

Stargardt’s Disease: Molecular Pathogenesis and Current Therapeutic Landscape


Abstract
1. Introduction
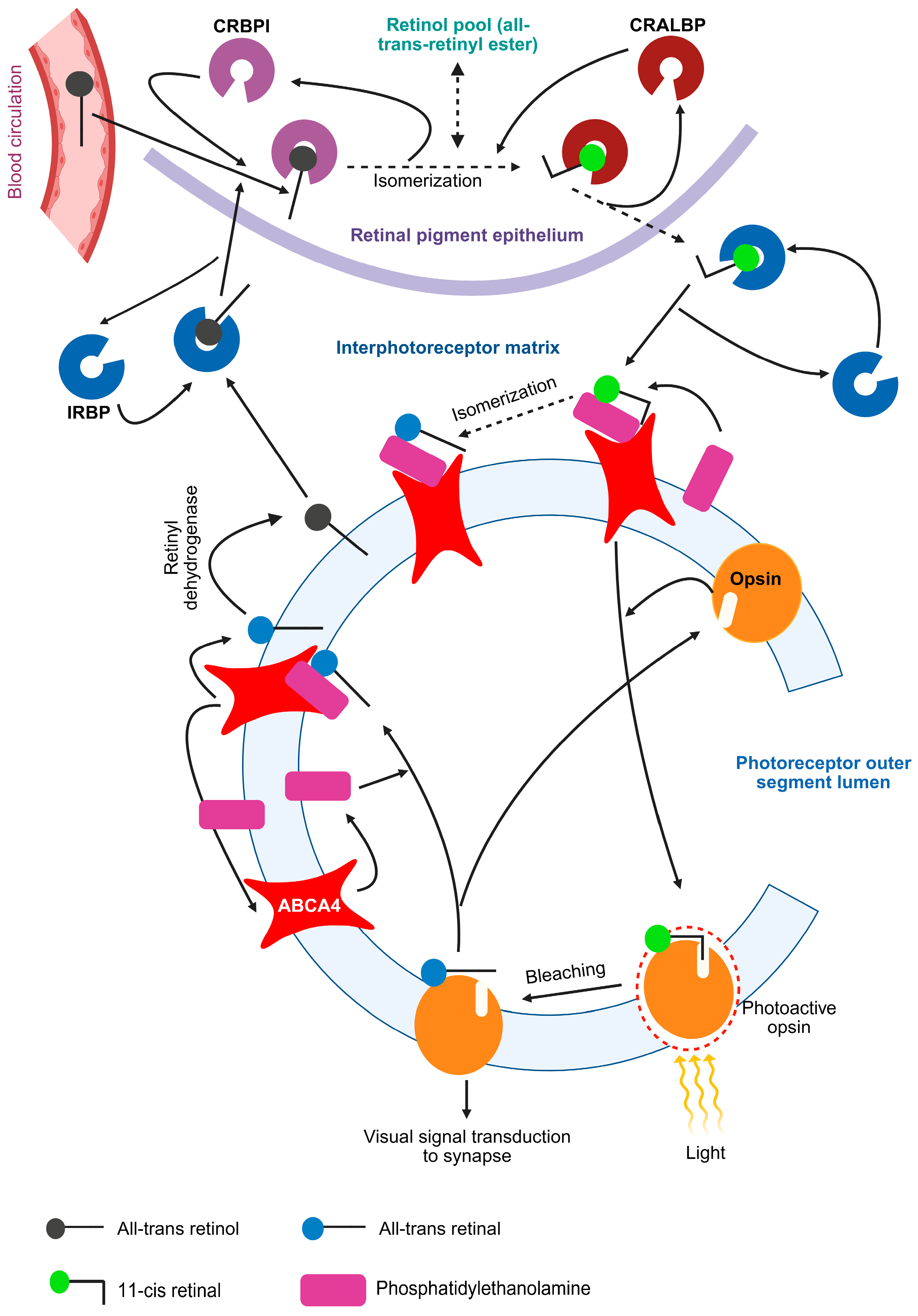
1.1. The Visual Cycle and ABCA4 Protein
1.2. Etiology of Stargardt’s Disease
1.2.1. Role of Complement System
1.2.2. Role of Oxidative Stress
1.3. Stargardt’s Disease: Pathophysiology and Clinical Presentation
1.4. Stargardt’s Disease: Diagnosis
- Visual acuity test: This test determines a patient’s ability to distinguish between two standard symbols or alphabets while viewing them from a standard distance (20 feet for US; 6 feet for UK). A visual acuity of x/y indicates that the patient can recognize symbols from a distance of x, whereas individuals with normal vision can identify the same symbols from a distance of y [75]. An age-related decline in the probability of maintaining a visual acuity of 20/40 has been observed in patients with Stargardt’s disease. A faster decline in visual acuity has also been reported in these patients after it dropped further from 20/40 and stabilized at 20/200 [76].
