
Chronic Morphine Treatment Leads to a Global DNA Hypomethylation via Active and Passive Demethylation Mechanisms in mESCs

 , , , and
, , , and
Abstract

1. Introduction
2. Results
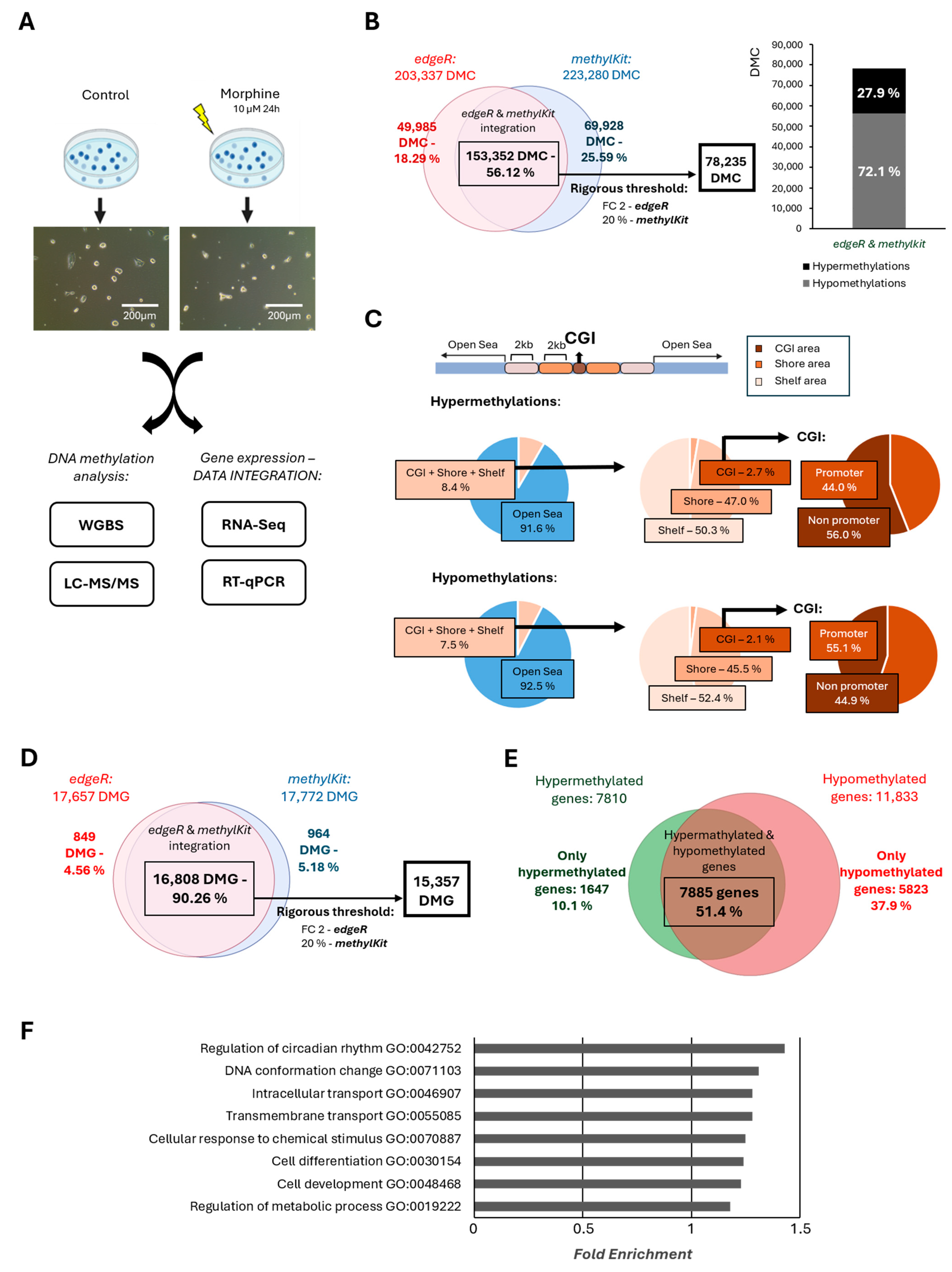
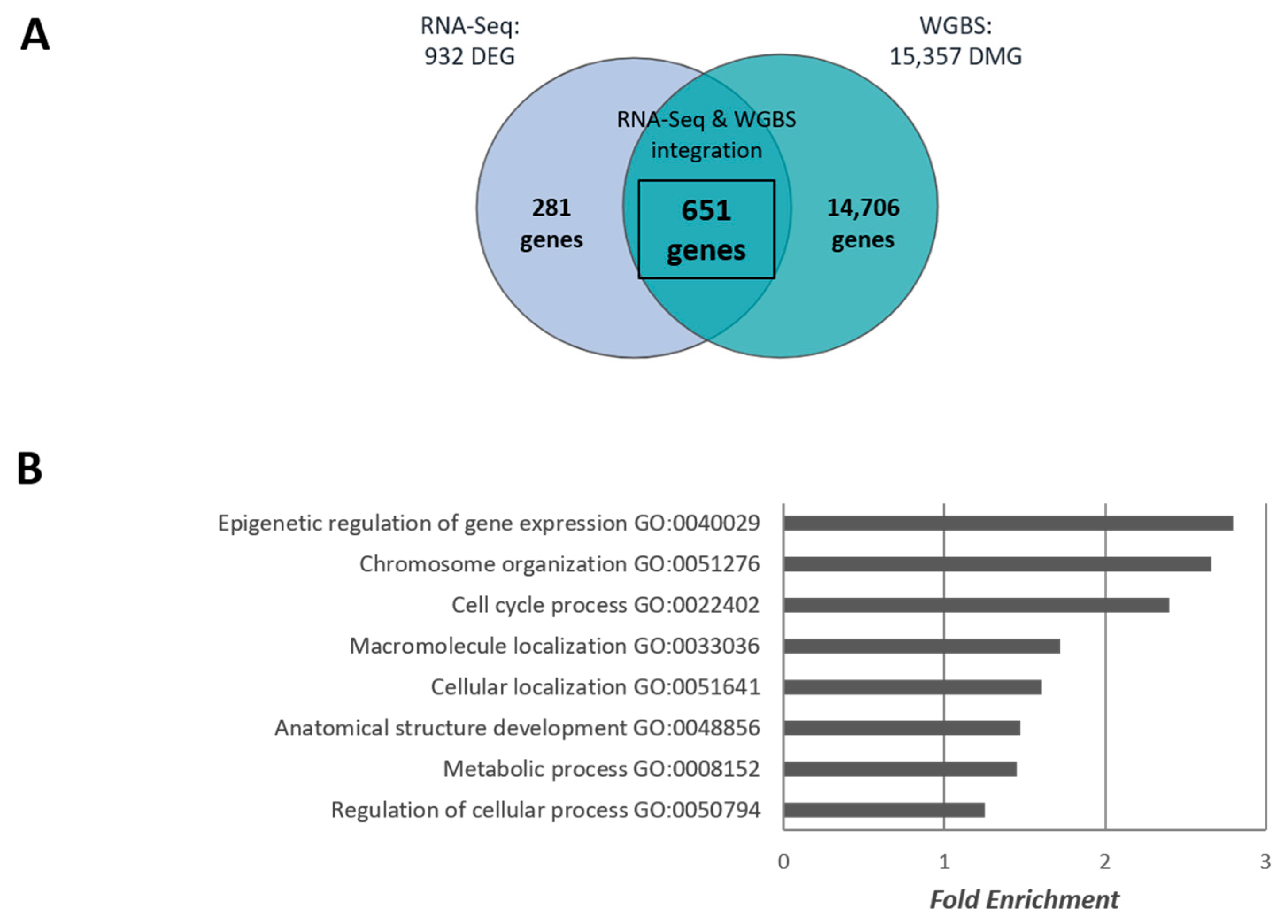
2.1. Effect of Chronic Morphine Treatment on DNA Methylation in mESCs by Whole Genome Bisulphite Sequencing (WGBS)
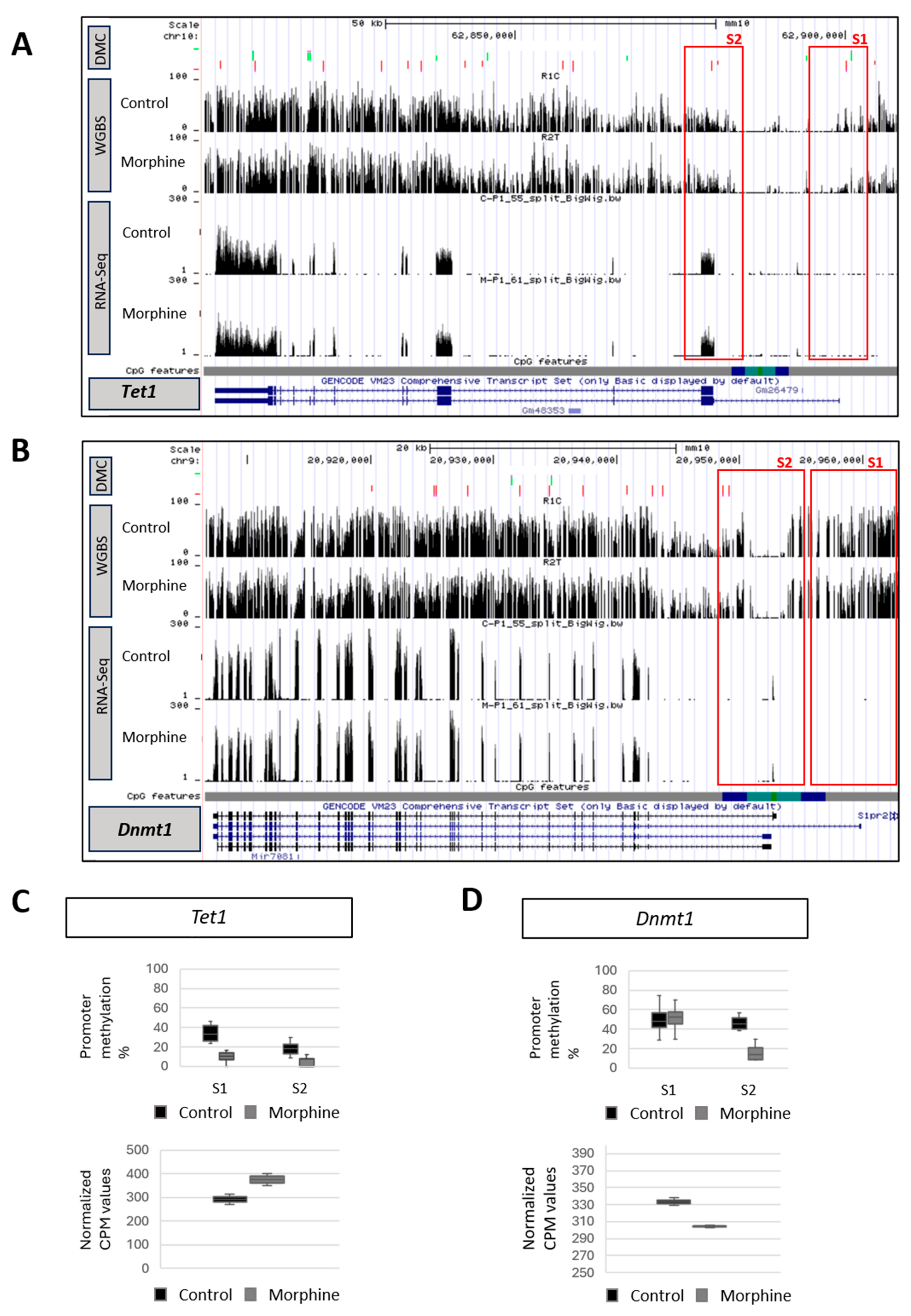
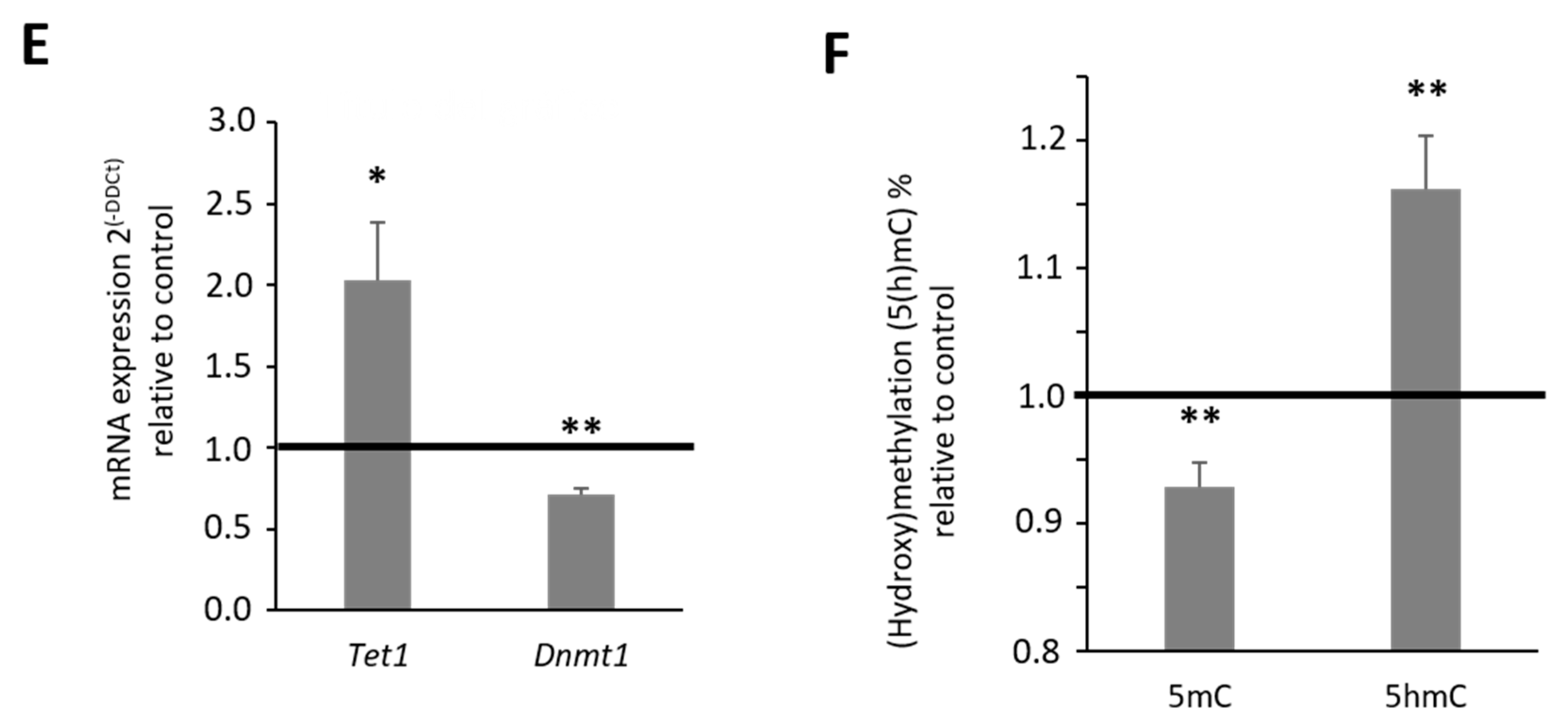
2.2. Effect of Chronic Morphine Treatment on DNA Methylation Machinery
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Cell Lysates, DNA Extraction and Quality Measurement
4.3. DNA Methylation Analysis by WGBS
4.4. Bioinformatics Analyses of WGBS’s Data, and WGBS and RNA-Seq Base Data Integrative Analyses
4.5. LC-MS/MS. Mass Spectrometry-Based Quantification of DNA
4.6. Real-Time PCR (RT-qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5hmC | Hydroxymethylcytosine |
| 5mC | Methylcytosine |
| cDNA | Complementary DNA |
| CGI | CpG Island |
| CPM | Counts Per Million |
| CT | Cycle quantification value |
| ddCT | 2(-Delta Delta C(T)) method |
| DEG | Differentially Expressed Gene |
| DMC | Differentially Methylated Cytosine |
| DMG | Differentially Methylated Gene |
| DMR | Differentially Methylated Region |
| DNA | Deoxyribonucleic acid |
| DNMT1/3A/3B/3L | DNA methyltransferase 1/3A/3B/3L |
| ESC | Embryonic Stem Cell |
| FDR | False Discovery Rate |
| GAPDH | Glyceraldehyde-3 phosphate dehydrogenase |
| GFP | Green Fluorescent Protein |
| GO | Gene Ontology |
| H3K27me3 | Trimethylation of lysine 27 on histone 3 protein subunit |
| ICR | Imprinting Control Region |
| KSR | KnockOut Serum Replacement |
| LC-MS/MS | Liquid Chromatography with tandem mass spectrometry |
| LIF | Leukemia Inhibitor Factor |
| mESC | mouse Embryonic Stem Cell |
| mRNA | messenger RNA |
| MS/MS | mass spectrometry |
| OCT4 (POU5F1) | Octamer-binding transcription factor 4 (POU Class 5 Homeobox 1) |
| PCA | Principal Component Analysis |
| PCR | Polymerase Chain Reaction |
| PCX | Pyruvate carboxylase |
| PE | Paired End |
| PSC | Pluripotent Stem Cells |
| RNA | Ribonucleic acid |
