
Design, Synthesis, and Anticancer Evaluation of New Small-Molecule EGFR Inhibitors Targeting NSCLC and Breast Cancer





Abstract
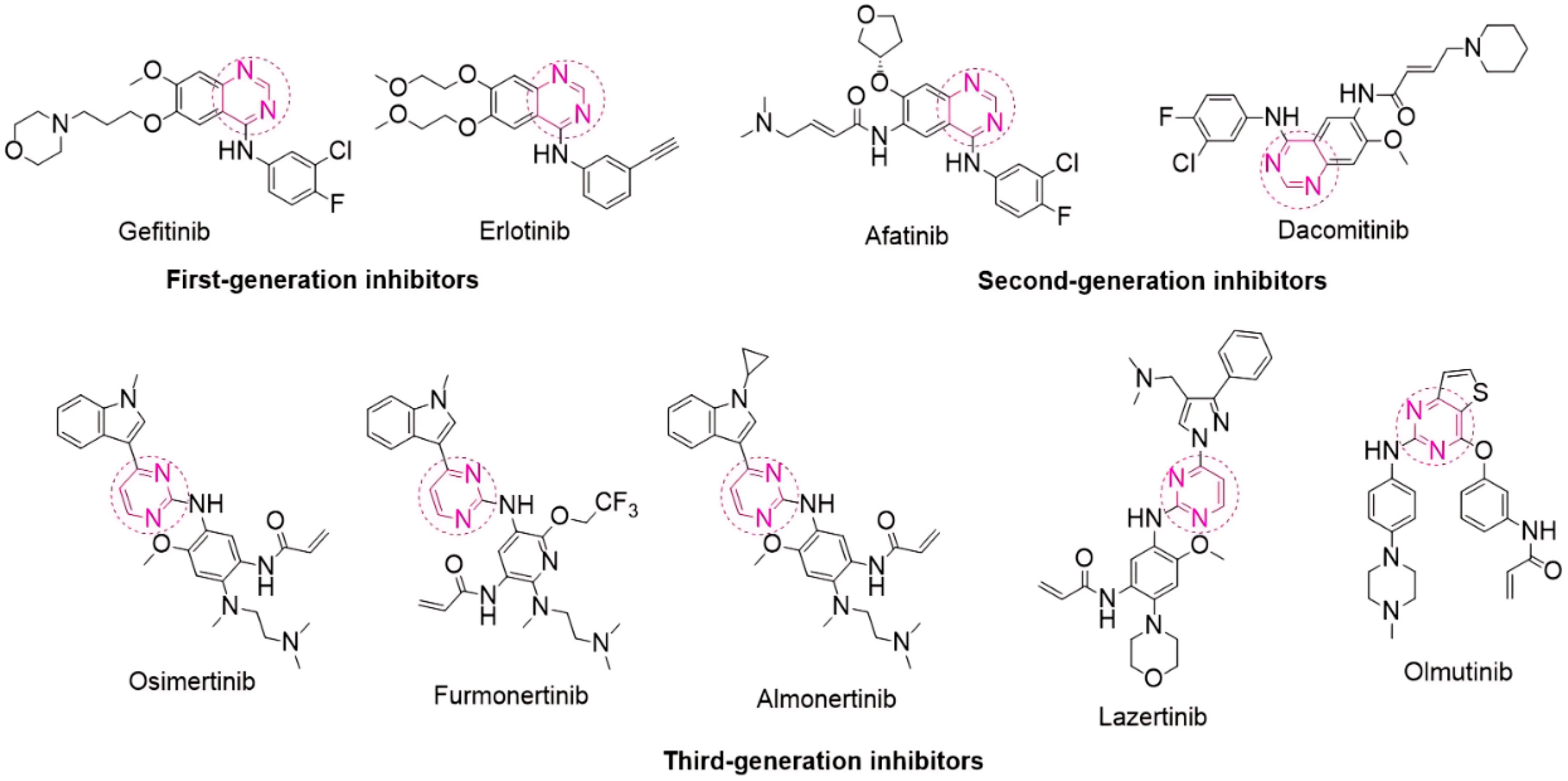
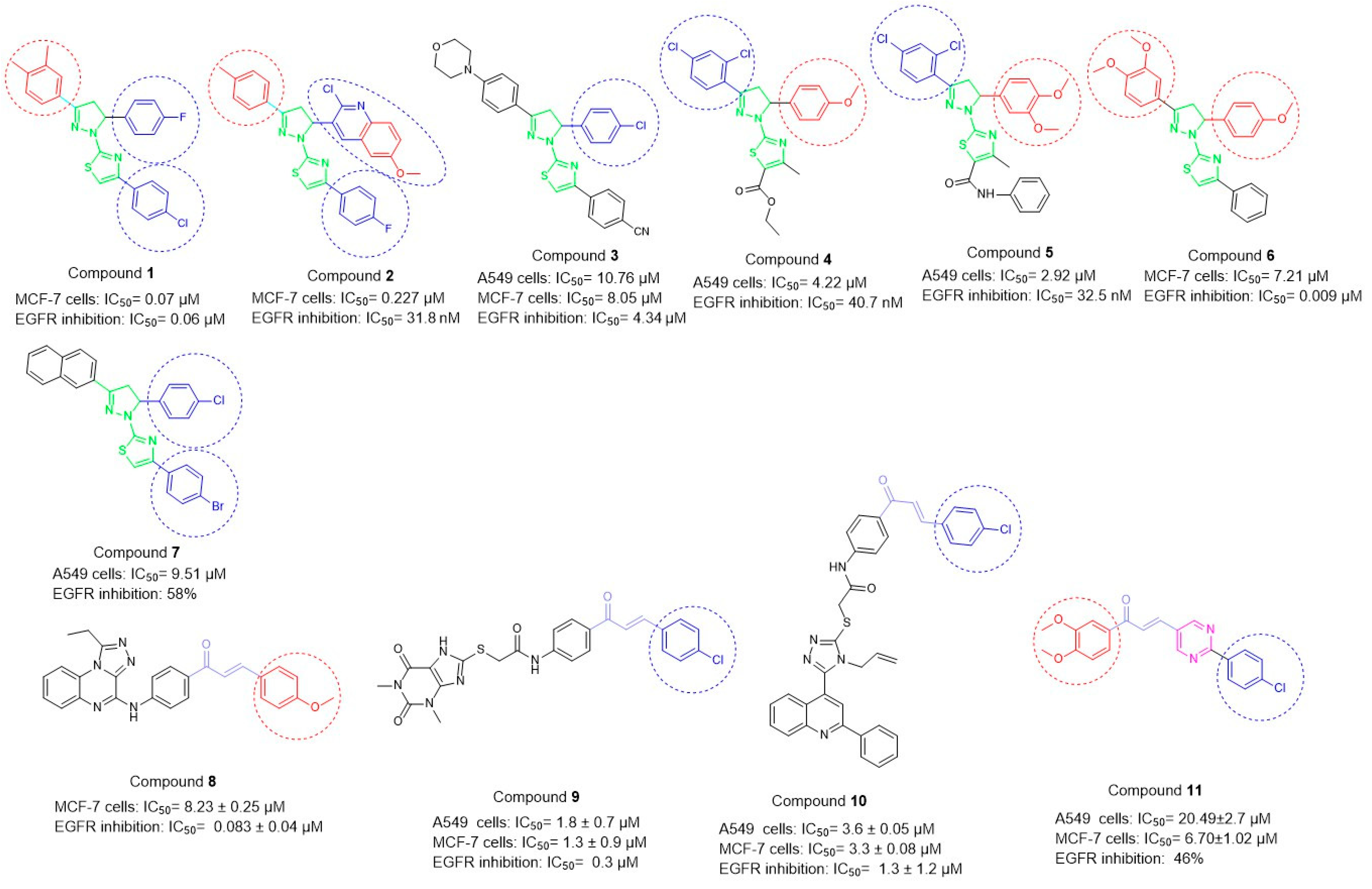
1. Introduction
2. Results
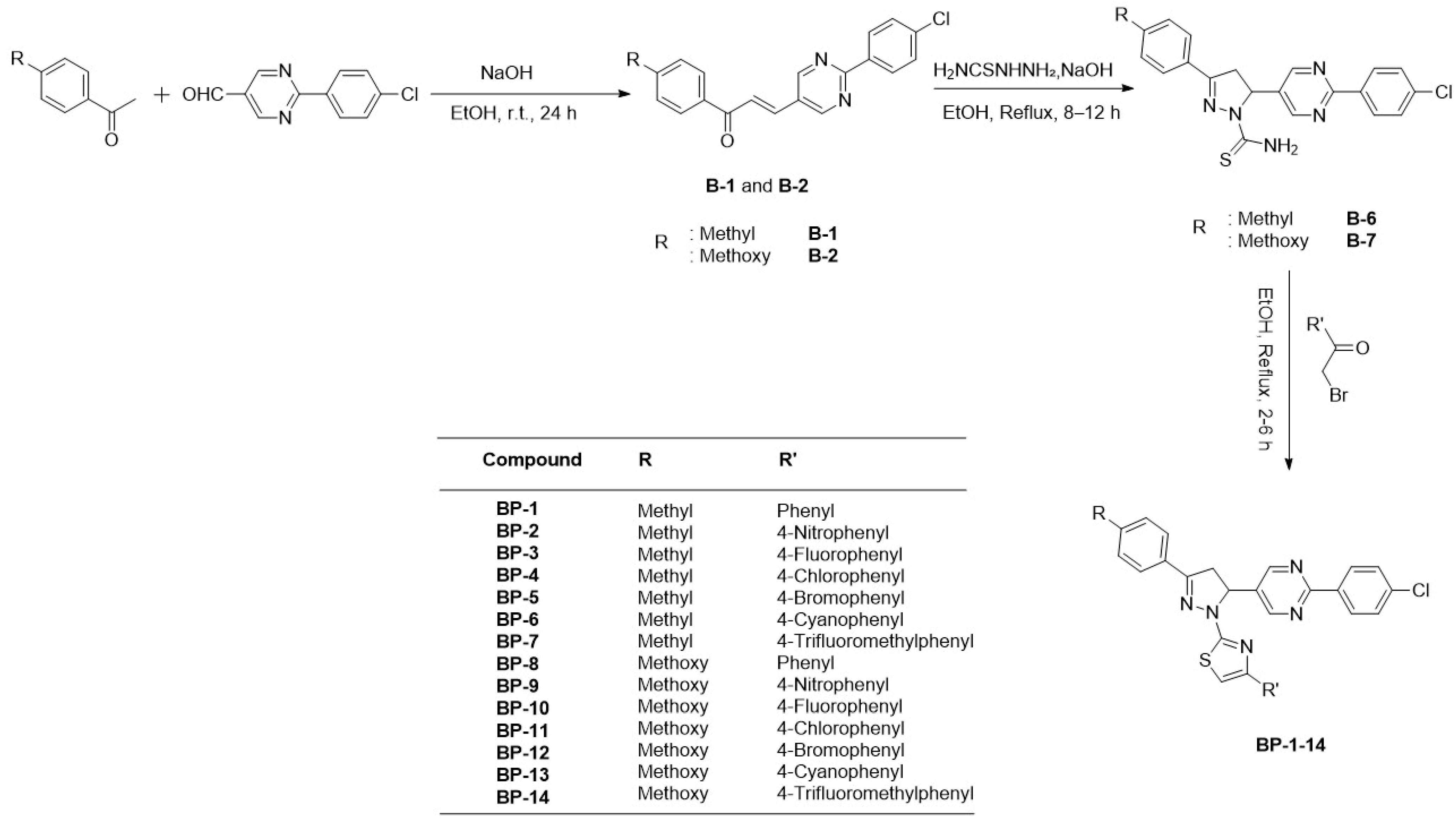
2.1. Chemistry
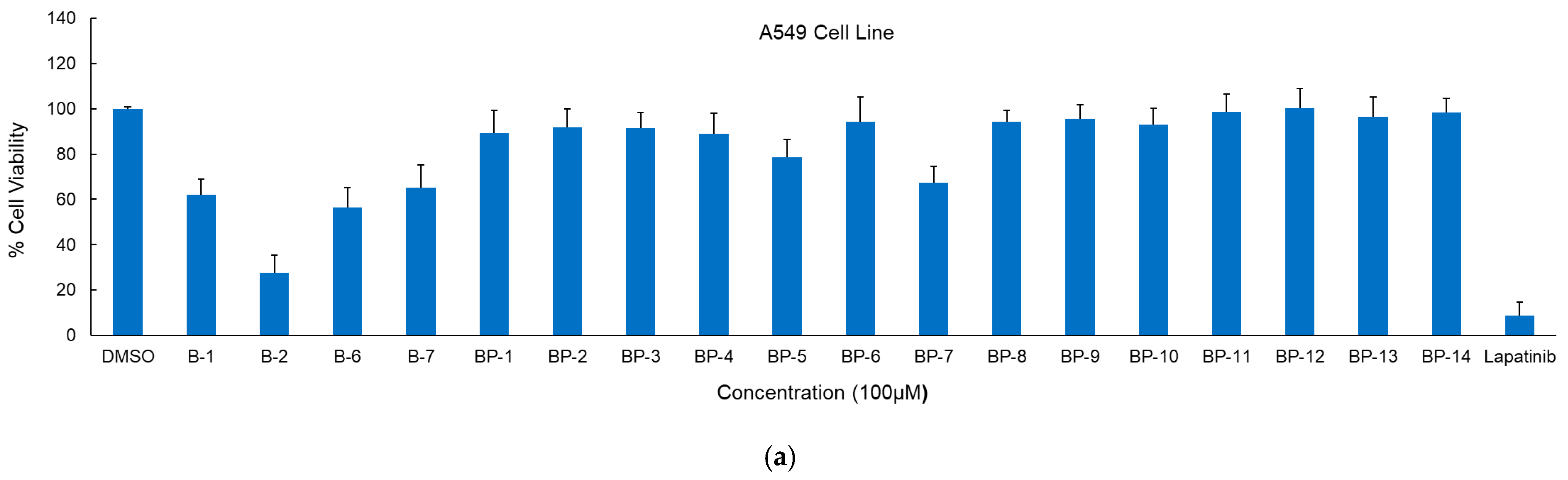
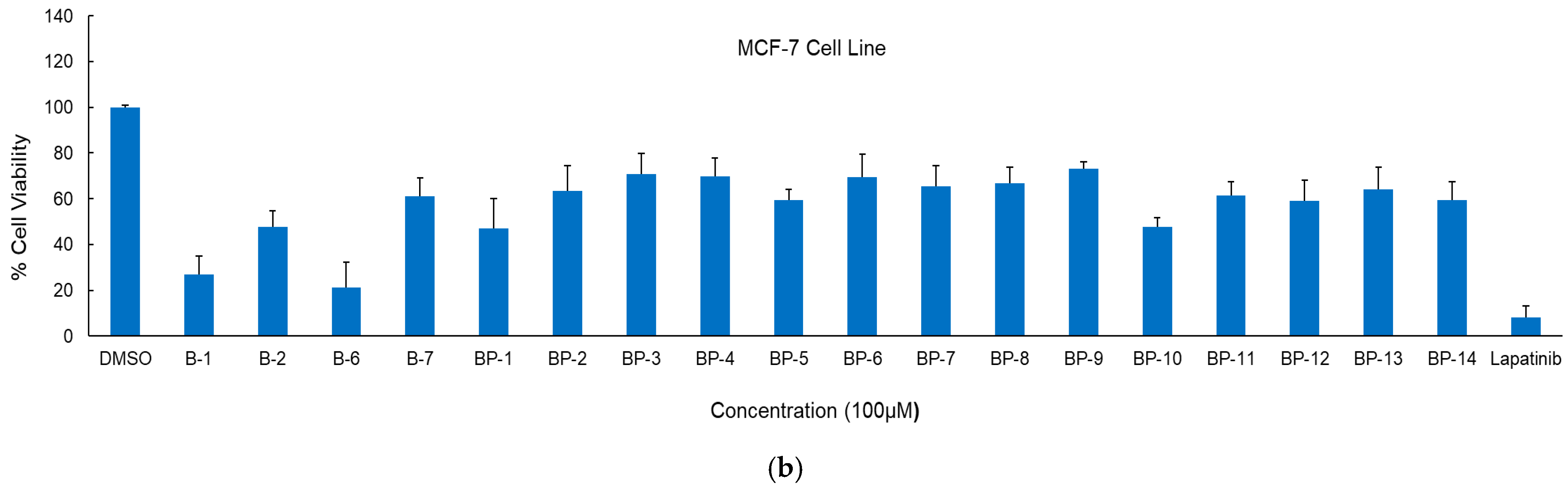
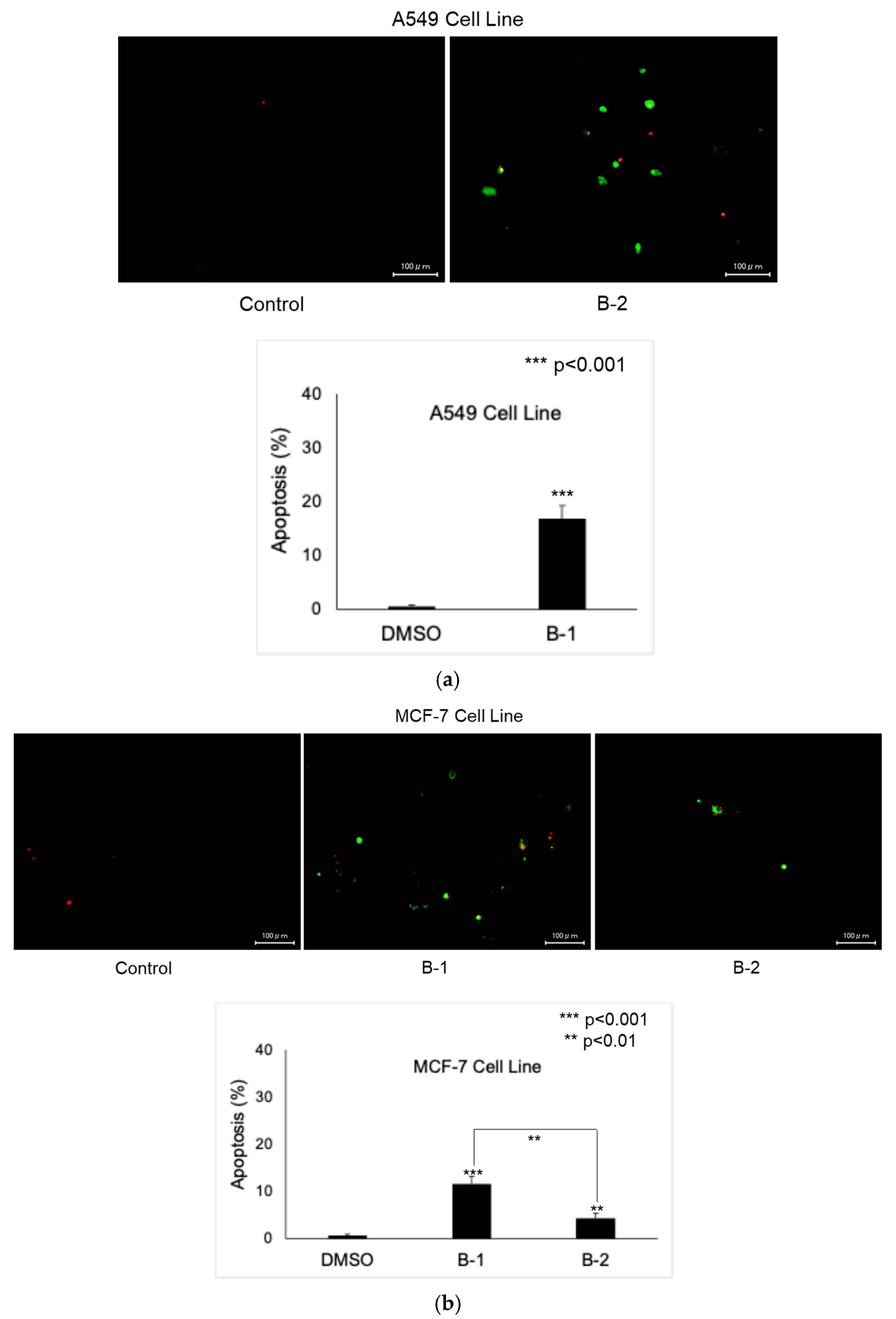
2.2. Anticancer Activity
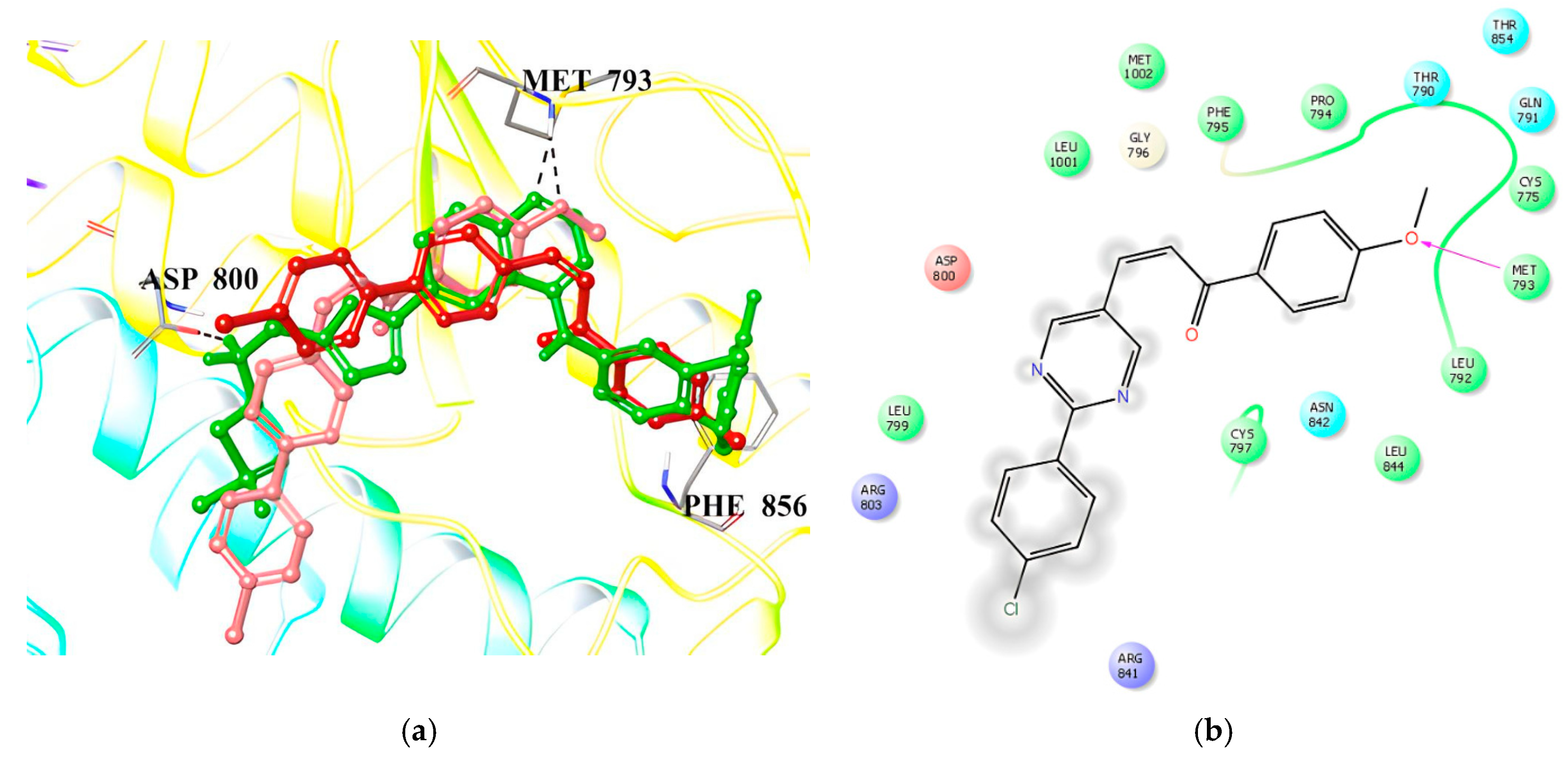
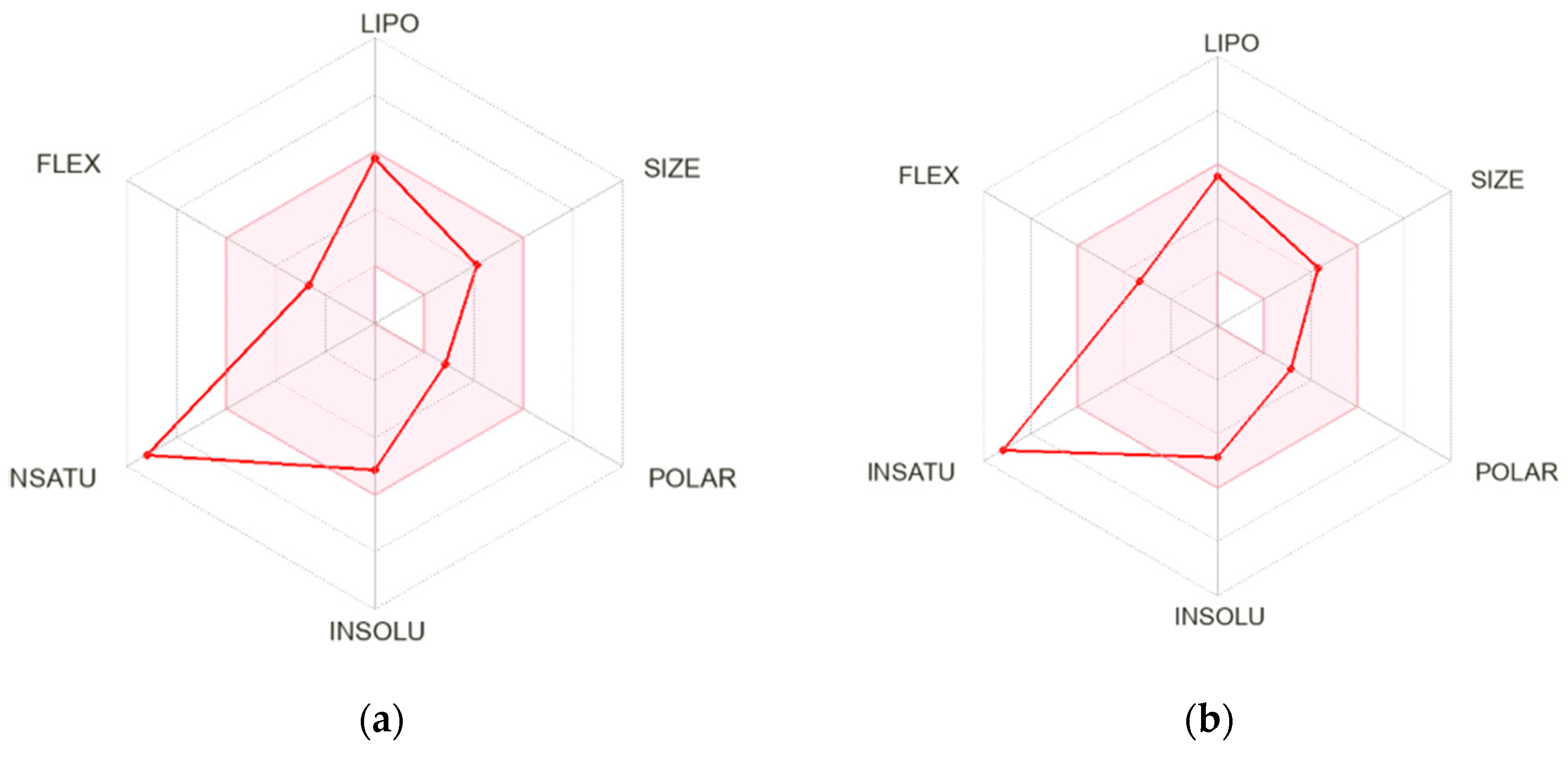
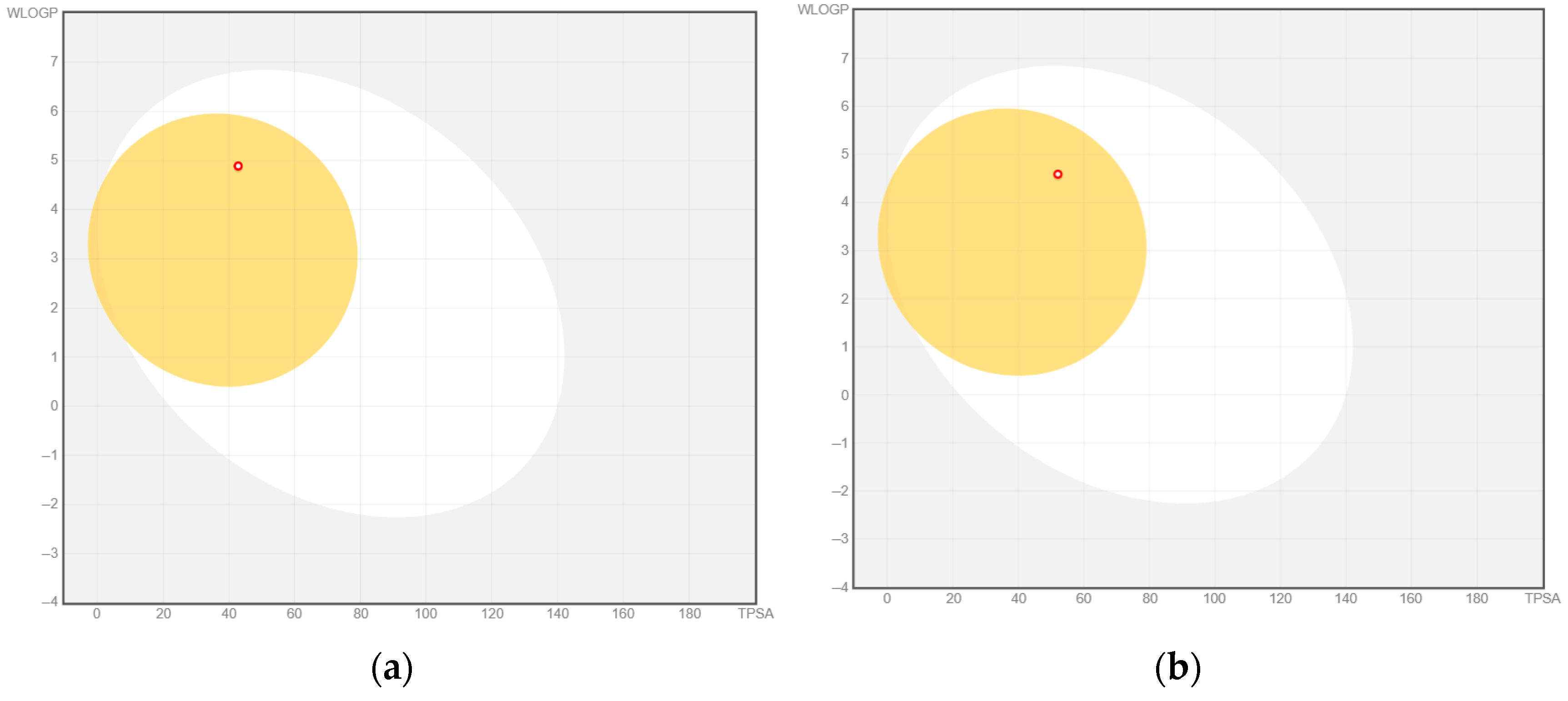