- Ophthalmoscopy: It is a clinical assessment to observe the internal structures of the eyes, specifically the fundus, of the patient through the dilated pupil using a ophthalmoscope [77]. The characteristic fundus appearance of Stargardt disease includes a macula with “beaten bronze” look, surrounded by distinct yellow flecks, either round or pisciform, located at the level of the retinal pigment epithelium [78].
- Fundus autofluorescence (FAF): This imaging technique detects the autofluorescence caused due to lipofuscin deposition in retinal pigment epithelium cells [79,80]. In the ProgStar study, which is a multinational, observational cohort study involving patients with Stargardt’s disease, around 250 patients each were analyzed retrospectively and prospectively with FAF as the primary outcome measure [81].
- Optical coherence tomography (OCT) and Spectral-domain OCT (SD-OCT): OCT uses light in the near-infrared region to examine the tissue. The delay in the reflected light provides a measure of the depth of reflection that determines its axial resolution. SD-OCT is an improved version of OCT with increased speed and image quality [82]. SD-OCT reveals outer retinal atrophy, including loss of the ellipsoid zone and thinning of the retinal pigment epithelium [83].
- Electroretinography (ERG): It can aid in the diagnosis and monitoring of Stargardt’s disease, but it is suggested to avoid these techniques as it involves bright light flashes that can exacerbate lipofuscin formation [84].
- Type 1: A localized low FAF signal at the fovea, set against a uniform background, with or without perifoveal foci of increased or decreased signal.
- Type 2: A localized low FAF signal at the macula, surrounded by a varied background, with widespread foci of increased or decreased FAF signal extending beyond the vascular arcades.
- Type 3: Multiple areas of low FAF signal at the posterior pole, accompanied by a heterogeneous background and/or foci of increased or decreased signal.
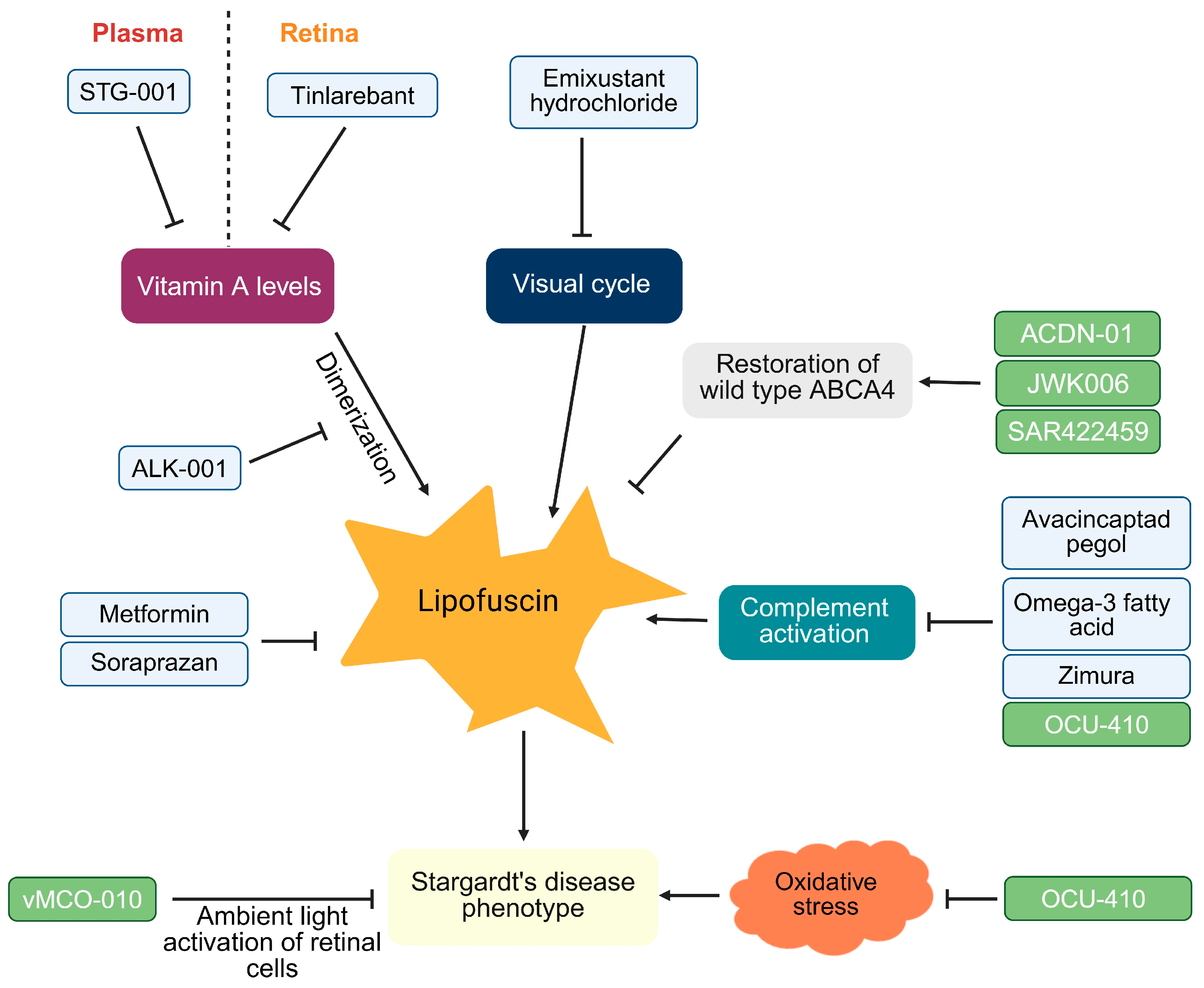
1.5. Stargardt’s Disease: Therapeutic Landscape
1.5.1. Small Molecules for Stargardt’s Disease
1.5.2. Gene Therapy for Stargardt’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | MOA | Outcomes Assessed | Phase | Sponsor | Reference |
|---|---|---|---|---|---|
| Small Molecules | |||||
| Tinlarebant | Retinol binding protein 4 antagonist | PK/PD, safety, change in atrophic lesion size | 1b, 2/3 | Mata Nathan | JPRN-jRCT2031240209 [124] |
| Change in atrophic lesion size | 3 | Belite Bio, Inc. | NCT05244304 [125] | ||
| Systemic and ocular safety and tolerability; the optimal dose for Phase 2 | 1/2 | RBP4 Pty Ltd. | NCT05266014 [126] | ||
| STG-001 | Reduces plasma levels of RBP4 | Safety and tolerability | 2 | Stargazer Pharmaceuticals, Inc. | NCT04489511 [96] |
