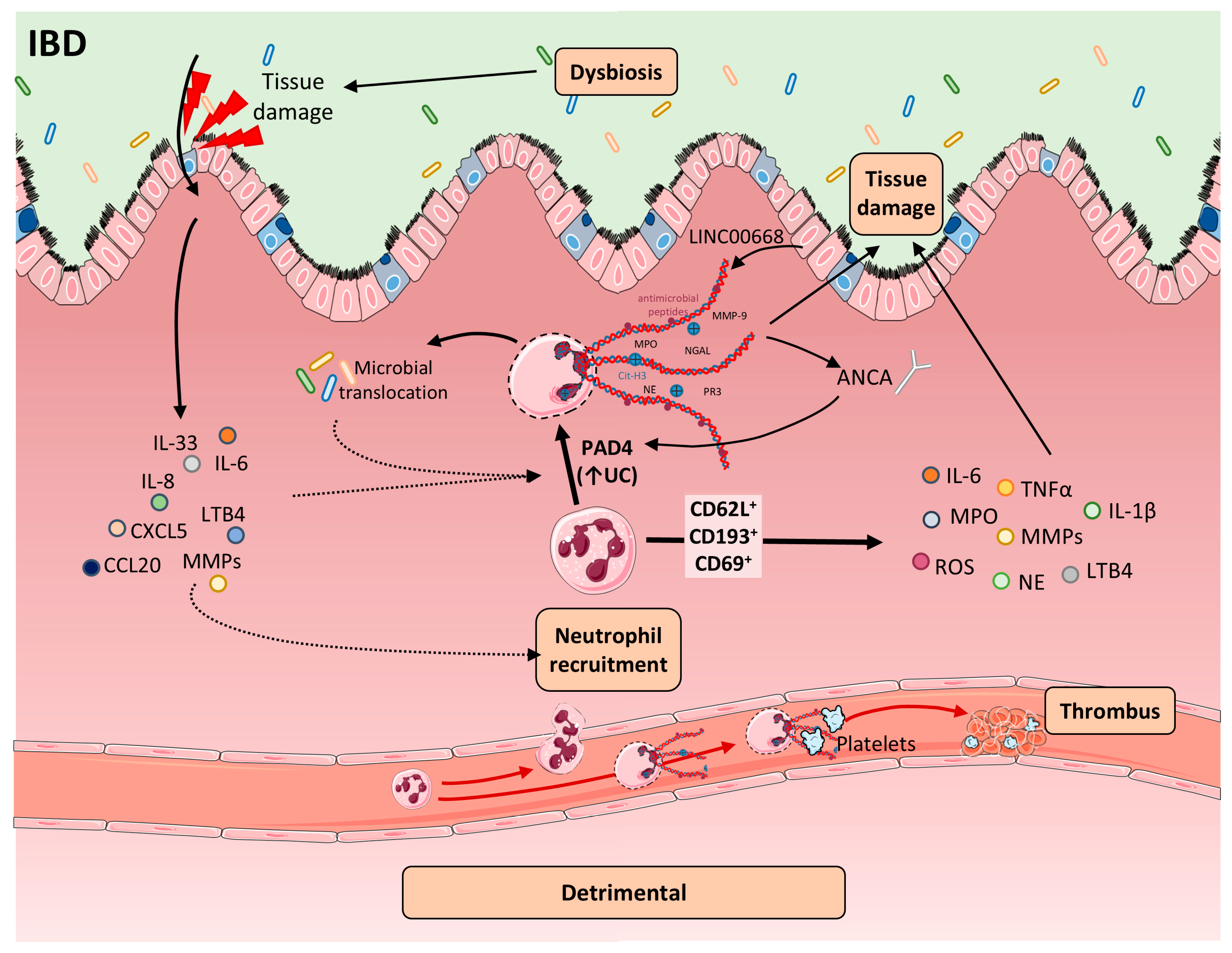
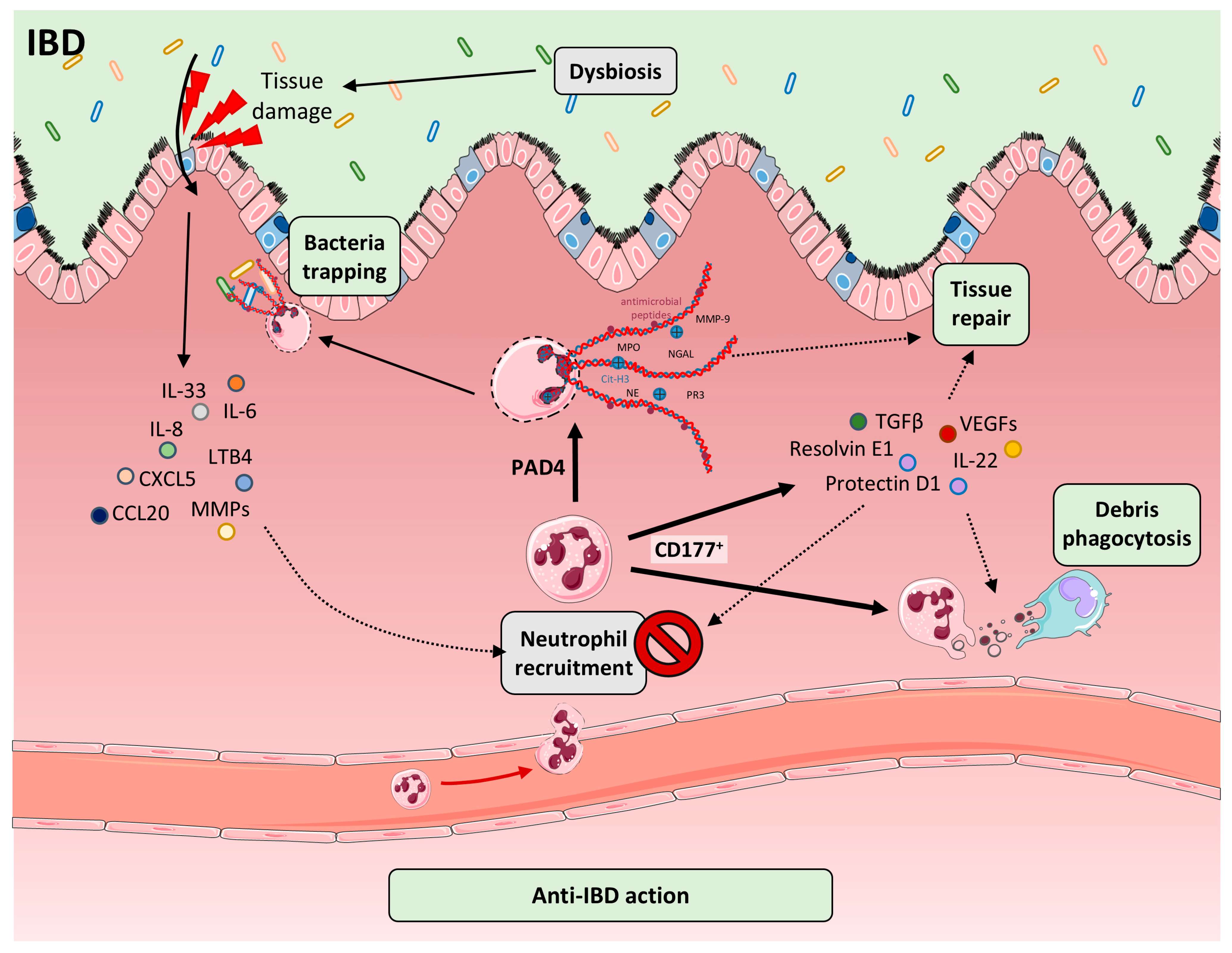
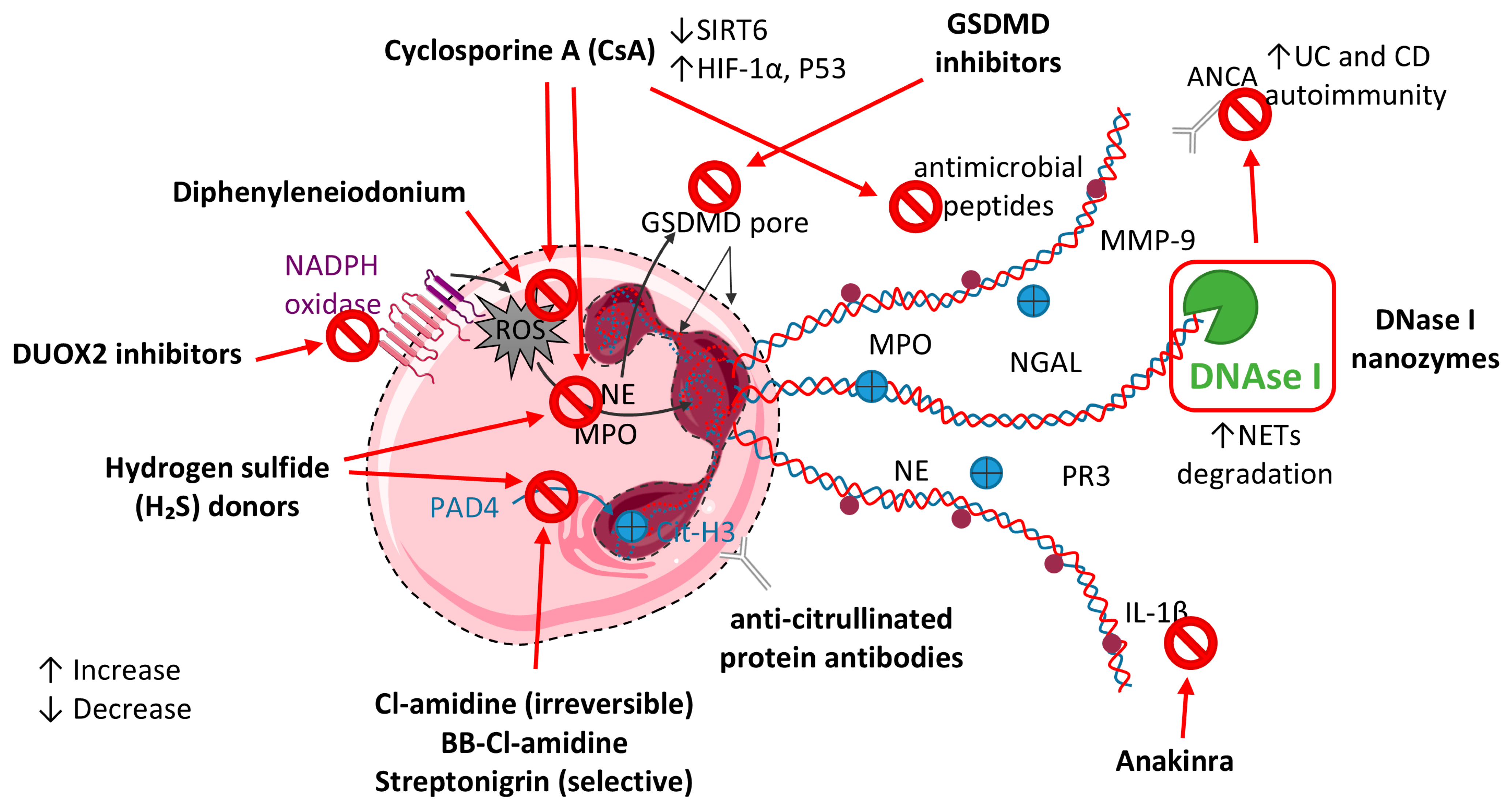
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega-Zapero, M.; Gomez-Bris, R.; Pascual-Laguna, I.; Saez, A.; Gonzalez-Granado, J.M. Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2025, 26, 7098. https://doi.org/10.3390/ijms26157098
Ortega-Zapero M, Gomez-Bris R, Pascual-Laguna I, Saez A, Gonzalez-Granado JM. Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2025; 26(15):7098. https://doi.org/10.3390/ijms26157098
Chicago/Turabian StyleOrtega-Zapero, Marina, Raquel Gomez-Bris, Ines Pascual-Laguna, Angela Saez, and Jose M. Gonzalez-Granado. 2025. "Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease" International Journal of Molecular Sciences 26, no. 15: 7098. https://doi.org/10.3390/ijms26157098
APA StyleOrtega-Zapero, M., Gomez-Bris, R., Pascual-Laguna, I., Saez, A., & Gonzalez-Granado, J. M. (2025). Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease. International Journal of Molecular Sciences, 26(15), 7098. https://doi.org/10.3390/ijms26157098