2.3. Molecular Docking and In Silico Pharmacokinetic Estimation
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Synthesis of (E)-3-(2-(4-Chlorophenyl)pyrimidin-5-yl)-1-(4-methylphenyl)-2-propen-1-one (B-1) and (E)-3-(2-(4-Chlorophenyl)pyrimidin-5-yl)-1-(4-methoxyphenyl)-2-propen-1-one (B-2)
4.1.2. Synthesis of 3-(4-Methylphenyl)-5-(2-(4-chlorophenyl)pyrimidin-5-yl)-1-thiocarbamoyl-2-pyrazoline (B-6) and 3-(4-methoxyphenyl)-5-(2-(4-chlorophenyl)pyrimidin-5-yl)-1-thiocarbamoyl-2- pyrazoline (B-7)
4.1.3. Synthesis of 1-(4-(Aryl)thiazol-2-yl)-3-(4-methyl/methoxyphenyl)-5-(2-(4-chlorophenyl)pyrimidin-5-yl)-2-pyrazoline derivatives (BP1–14)
4.2. Cytotoxicity
4.3. Apoptosis
4.4. EGFR Inhibition
4.5. In Silico Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castelo-Soccio, L.; Kim, H.; Gadina, M.; Schwartzberg, P.L.; Laurence, A.; O’Shea, J.J. Protein kinases: Drug targets for immunological disorders. Nat. Rev. Immunol. 2023, 23, 787–806. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Mursal, M.; Verma, G.; Hasan, S.M.; Khan, M.F. Targeting oncogenic kinases: Insights on FDA approved tyrosine kinase inhibitors. Eur. J. Pharmacol. 