| Metformin | targeting lysosomal and fatty acid oxidation pathways in retinal pigment epithelium cells | Change in atrophic lesion size in ellipsoid zone band | 1/2 | National Eye Institute | NCT04545736 [99] |
| ALK001 | Deuterated vitamin A; slows down the vitamin A dimer formation | Safety and tolerability, change in atrophic lesion size, PK | 2 | Alkeus Pharmaceuticals, Inc. | NCT02402660 [127] |
| Safety and tolerability, PK | NCT04239625 [128] | ||||
| Emixustat Hydrochloride | Inhibition of RPE65 and reducing 11 cis-retinal production | Change in retina electroretinogram; safety | 2 | Kubota Vision Inc. | NCT03033108 [103] |
| Change in macular atrophic lesion | 3 | Kubota Vision Inc. | NCT03772665 [104] | ||
| Soraprazan | removal of lipofuscin in retinal pigment epithelium cells | Change in quantitative fundus autofluorescence | 2 | Katairo GmbH | NL-OMON48130 [110] |
| Omega-3 Fatty Acids Supplementation | Reducing complement C3 and accumulation of lipofuscin in retinal pigment epithelium cells | Visual acuity | N/A | Ophthalmos Research and Education Institute | NCT03297515 [129] |
| Zimura (Aptamer) | Complement C5 inhibition | Change in atrophic lesion size in ellipsoid zone | 2 | Astellas Pharma Global Development, Inc. | NCT03364153 [113] |
| Gene Therapies | |||||
| ACDN-01 RNA exon editor | AAV-based vector carrying a DNA construct encoding for an ABCA4 RNA exon editor | Safety and tolerability | 1/2 | Ascidian Therapeutics, Inc. | NCT06467344 [116] |
| JWK006 | ABCA4 gene expression via adeno-associated virus | Safety and visual acuity | 1/2 | West China Hospital | NCT06300476 [117] |
| OCU-410 | RORA expression using Adeno-Associated Virus serotype 5 | Safety and visual acuity | 1/2 | Ocugen | NCT05956626 [123] |
| SAR422459 | Expression of ABCA4 using recombinant equine infectious anemia virus | Safety and tolerability | 1/2 | Sanofi | NCT01736592 [130] |
| vMCO-010 | Viral expression of Opsin activated by ambient light | Safety, visual acuity, light-guided mobility, determination of shape and optical flow | 2 | Nanoscope Therapeutics Inc. | NCT05417126 [131] |
| Stem Cell Therapies | |||||
| autologous bone marrow-derived stem cells | Treatment of retinal and optic nerve damage | Safety and neuronal degradation in the eye | 1 | Pomeranian Medical University Szczecin | NCT03772938 [132] |
| Visual acuity | N/A | MD Stem Cells | NCT01920867 [133] | ||
| NCT03011541 [134] | |||||
1.5.3. Stem Cell Therapy for Stargardt’s Disease
2. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Stargardt, K. Über Familiäre, progressive degeneration in der maculagegend des auges. Graefe’s Arch. Clin. Exp. Ophthalmol. 1909, 71, 534–550. [Google Scholar] [CrossRef]