| RNA-Seq | RNA-Sequencing |
| RT-qPCR | Real Time Quantitative Polymerase Chain Reaction |
| TET1/2/3 | Ten eleven translocation 1/2/3 |
| TSS | Transcription start site |
| UCSC | University of California Santa Cruz |
| WGBS | Whole Genome Bisulfite Sequencing |
References
- de Gonzalo-Calvo, D.; Iglesias-Gutiérrez, E.; Llorente-Cortés, V. Epigenetic Biomarkers and Cardiovascular Disease: Circulating MicroRNAs. Rev. Española Cardiol. (Engl. Ed.) 2017, 70, 763–769. [Google Scholar] [CrossRef]
- Moosavi, A.; Motevalizadeh Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.B.; Hwang, K.A.; Choi, K.C. Prenatal Toxicity of the Environmental Pollutants on Neuronal and Cardiac Development Derived from Embryonic Stem Cells. Reprod. Toxicol. 2019, 90, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Matthes, H.W.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dollé, P.; et al. Loss of Morphine-Induced Analgesia, Reward Effect and Withdrawal Symptoms in Mice Lacking the Mu-Opioid-Receptor Gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal Structure of the μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T. Opioid Therapy and Management of Side Effects Associated with Opioids. Gan Kagaku Ryoho 2017, 44, 294–297. [Google Scholar]
- Kazemi, M.; Sahraei, H.; Azarnia, M.; Dehghani, L.; Bahadoran, H.; Tekieh, E. The Effect of Morphine Consumption on Plasma Corticosteron Concentration and Placenta Development in Pregnant Rats. Iran. J. Reprod. Med. 2011, 9, 71–76. [Google Scholar] [PubMed]
- Levitt, P. Prenatal Effects of Drugs of Abuse on Brain Development. Drug Alcohol. Depend. 1998, 51, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.S.; Rönnbäck, L. Effects of Prenatal Morphine Treatment of Rats on Mortality, Bodyweight and Analgesic Response in the Offspring. Drug Alcohol. Depend. 1989, 24, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Niknam, N.A.; Azarnia, M.; Bahadoran, H.; Kazemi, M.; Tekieh, E.; Ranjbaran, M.; Sahraei, H. Evaluating the Effects of Oral Morphine on Embryonic Development of Cerebellum in Wistar Rats. Basic Clin. Neurosci. 2013, 4, 130–135. [Google Scholar] [PubMed]