2024, 970, 176484. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Tigu, A.B.; Munteanu, R.; Moldovan, C.S.; Kegyes, D.; Onaciu, A.; Gulei, D.; Ghiaur, G.; Einsele, H.; Croce, C.M. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Sig. Transduct. Target Ther. 2024, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, E.A.; Kahler, S.; Tefay, J.; Joseph, S.R.; Simpson, F. Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review. Cells 2021, 10, 1206. [Google Scholar] [CrossRef] [PubMed]
- Shaban, N.; Kamashev, D.; Emelianova, A.; Buzdin, A. Targeted Inhibitors of EGFR: Structure, Biology, Biomarkers, and Clinical Applications. Cells 2023, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Hirsch, F.R.; Pirker, R.; Felip, E.; Valencia, C.; Smit, E.F. The role of anti-EGFR therapies in EGFR-TKI-resistant advanced non-small cell lung cancer. Cancer Treat. Rev. 2024, 122, 102664. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Perol, M.; Banna, G.L.; Parikh, K.; Addeo, A. Oncogenic alterations in advanced NSCLC: A molecular super-highway. Biomark. Res. 2024, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.Q.; Abughanimeh, O. Current management of uncommon EGFR mutations in non-small cell lung cancer. Curr. Probl. Cancer 2024, 49, 101064. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, W.; Liu, T.; Li, X.; Yu, Y.; Wu, J. Annual review of EGFR inhibitors in 2024. Eur. J. Med. Chem. 2025, 292, 117677. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. International Agency for Research on Cancer: Lyon, France, 2024. Available online: https://gco.iarc.who.int/today (accessed on 5 June 2025).