- Glazer, L.C.; Dryja, T.P. Understanding the etiology of Stargardt’s disease. Ophthalmol. Clin. N. Am. 2002, 15, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The genetics of inherited macular dystrophies. J. Med. Genet. 2003, 40, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, J.J.; Jensen, A.M. Abca4, mutated in Stargardt disease, is required for structural integrity of cone outer segments. Dis. Model. Mech. 2025, 18, DMM052052. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, P.S.; Tammur, J.; Singh, N.; Hutchinson, A.; Dixon, M.; Pappas, C.M.; Zabriskie, N.A.; Zhang, K.; Petrukhin, K.; Leppert, M.; et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3331–3336. [Google Scholar]
- Imani, S.; Cheng, J.; Dehghan Shasaltaneh, M.; Wei, C.; Yang, L.; Fu, S.; Zou, H.; Khan, M.A.; Zhang, X.; Chen, H.; et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget 2018, 9, 122–141. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Shalom, S.; Ben-Yosef, T.; Sher, I.; Zag, A.; Rotenstreich, Y.; Poleg, T.; Birk, O.S.; Gradstein, L.; Ehrenberg, M.; Deitch, I.; et al. Nationwide Prevalence of Inherited Retinal Diseases in the Israeli Population. JAMA Ophthalmol. 2024, 142, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Runhart, E.H.; Dhooge, P.; Meester-Smoor, M.; Pas, J.; Pott, J.W.R.; van Leeuwen, R.; Kroes, H.Y.; Bergen, A.A.B.; de Jong-Hesse, Y.; Thiadens, A.A.; et al. Stargardt disease: Monitoring incidence and diagnostic trends in the Netherlands using a nationwide disease registry. Acta Ophthalmol. 2022, 100, 395–402. [Google Scholar] [CrossRef] [PubMed]
- EMA. EU/3/19/2162-Orphan Designation for Treatment of Stargardt’s Disease. Emixustat Hydrochloride. 2025. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-19-2162 (accessed on 15 July 2025).
- Heath Jeffery, R.C.; Mukhtar, S.A.; McAllister, I.L.; Morgan, W.H.; Mackey, D.A.; Chen, F.K. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021, 42, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Runhart, E.H.; Khan, M.; Cornelis, S.S.; Roosing, S.; Del Pozo-Valero, M.; Lamey, T.M.; Liskova, P.; Roberts, L.; Stöhr, H.; Klaver, C.C.W.; et al. Association of Sex With Frequent and Mild ABCA4 Alleles in Stargardt Disease. JAMA Ophthalmol. 2020, 138, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Sassone, F.; Estay-Ahumada, C.; Roux, M.J.; Ciocca, D.; Rossolillo, P.; Birling, M.-C.; Sparrow, J.R.; Montenegro, D.; Hicks, D. Interruption of the visual cycle in a novel animal model induces progressive vision loss resembling Stargardts Disease. Sci. Rep. 2024, 14, 30880. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Biswas-Fiss, E.E.; Kurpad, D.S.; Joshi, K.; Biswas, S.B. Interaction of extracellular domain 2 of the human retina-specific ATP-binding cassette transporter (ABCA4) with all-trans-retinal. J. Biol. Chem. 2010, 285, 19372–19383. [Google Scholar] [CrossRef] [PubMed]
- UniProt. P78363·ABCA4_HUMAN. June 2024. Available online: https://www.uniprot.org/uniprotkb/P78363/entry (accessed on 15 July 2025).