- Byrnes, J.J.; Babb, J.A.; Scanlan, V.F.; Byrnes, E.M. Adolescent Opioid Exposure in Female Rats: Transgenerational Effects on Morphine Analgesia and Anxiety-like Behavior in Adult Offspring. Behav. Brain Res. 2011, 218, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Gapp, K.; Jawaid, A.; Sarkies, P.; Bohacek, J.; Pelczar, P.; Prados, J.; Farinelli, L.; Miska, E.; Mansuy, I.M. Implication of Sperm RNAs in Transgenerational Inheritance of the Effects of Early Trauma in Mice. Nat. Neurosci. 2014, 17, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Subirán, N.; Casis, L.; Irazusta, J. Regulation of Male Fertility by the Opioid System. Mol. Med. 2011, 17, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Correa, L.O.; Jordan, M.S.; Carty, S.A. DNA Methylation in T-Cell Development and Differentiation. Crit. Rev. Immunol. 2020, 40, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, Y. Pluripotent Stem Cells in the Research for Extraembryonic Cell Differentiation. Dev. Growth Differ. 2021, 63, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Howell, C.Y.; Steptoe, A.L.; Miller, M.W.; Chaillet, J.R. Cis-Acting Signal for Inheritance of Imprinted DNA Methylation Patterns in the Preimplantation Mouse Embryo. Mol. Cell. Biol. 1998, 18, 4149–4156. [Google Scholar] [CrossRef] [PubMed]
- Howell, C.Y.; Bestor, T.H.; Ding, F.; Latham, K.E.; Mertineit, C., II; Trasler, J.M., II; Chaillet, J.R. Genomic Imprinting Disrupted by a Maternal Effect Mutation in the Dnmt1 Gene. Cell 2001, 104, 829–838. [Google Scholar] [CrossRef] [PubMed]
- McGraw, S.; Oakes, C.C.; Martel, J.; Cirio, M.C.; de Zeeuw, P.; Mak, W.; Plass, C.; Bartolomei, M.S.; Chaillet, J.R.; Trasler, J.M. Loss of DNMT1o Disrupts Imprinted X Chromosome Inactivation and Accentuates Placental Defects in Females. PLoS Genet. 2013, 9, e1003873. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted Mutation of the DNA Methyltransferase Gene Results in Embryonic Lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Shen, L.; Dai, Q.; He, C.; Zhang, Y. Generation and Replication-Dependent Dilution of 5fC and 5caC during Mouse Preimplantation Development. Cell Res. 2011, 21, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, T. Tet Family of 5-Methylcytosine Dioxygenases in Mammalian Development. J. Hum. Genet. 