- Reymova, F.; Sever, B.; Topalan, E.; Sevimli-Gur, C.; Can, M.; Tuyun, A.F.; Başoğlu, F.; Ece, A.; Otsuka, M.; Fujita, M.; et al. Design, Synthesis, and Mechanistic Anticancer Evaluation of New Pyrimidine-Tethered Compounds. Pharmaceuticals 2025, 18, 270. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhong, C.; Wang, J.; Chen, J.; Zhou, T. Current status and breakthroughs in treating advanced non-small cell lung cancer with EGFR exon 20 insertion mutations. Front. Immunol. 2024, 15, 1399975. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, K.W.; Urul, D.A.; Ogboo, B.C.; Wittlinger, F.; Laufer, S.A.; Schaefer, E.M.; May, E.W.; Heppner, D.E. Pitfalls and Considerations in Determining the Potency and Mutant Selectivity of Covalent Epidermal Growth Factor Receptor Inhibitors. J. Med. Chem. 2024, 67, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Cui, H.; Wang, Y.; Yang, J.; Lin, C.; Shi, X.; Zou, Y.; Chen, J.; Jia, X.; Su, L. The advance of the third generation EGFR TKI in the treatment of non small cell lung cancer (Review). Oncol. Rep. 2024, 51, 16. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, H.; Diab, A.; Al Musaimi, O. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects. Int. J. Mol. Sci. 2024, 25, 10008. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gong, C.; Zhou, H.; Liu, J.; Xia, X.; Ha, W.; Jiang, Y.; Liu, Q.; Xiong, H. Kinase Inhibitors and Kinase-Targeted Cancer Therapies: Recent Advances and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 5489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.; Zhu, Y.; Dong, T.; Liu, Z. Understanding the treatment response and resistance to targeted therapies in non-small cell lung cancer: Clinical insights and perspectives. Front. Oncol. 2024, 14, 1387345. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Luo, J.; Zhang, D.; Lu, Y.; Liao, W.; Zhang, J. Innovative design and potential applications of covalent strategy in drug discovery. Eur. J. Med. Chem. 2025, 284, 117202. [Google Scholar] [CrossRef] [PubMed]
- Topalan, E.; Büyükgüngör, A.; Çiğdem, M.; Güra, S.; Sever, B.; Otsuka, M.; Fujita, M.; Demirci, H.; Ciftci, H. A Structural Insight Into Two Important ErbB Receptors (EGFR and HER2) and Their Relevance to Non-Small Cell Lung Cancer. Arch. Pharm. 2025, 58, e2400992. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Olmutinib: First Global Approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Abdelhameed, A.S. Investigation of the metabolic stability of olmutinib by validated LC-MS/MS: Quantification in human plasma. RSC Adv. 2018, 8, 40387–40394. [Google Scholar] [CrossRef] [PubMed]
- Tariq, A.; Shoaib, M.; Qu, L.; Shoukat, S.; Nan, X.; Song, J. Exploring 4th generation EGFR inhibitors: A review of clinical outcomes and structural binding insights. Eur. J. Pharmacol. 2025, 997, 177608. [Google Scholar] [CrossRef] [PubMed]
- Babak, M.V.; Zalutsky, M.R.; Balyasnikova, I.V. Heterogeneity and vascular permeability of breast cancer brain metastases. Cancer Lett. 2020, 489, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.R.; Kar, T.; Das, A. Epidermal growth factor receptor mutations in breast Cancer: Therapeutic challenges and way forward. Bioorg. Chem. 2025, 154, 108037. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Amorim, I.; Gärtner, F.; Vale, N. Understanding Breast cancer: From conventional therapies to repurposed drugs. Eur. J. Pharm. Sci. 2020, 151, 105401. [Google Scholar] [CrossRef] [PubMed]
- Sankarapandian, V.; Rajendran, R.L.; Miruka, C.O.; Sivamani, P.; Maran, B.A.V.; Krishnamoorthy, R.; Gangadaran, P.; Ahn, B.C. A review on tyrosine kinase inhibitors for targeted breast cancer therapy. Pathol. Res. Pract. 2024, 263, 155607. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.L.; Lam, T.C.; Lam, K.O.; Luk, M.Y.; Kai-Cheong, R.N.; Kwong, L.D. Local and systemic treatment for HER2-positive breast cancer with brain metastases: A comprehensive review. Ther. Adv. Med. Oncol. 2020, 12, 1758835920953729. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.; Oral, A.; Coskun, Y.; Renda, G.; Otsuka, M.; Fujita, M.; Sever, B. Assessment of the anticancer function of Coronilla orientalis MILLER through comprehensive in vitro and computational studies. Turk. J. Biochem. 2025, in press. [CrossRef]
- Li, X.; Zhao, L.; Chen, C.; Nie, J.; Jiao, B. Can EGFR be a therapeutic target in breast cancer? Biochim. Biophys. Acta Rev. Cancer. 2022, 1877, 188789. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.M.; Joy, A.J.; Manjunathaiah, R.N.; Justin, A.; Kumar, B.R.P. EGFR as therapeutic target to develop new generation tyrosine kinase inhibitors against breast cancer: A critical review. Results Chem. 2024, 7, 101490. [Google Scholar] [CrossRef]