- Patel, M.J.; Biswas, S.B.; Biswas-Fiss, E.E. Functional significance of the conserved C-Terminal VFVNFA motif in the retina-specific ABC transporter, ABCA4, and its role in inherited visual disease. Biochem. Biophys. Res. Commun. 2019, 519, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ebrey, T.; Koutalos, Y. Vertebrate photoreceptors. Prog. Retin. Eye Res. 2001, 20, 49–94. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Moritz, O.L. Photoreceptors at a glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.F.; Moritz, O.L.; Williams, D.S. Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res. 2016, 55, 52–81. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.G.; Xue, T.; Yau, K.W. How vision begins: An odyssey. Proc. Natl. Acad. Sci. USA 2008, 105, 9855–9862. [Google Scholar] [CrossRef] [PubMed]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Molday, R.S. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl. Acad. Sci. USA 2014, 111, 5024–5029. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Lenevich, S.; Molday, R.S. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012, 3, 925. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Adler, L.; Goletz, P.; Gonzalez-Fernandez, F.; Thompson, D.A.; Koutalos, Y. Interphotoreceptor retinoid-binding protein removes all-trans-retinol and retinal from rod outer segments, preventing lipofuscin precursor formation. J. Biol. Chem. 2017, 292, 19356–19365. [Google Scholar] [CrossRef] [PubMed]
- Sears, A.E.; Bernstein, P.S.; Cideciyan, A.V.; Hoyng, C.B.; Charbel Issa, P.; Palczewski, K.; Rosenfeld, P.J.; Sadda, S.; Schraermeyer, U.; Sparrow, J.R.; et al. Towards Treatment of Stargardt Disease: Workshop Organized and Sponsored by the Foundation Fighting Blindness. Transl. Vis. Sci. Technol. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Garces, F.A.; Scortecci, J.F.; Molday, L.L. Structure and function of ABCA4 and its role in the visual cycle and Stargardt macular degeneration. Prog. Retin. Eye Res. 2022, 89, 101036. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Kim, H.J.; Zhao, J.; Sparrow, J.R. Bisretinoid Photodegradation Is Likely Not a Good Thing. Adv. Exp. Med. Biol. 2018, 1074, 395–401. [Google Scholar] [PubMed]
- Zhang, D.; Robinson, K.; Saad, L.; Washington, I. Vitamin A cycle byproducts impede dark adaptation. J. Biol. Chem. 2021, 297, 101074. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.C.; Bauwens, M.; haer-Wigman, L.; de Bruyne, M.; Pantrani, M.; de Baere, E.; Hufnagel, R.B.; Cremers, F.P.M. Compendium of Clinical Variant Classification for 2246 Unique ABCA4 Variants to Clarify Variant Pathogenicity in Stargardt Disease Using a Modified ACMG/AMP Framework. Hum. Mutat. 2023, 6815504. [Google Scholar]
- Lewis, R.A.; Shroyer, N.F.; Singh, N.; Allikmets, R.; Hutchinson, A.; Li, Y.; Lupski, J.R.; Leppert, M.; Dean, M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 1999, 64, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Zernant, J.; Nagasaki, T.; Fishman, G.A.; Tsang, S.H.; Allikmets, R. Clinical and Genetics Characterization of Hypomorphic Variants in Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2021, 62, 2171. [Google Scholar]
- Nassisi, M.; Mohand-Saïd, S.; Dhaenens, C. -M.; Boyard, F.; Démontant, V.; Andrieu, C.; Antonio, A.; Condroyer, C.; Foussard, M.; Méjécase, C.; et al. Expanding the Mutation Spectrum in ABCA4: Sixty Novel Disease Causing Variants and Their Associated Phenotype in a Large French Stargardt Cohort. Int. J. Mol. Sci. 2018, 19, 2196. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Alvarez, R.; Aguirre-Lamban, J.; Lopez-Martinez, M.A.; Trujillo-Tiebas, M.J.; Cantalapiedra, D.; Vallespin, E.; Avila-Fernandez, A.; Ramos, C.; Ayuso, C. Frequency of ABCA4 mutations in 278 Spanish controls: An insight into the prevalence of autosomal recessive Stargardt disease. Br. J. Ophthalmol. 2009, 93, 1359–1364. [Google Scholar] [CrossRef] [PubMed]
- Mena, M.D.; Moresco, A.A.; Vidal, S.H.; Aguilar-Cortes, D.; Obregon, M.G.; Fandiño, A.C.; Sendoya, J.M.; Llera, A.S.; Podhajcer, O.L. Clinical and Genetic Spectrum of Stargardt Disease in Argentinean Patients. Front. Genet. 2021, 12, 646058. [Google Scholar] [CrossRef] [PubMed]
- Passerini, I.; Sodi, A.; Giambene, B.; Mariottini, A.; Menchini, U.; Torricelli, F. Novel mutations in of the ABCR gene in Italian patients with Stargardt disease. Eye 2010, 24, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Strauss, R.W.; Chiang, J.P.; Audo, I.S.; Bernstein, P.S.; Birch, D.G.; Bomotti, S.M.; Cideciyan, A.V.; Ervin, A.M.; Marino, M.J.; et al. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol. 2019, 103, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, Z.; Cui, J.; Tan, C.; Sun, W.; Lin, Y. Clinical and Genetic Characteristics of 18 Patients from Southeast China with ABCA4-Associated Stargardt Disease. Int. J. Mol. Sci. 2025, 26, 3354. [Google Scholar] [CrossRef] [PubMed]