2013, 58, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Bagci, H.; Fisher, A.G. Dna Demethylation in Pluripotency and Reprogramming: The Role of Tet Proteins and Cell Division. Cell Stem Cell 2013, 13, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, J.; Chen, H.; Wang, F.; Li, Y.; Wang, H.; Qu, T. Mesenchymal Stem Cells from Bone Marrow Attenuated the Chronic Morphine-Induced CAMP Accumulation In Vitro. Neurosci. Lett. 2019, 698, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Cain, J.A.; Montibus, B.; Oakey, R.J. Intragenic CpG Islands and Their Impact on Gene Regulation. Front. Cell Dev. Biol. 2022, 10, 832348. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG Islands and the Regulation of Transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. CpG Islands—“A Rough Guide”. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, Silencing Potential and Evolutionary Impact of Promoter DNA Methylation in the Human Genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Muñoa-Hoyos, I.; Halsall, J.A.; Araolaza, M.; Ward, C.; Garcia, I.; Urizar-Arenaza, I.; Gianzo, M.; Garcia, P.; Turner, B.; Subirán, N. Morphine Leads to Global Genome Changes in H3K27me3 Levels via a Polycomb Repressive Complex 2 (PRC2) Self-Regulatory Mechanism in MESCs. Clin. Epigenetics 2020, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Kareta, M.S.; Botello, Z.M.; Ennis, J.J.; Chou, C.; Chédin, F. Reconstitution and Mechanism of the Stimulation of de Novo Methylation by Human DNMT3L. J. Biol. Chem. 2006, 281, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Guenatri, M.; Duffié, R.; Iranzo, J.; Fauque, P.; Bourc’his, D. Plasticity in Dnmt3L-Dependent and -Independent Modes of de Novo Methylation in the Developing Mouse Embryo. Development 2013, 140, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Gepstein, L. Derivation and Potential Applications of Human Embryonic Stem Cells. Circ. Res. 2002, 91, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Dvash, T.; Benvenisty, N. Human Embryonic Stem Cells as a Model for Early Human Development. Best Pract. Res. Clin. Obstet. Gynaecol. 