- Lv, P.C.; Li, D.D.; Li, Q.S.; Lu, X.; Xiao, Z.P.; Zhu, H.L. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives as EGFR TK inhibitors and potential anticancer agents. Bioorg. Med. Chem. Lett. 2011, 21, 5374–5377. [Google Scholar] [CrossRef] [PubMed]
- George, R.F.; Samir, E.M.; Abdelhamed, M.N.; Abdel-Aziz, H.A.; Abbas, S.E. Synthesis and anti-proliferative activity of some new quinoline based 4,5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem. 2019, 83, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Sever, B.; Altıntop, M.D.; Radwan, M.O.; Özdemir, A.; Otsuka, M.; Fujita, M.; Ciftci, H.I. Design, synthesis and biological evaluation of a new series of thiazolyl-pyrazolines as dual EGFR and HER2 inhibitors. Eur. J. Med. Chem. 2019, 182, 111648. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, E.A.; Abd El-Hafeez, A.A.; Eldehna, W.M.; El Hassab, M.A.; Marzouk, H.M.M.; Elaasser, M.M.; Abou Taleb, N.A.; Amin, K.M.; Abdel-Aziz, H.A.; Ghosh, P.; et al. Discovery of novel thiazolyl-pyrazolines as dual EGFR and VEGFR-2 inhibitors endowed with in vitro antitumor activity towards non-small lung cancer. J. Enzyme Inhib. Med. Chem. 2022, 37, 2265–2282. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, M.M.; Mahmoud, K.; Nafie, M.S.; Noor, A.O.; Hareeri, R.H.; Salama, I.; Kishk, S.M. Rational Design, Synthesis and Biological Evaluation of Novel Pyrazoline-Based Antiproliferative Agents in MCF-7 Cancer Cells. Pharmaceuticals 2022, 15, 1245. [Google Scholar] [CrossRef] [PubMed]
- Çiftçi, H.; Otsuka, M.; Fujita, M.; Sever, B. New naphthalene-linked pyrazoline-thiazole hybrids as prominent antilung and antibreast cancer inhibitors. Turk. J. Chem. 2024, 48, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo [4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Youssif, B.G.M.; Tateishi, H.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg. Chem. 2021, 106, 104510. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Odintsov, I.; Sholl, L.M. Prognostic and predictive biomarkers in non-small cell lung carcinoma. Pathology 2024, 56, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2016-2; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Schrödinger. Schrödinger Release 2016-2: QikProp; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- ADMETlab 3.0. Available online: https://admetlab3.scbdd.com (accessed on 6 June 2025).
- SwissADME. Available online: http://www.swissadme.ch (accessed on 7 June 2025).
- Bayrak, N.; Ciftci, H.I.; Yıldız, M.; Yıldırım, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Structure based design, synthesis, and evaluation of anti-CML activity of the quinolinequinones as LY83583 analogs. Chem. Biol. Interact. 2021, 345, 109555. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.; Sever, B.; Ocak, F.; Bayrak, N.; Yıldız, M.; Yıldırım, H.; DeMirci, H.; Tateishi, H.; Otsuka, M.; Fujita, M.; et al. In Vitro and In Silico Study of Analogs of Plant Product Plastoquinone to Be Effective in Colorectal Cancer Treatment. Molecules 2022, 27, 693. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 Values (μM) | SI * | |||
|---|---|---|---|---|---|
| A549 Cells | MCF-7 Cells | Jurkat Cells | PBMCs | ||
| B-1 | >100 | 6.10 ± 1.26 | 2.12 ± 0.48 | 121.02 ± 14.75 | 57.08 |
| B-2 | 2.14 ± 0.83 | 8.91 ± 1.38 | 2.96 ± 0.44 | 68.67 ± 9.82 | 23.20 |
| B-6 | >100 | 6.52 ± 0.97 | 15.06 ± 2.85 | 112.73 ± 15.07 | 7.49 |
| BP-1 | >100 | 91.58 ± 10.20 | >100 | ||
| BP-10 | >100 | 76.04 ± 9.91 | >100 | ||
| Lapatinib | 18.21 ± 3.25 | 9.71 ± 1.12 | 1.43 ± 0.35 | 11.04 ± 2.47 | 7.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sever, B.; Otsuka, M.; Fujita, M.; Ciftci, H. Design, Synthesis, and Anticancer Evaluation of New Small-Molecule EGFR Inhibitors Targeting NSCLC and Breast Cancer. Int. J. Mol. Sci. 2025, 26, 7065. https://doi.org/10.3390/ijms26157065
Sever B, Otsuka M, Fujita M, Ciftci H. Design, Synthesis, and Anticancer Evaluation of New Small-Molecule EGFR Inhibitors Targeting NSCLC and Breast Cancer. International Journal of Molecular Sciences. 2025; 26(15):7065. https://doi.org/10.3390/ijms26157065
Chicago/Turabian StyleSever, Belgin, Masami Otsuka, Mikako Fujita, and Halilibrahim Ciftci. 2025. "Design, Synthesis, and Anticancer Evaluation of New Small-Molecule EGFR Inhibitors Targeting NSCLC and Breast Cancer" International Journal of Molecular Sciences 26, no. 15: 7065. https://doi.org/10.3390/ijms26157065
APA StyleSever, B., Otsuka, M., Fujita, M., & Ciftci, H. (2025). Design, Synthesis, and Anticancer Evaluation of New Small-Molecule EGFR Inhibitors Targeting NSCLC and Breast Cancer. International Journal of Molecular Sciences, 26(15), 7065. https://doi.org/10.3390/ijms26157065