- Pas, J.; Li, C.H.Z.; Van den Broeck, F.; Dhooge, P.P.A.; De Zaeytijd, J.; Collin, R.W.J.; Leroy, B.P.; Hoyng, C.B. Progression of Atrophy as a Function of ABCA4 Variants and Age of Onset in Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2025, 66, 76. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, M.; Iglesias-Romero, A.B.; Cancellieri, F.; Kaminska, K.; Scholl, H.P.N.; Pfau, M.; Rivolta, C. ABCA4 c.5461-6T>C Causes Stargardt Disease Through Exon Skipping. Adv. Exp. Med. Biol. 2025, 1468, 57–62. [Google Scholar] [PubMed]
- Zernant, J.; Xie, Y.A.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.H.J.; Hoefsloot, L.H.; van den Born, L.I.; Phan, M.; Klevering, B.J.; Westeneng-van Haaften, C.; Braun, T.A.; et al. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat. 2015, 36, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, M.; De Zaeytijd, J.; Weisschuh, N.; Kohl, S.; Meire, F.; Dahan, K.; Depasse, F.; De Jaegere, S.; De Ravel, T.; De Rademaeker, M.; et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum. Mutat. 2015, 36, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Del Pozo-Valero, M.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, P.; Yi, Z.; Ouyang, J.; Jiang, Y.; Li, S.; Jia, X.; Xiao, X.; Hejtmancik, J.F.; Sun, W.; et al. ABCA4 Deep Intronic Variants Contributed to Nearly Half of Unsolved Stargardt Cases With a Milder Phenotype. Investig. Ophthalmol. Vis. Sci. 2025, 66, 65. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Zernant, J.; Nagasaki, T.; Molday, L.L.; Su, P.-Y.; Fishman, G.A.; Tsang, S.H.; Molday, R.S.; Allikmets, R. Cis-acting modifiers in the ABCA4 locus contribute to the penetrance of the major disease-causing variant in Stargardt disease. Hum. Mol. Genet. 2021, 30, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Guymer, R.H.; Heon, E.; Lotery, A.J.; Munier, F.L.; Schorderet, D.F.; Baird, P.N.; McNeil, R.J.; Haines, H.; Sheffield, V.C.; Stone, E.M. Variation of codons 1961 and 2177 of the Stargardt disease gene is not associated with age-related macular degeneration. Arch. Ophthalmol. 2001, 119, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold. Spring. Harb. Mol. Case. Stud. 2018, 4, a002733. [Google Scholar] [CrossRef] [PubMed]
- Zaneveld, J.; Siddiqui, S.N.; Li, H.; Wang, X.; Wang, H.; Wang, K.; Li, H.; Ren, H.; Lopez, I.; Dorfman, A.; et al. Comprehensive analysis of patients with Stargardt macular dystrophy reveals new genotype-phenotype correlations and unexpected diagnostic revisions. Genet. Med. 2015, 17, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Pauer, G.J.; Hagstrom, S.A.; Bok, D.; DeBenedictis, M.J.; Bonilha, V.L.; Hollyfield, J.G.; Radu, R.A. Evidence of complement dysregulation in outer retina of Stargardt disease donor eyes. Redox Biol. 2020, 37, 101787. [Google Scholar] [CrossRef] [PubMed]
- Lenis, T.L.; Sarfare, S.; Jiang, Z.; Lloyd, M.B.; Bok, D.; Radu, R.A. Complement modulation in the retinal pigment epithelium rescues photoreceptor degeneration in a mouse model of Stargardt disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3987–3992. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jang, Y.P.; Kim, S.R.; Sparrow, J.R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2006, 103, 16182–16187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kim, S.R.; Westlund, B.S.; Sparrow, J.R. Complement activation by bisretinoid constituents of RPE lipofuscin. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.V.; Liles, M.R.; Newsome, D.A. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp. Cell Res. 1994, 214, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Boulton, M.E.; Gottsch, J.D.; Sternberg, P. Oxidative damage and age-related macular degeneration. Mol. Vis. 1999, 5, 32. [Google Scholar] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Taubitz, T.; Tschulakow, A.V.; Tikhonovich, M.; Illing, B.; Fang, Y.; Biesemeier, A.; Julien-Schraermeyer, S.; Schraermeyer, U. Ultrastructural alterations in the retinal pigment epithelium and photoreceptors of a Stargardt patient and three Stargardt mouse models: Indication for the central role of RPE melanin in oxidative stress. PeerJ 2018, 6, e5215. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Mata, N.L.; Bagla, A.; Travis, G.H. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 5928–5933. [Google Scholar] [CrossRef] [PubMed]
- Clinic, C. Macula. 26 January 2025. Available online: https://my.clevelandclinic.org/health/body/23185-macula (accessed on 15 July 2025).