2004, 18, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Dvash, T.; Ben-Yosef, D.; Eiges, R. Human Embryonic Stem Cells as a Powerful Tool for Studying Human Embryogenesis. Pediatr. Res. 2006, 60, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhao, M.; Li, M.; Song, T.; Zhang, M.; Quan, L.; Li, S.; Sun, Z.S. Reversal of Cocaine-Conditioned Place Preference through Methyl Supplementation in Mice: Altering Global DNA Methylation in the Prefrontal Cortex. PLoS ONE 2012, 7, e33435. [Google Scholar] [CrossRef] [PubMed]
- Novikova, S.I.; He, F.; Bai, J.; Cutrufello, N.J.; Lidow, M.S.; Undieh, A.S. Maternal Cocaine Administration in Mice Alters DNA Methylation and Gene Expression in Hippocampal Neurons of Neonatal and Prepubertal Offspring. PLoS ONE 2008, 3, e1919. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Szutorisz, H.; Garg, P.; Martin, Q.; Landry, J.A.; Sharp, A.J.; Hurd, Y.L. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated With Cross-Generational Effects of Adolescent THC Exposure. Neuropsychopharmacology 2015, 40, 2993–3005. [Google Scholar] [CrossRef] [PubMed]
- Dholakiya, S.L.; Aliberti, A.; Barile, F.A. Morphine Sulfate Concomitantly Decreases Neuronal Differentiation and Opioid Receptor Expression in Mouse Embryonic Stem Cells. Toxicol. Lett. 2016, 247, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.W.; Jagwani, S.; Kim, E.; Rendell, V.R.; He, J.; Ezerskiy, L.A.; Wesselschmidt, R.; Coscia, C.J.; Belcheva, M.M. Mu and Kappa Opioids Modulate Mouse Embryonic Stem Cell-Derived Neural Progenitor Differentiation via MAP Kinases. J. Neurochem. 2010, 112, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Bansal, P.; Ahern, D.T.; Kondaveeti, Y.; Qiu, C.W.; Pinter, S.F. Contiguous Erosion of the Inactive X in Human Pluripotency Concludes with Global DNA Hypomethylation. Cell Rep. 2021, 35, 109215. [Google Scholar] [CrossRef] [PubMed]