- The Human Protein Atlas. Tissue Expression of ABCA4-Summary-The Human Protein Atlas. 2025. Available online: https://www.proteinatlas.org/ENSG00000198691-ABCA4/tissue (accessed on 15 July 2025).
- Sparrow, J.R.; Marsiglia, M.; Allikmets, R.; Tsang, S.; Lee, W.; Duncker, T.; Zernant, J. Flecks in Recessive Stargardt Disease: Short-Wavelength Autofluorescence, Near-Infrared Autofluorescence, and Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5029–5039. [Google Scholar] [CrossRef] [PubMed]
- Zaydon, Y.A.; Tsang, S.H. The ABCs of Stargardt disease: The latest advances in precision medicine. Cell Biosci. 2024, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Rotenstreich, Y.; Fishman, G.A.; Anderson, R.J. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology 2003, 110, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Tracewska, A.M.; Kocyła-Karczmarewicz, B.; Rafalska, A.; Murawska, J.; Jakubaszko-Jablonska, J.; Rydzanicz, M.; Stawiński, P.; Ciara, E.; Khan, M.I.; Henkes, A.; et al. Genetic Spectrum of ABCA4-Associated Retinal Degeneration in Poland. Genes 2019, 10, 959. [Google Scholar] [CrossRef] [PubMed]
- Lambertus, S.; van Huet, R.A.; Bax, N.M.; Hoefsloot, L.H.; Cremers, F.P.; Boon, C.J.; Klevering, B.J.; Hoyng, C.B. Early-onset stargardt disease: Phenotypic and genotypic characteristics. Ophthalmology 2015, 122, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Zernant, J.; Su, P.Y.; Nagasaki, T.; Tsang, S.H.; Allikmets, R. A genotype-phenotype correlation matrix for ABCA4 disease based on long-term prognostic outcomes. JCI Insight 2022, 7, e156154. [Google Scholar] [CrossRef]
- Li, C.H.Z.; Pas, J.A.A.H.; Corradi, Z.; Hitti-Malin, R.J.; Hoogstede, A.; Runhart, E.H.; Dhooge, P.P.A.; Collin, R.W.J.; Cremers, F.P.M.; Hoyng, C.B. Study of Late-Onset Stargardt Type 1 Disease: Characteristics, Genetics, and Progression. Ophthalmology 2024, 131, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Daiber, H.F.; Gnugnoli, D.M. Visual Acuity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Fishman, G.A.; Farber, M.; Patel, B.S.; Derlacki, D.J. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology 1987, 94, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Sutradhar, S. Direct Ophthalmoscope-Everything You Need to Know. 13 May 2024. Available online: https://smartoptometryacademy.com/direct-ophthalmoscope-all-u-need-to-know/ (accessed on 15 July 2025).
- Mukherjee, N.; Schuman, S. Diagnosis and Management of Stargardt Disease. EyeNet Magazine, 1 December 2014; pp. 29–31. [Google Scholar]
- Delori, F.C.; Dorey, C.K.; Staurenghi, G.; Arend, O.; Goger, D.G.; Weiter, J.J. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Investig. Ophthalmol. Vis. Sci. 1995, 36, 718–729. [Google Scholar]
- von Ruckmann, A.; Fitzke, F.W.; Bird, A.C. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br. J. Ophthalmol. 1995, 79, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.W.; Muñoz, B.; Ho, A.; Jha, A.; Michaelides, M.; Cideciyan, A.V.; Audo, I.; Birch, D.G.; Hariri, A.H.; Nittala, M.G.; et al. Progression of Stargardt Disease as Determined by Fundus Autofluorescence in the Retrospective Progression of Stargardt Disease Study (ProgStar Report No. 9). JAMA Ophthalmol. 2017, 135, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Aumann, S.; Donner, S.; Fischer, J.; Muller, F. Optical Coherence Tomography (OCT): Principle and Technical Realization. In High Resolution Imaging in Microscopy and Ophthalmology: New Frontiers in Biomedical Optics; Bille, J.F., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 59–85. [Google Scholar]
- Strauss, R.W.; Lang, L.; Ho, A.; Jha, A.; Ip, M.; Bernstein, P.S.; Birch, D.G.; Cideciyan, A.V.; Michaelides, M.; Audo, I.; et al. The Progression of Stargardt Disease as Determined by Spectral-Domain Optical Coherence Tomography over a 24-Month Period (ProgStar Report No. 18). Ophthalmic Res. 2024, 67, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, J.C.; Scruggs, B.A. The Pathology of Stargardt Disease and Future Treatment Horizons. Retin. Physician 2024, 21, 8–12. [Google Scholar]
- Fishman, G.A. Fundus flavimaculatus: A clinical classification. Arch. Ophthalmol. 1976, 94, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Fujinami, K.; Lois, N.; Mukherjee, R.; McBain, V.A.; Tsunoda, K.; Tsubota, K.; Stone, E.M.; Fitzke, F.W.; Bunce, C.; et al. A longitudinal study of Stargardt disease: Quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8181–8190. [Google Scholar] [CrossRef] [PubMed]
- Wabbels, B.; Demmler, A.; Paunescu, K.; Wegscheider, E.; Preising, M.N.; Lorenz, B. Fundus autofluorescence in children and teenagers with hereditary retinal diseases. Graefes Arch. Clin. Exp. Ophthalmol. 2006, 244, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fallon, J. Belite Bio Announces First Patient Dosed in Phase 2/3 DRAGON II Trial of Tinlarebant for the Treatment of Stargardt Disease. 10 September 2024. Available online: https://www.globenewswire.com/news-release/2024/09/10/2943626/0/en/Belite-Bio-Announces-First-Patient-Dosed-in-Phase-2-3-DRAGON-II-Trial-of-Tinlarebant-for-the-Treatment-of-Stargardt-Disease.html (accessed on 15 July 2025).