- von Meyenn, F.; Iurlaro, M.; Habibi, E.; Liu, N.Q.; Salehzadeh-Yazdi, A.; Santos, F.; Petrini, E.; Milagre, I.; Yu, M.; Xie, Z.; et al. Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells. Mol. Cell 2016, 62, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Leitch, H.G.; Mcewen, K.R.; Turp, A.; Encheva, V.; Carroll, T.; Grabole, N.; Mansfield, W.; Nashun, B.; Knezovich, J.G.; Smith, A.; et al. Naive pluripotency is associated with global DNA hypomethylation. Nat. Struct. Mol. Biol. 2013, 20, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.; Uh, K.; Lee, K. TET Enzyme Driven Epigenetic Reprogramming in Early Embryos and Its Implication on Long-Term Health. Front. Cell Dev. Biol. 2024, 12, 1358649. [Google Scholar] [CrossRef] [PubMed]
- Bartoccetti, M.; van der Veer, B.K.; Luo, X.; Khoueiry, R.; She, P.; Bajaj, M.; Xu, J.; Janiszewski, A.; Thienpont, B.; Pasque, V.; et al. Regulatory Dynamics of Tet1 and Oct4 Resolve Stages of Global DNA Demethylation and Transcriptomic Changes in Reprogramming. Cell Rep. 2020, 30, 2150–2169.e9. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.J.; Lodh, A.; Choi, J.H.; van Riggelen, J. MYC deregulates TET1 and TET2 expression to control global DNA (hydroxy)methylation and gene expression to maintain a neoplastic phenotype in T-ALL. Epigenetics Chromatin 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jimenez-Gonzalez, A.; García-Concejo, A.; León-Lobera, F.; Rodriguez, R.E. Morphine delays neural stem cells differentiation by facilitating Nestin overexpression. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA Methylation and Demethylation in Mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sun, H.; Lin, L.; Zhang, Y.; Chen, J.; Liang, L.; Li, Y.; Zhang, M.; Yang, X.; Wang, X.; et al. Passive DNA Demethylation Preferentially Up-Regulates Pluripotency-Related Genes and Facilitates the Generation of Induced Pluripotent Stem Cells. J. Biol. Chem. 2017, 292, 18542–18555. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, X.; Rosenfeld, M.G. Histone H2A Ubiquitination in Transcriptional Regulation and DNA Damage Repair. Int. J. Biochem. Cell Biol. 2009, 41, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.Y.; Shi, G.; Zhao, P. Reversal of Oxycodone Conditioned Place Preference by Oxytocin: Promoting Global DNA Methylation in the Hippocampus. Neuropharmacology 2019, 160, 107778. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.Y.; Shi, G.; He, X.J.; Li, X.Y.; Wan, Y.X.; Jian, L.Y. Oxytocin Prevents Cue-Induced Reinstatement of Oxycodone Seeking: Involvement of DNA Methylation in the Hippocampus. Addict. Biol. 2021, 26, e13025. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Gu, X.; Pan, Z.; Guo, X.; Liu, J.; Atianjoh, F.E.; Wu, S.; Mo, K.; Xu, B.; Liang, L.; et al. Contribution of DNMT1 to Neuropathic Pain Genesis Partially through Epigenetically Repressing Kcna2 in Primary Afferent Neurons. J. Neurosci. 2019, 39, 6595–6607. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barrot, M.; Olivier, J.D.; Perrotti, L.I.; DiLeone, R.J.; Berton, O.; Eisch, A.J.; Impey, S.; Storm, D.R.; Neve, R.L.; Yin, J.C.; et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc. Natl. Acad. Sci. USA 2002, 99, 11435–11440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of Novel Families of Enzymes Involved in Oxidative and Other Complex Modifications of Bases in Nucleic Acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic Shift: Biological Roles of TET Proteins in DNA Demethylation and Transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The Polycomb Group Protein EZH2 Is Involved in Progression of Prostate Cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Karanikolas, B.D.W.; Figueiredo, M.L.; Wu, L. Polycomb Group Protein Enhancer of Zeste 2 Is an Oncogene That Promotes the Neoplastic Transformation of a Benign Prostatic Epithelial Cell Line. Mol. Cancer Res. 2009, 7, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gonzalez, M.E.; Toy, K.; Filzen, T.; Merajver, S.D.; Kleer, C.G. Targeted Overexpression of EZH2 in the Mammary Gland Disrupts Ductal Morphogenesis and Causes Epithelial Hyperplasia. Am. J. Pathol. 2009, 175, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 January 2020).
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. FelixKrueger/: v0.6.2. 2019. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 26 January 2020).
- Granlund, T.; Stallman, R.M. Cat. 2018. Available online: https://github.com/Batch-Man/cat (accessed on 26 January 2020).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krueger, F.; Kreck, B.; Franke, A.; Andrews, S.R. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods. 2012, 9, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Y.; Pal, B.; Visvader, J.E.; Smyth, G.K. Differential methylation analysis of reduced representation bisulfite sequencing experiments using edgeR. F1000Research 2017, 6, 2055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 25 May 2025).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yohn, N.L.; Bartolomei, M.S.; Blendy, J.A. Multigenerational and Transgenerational Inheritance of Drug Exposure: The Effects of Alcohol, Opiates, Cocaine, Marijuana, and Nicotine. Prog. Biophys. Mol. Biol. 2015, 118, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Sarkaki, A.; Assaei, R.; Motamedi, F.; Badavi, M.; Pajouhi, N. Effect of Parental Morphine Addiction on Hippocampal Long-Term Potentiation in Rats Offspring. Behav. Brain Res. 2008, 186, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Chorbov, V.M.; Todorov, A.A.; Lynskey, M.T.; Cicero, T.J. Elevated Levels of DNA Methylation at the OPRM1 Promoter in Blood and Sperm from Male Opioid Addicts. J. Opioid Manag. 2011, 7, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Saeidinezhad, M.; Razban, V.; Safizadeh, H.; Ezzatabadipour, M. Effects of Maternal Consumption of Morphine on Rat Skeletal System Development. BMC Musculoskelet. Disord. 2021, 22, 435. [Google Scholar] [CrossRef] [PubMed]
- Sadraie, S.H.; Kaka, G.R.; Sahraei, H.; Dashtnavard, H.; Bahadoran, H.; Mofid, M.; Nasab, H.M.; Jafari, F. Effects of Maternal Oral Administration of Morphine Sulfate on Developing Rat Fetal Cerebrum: A Morphometrical Evaluation. Brain Res. 2008, 1245, 36–40. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Full Gene Name |
|---|---|
| Glmn | Glomulin |
| Zfp445 | Zinc finger protein 445 |
| Suz12 | Polycomb protein Suz12 |
| Smarcad1 | SWI_SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A containing DEAD_H box 1 |
| Tex15 | Testis-expressed protein 15 |
| Cbx5 | Chromobox protein homologue 5 |
| Dnmt3l | DNA (cytosine-5)-methyltransferase 3-like |
| Smchd1 | Structural maintenance of chromosomes flexible hinge domain-containing protein 1 |
| Zfp869 | Zinc finger protein 869 |
| Dicer1 | Endoribonuclease Dicer |
| Atad2 | ATPase family AAA domain-containing protein 2 |
| Smarca5 | SWI_SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 |
| Tet1 | Methylcytosine dioxygenase TET1 |
| Klf2 | Krueppel-like factor 2 |
| Trip12 | E3 ubiquitin-protein ligase TRIP12 |
| Sirt1 | NAD-dependent protein deacetylase sirtuin-1 |
| Kdm5a | Lysine-specific demethylase 5A |
| Mettl4 | N(6)-adenine-specific methyltransferase METTL4 |
| Cbx3 | Chromobox protein homologue 3 |
| Wbp2 | WW domain-binding protein 2 |
| Rif1 | Telomere-associated protein RIF1 |
| Myc | Myc proto-oncogene protein |
| Hells | Lymphocyte-specific helicase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araolaza, M.; Muñoa-Hoyos, I.; Urizar-Arenaza, I.; Calzado, I.; Subirán, N. Chronic Morphine Treatment Leads to a Global DNA Hypomethylation via Active and Passive Demethylation Mechanisms in mESCs. Int. J. Mol. Sci. 2025, 26, 7056. https://doi.org/10.3390/ijms26157056
Araolaza M, Muñoa-Hoyos I, Urizar-Arenaza I, Calzado I, Subirán N. Chronic Morphine Treatment Leads to a Global DNA Hypomethylation via Active and Passive Demethylation Mechanisms in mESCs. International Journal of Molecular Sciences. 2025; 26(15):7056. https://doi.org/10.3390/ijms26157056
Chicago/Turabian StyleAraolaza, Manu, Iraia Muñoa-Hoyos, Itziar Urizar-Arenaza, Irune Calzado, and Nerea Subirán. 2025. "Chronic Morphine Treatment Leads to a Global DNA Hypomethylation via Active and Passive Demethylation Mechanisms in mESCs" International Journal of Molecular Sciences 26, no. 15: 7056. https://doi.org/10.3390/ijms26157056
APA StyleAraolaza, M., Muñoa-Hoyos, I., Urizar-Arenaza, I., Calzado, I., & Subirán, N. (2025). Chronic Morphine Treatment Leads to a Global DNA Hypomethylation via Active and Passive Demethylation Mechanisms in mESCs. International Journal of Molecular Sciences, 26(15), 7056. https://doi.org/10.3390/ijms26157056