- Steinhoff, J.S.; Lass, A.; Schupp, M. Biological Functions of RBP4 and Its Relevance for Human Diseases. Front. Physiol. 2021, 12, 659977. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; McCarthy, T. Belite Bio Receives FDA Fast Track Designation for LBS-008. 3 May 2022. Available online: https://www.globenewswire.com/news-release/2022/05/03/2434325/0/en/Belite-Bio-Receives-FDA-Fast-Track-Designation-For-LBS-008.html (accessed on 15 July 2025).
- Wu, J.; Fallon, J. Belite Bio Receives Sakigake (Pioneer Drug) Designation of Tinlarebant for Stargardt Disease in Japan. 12 June 2024. Available online: https://www.globenewswire.com/news-release/2024/06/12/2897299/0/en/Belite-Bio-Receives-Sakigake-Pioneer-Drug-Designation-of-Tinlarebant-for-Stargardt-Disease-in-Japan.html (accessed on 15 July 2025).
- Belite Bio. Belite Bio Presents Results from a 24-Month, Phase 2 Study of Tinlarebant in Childhood-Onset Stargardt Disease at the AAO Annual Meeting. June 2023. Available online: https://investors.belitebio.com/news-releases/news-release-details/belite-bio-presents-results-24-month-phase-2-study-tinlarebant (accessed on 15 July 2025).
- Belite Bio. Belite Bio Announces Interim Analysis Results from the Pivotal Global Phase 3 DRAGON Trial of Tinlarebant in Adolescent Stargardt Disease Subjects. 27 February 2025. Available online: https://investors.belitebio.com/news-releases/news-release-details/belite-bio-announces-interim-analysis-results-pivotal-global (accessed on 15 July 2025).
- Retinal Degeneration Fund; Stargazer Pharmaceuticals, Inc. Announces $57 Million Series A Financing and Initiation of a Phase 2a Clinical Study of STG-001 in Stargardt Disease Patients. 9 November 2020. Available online: https://www.retinaldegenerationfund.org/news/news-posts/stargazer-pharmaceuticals-inc-announces-57-million-series-a-financing-and-initiation-of-a-phase-2a-clinical-study-of-stg-001-in-stargardt-disease-patients/ (accessed on 15 July 2025).
- U.S. Food and Drug Administration. Search Orphan Drug Designations and Approvals: 3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-pyrazol-1-yl) Propanoic Acid. 22 April 2019. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=681519&utm (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase 2a Study of the Safety, Pharmacokinetics and Pharmacodynamics of STG-001 in Subjects With Stargardt Disease (STGD1) Caused by Autosomal Recessive Mutation in ATP Binding Cassette Subfamily A Member 4 (ABCA4) Gene. 27 April 2021. Available online: https://clinicaltrials.gov/study/NCT04489511 (accessed on 15 July 2025).
- Farnoodian, M.; Barone, F.; Boyle, M.; Gupta, R.; Nelson, L.M.; Bose, D.A.; Jun, B.; Gordon, W.C.; Do, K.V.; Guerin, M.A.; et al. Metformin Attenuates the Hallmarks of Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2023, 64, 476. [Google Scholar]
- Bose, D.A.; Farnoodian, M.; Sharma, R.; McGaughey, D.; Bharti, K. Use of Metformin as a Potential Treatment for Stargardt Maculopathy. Investig. Ophthalmol. Vis. Sci. 2024, 65, 2203. [Google Scholar]
- ClinicalTrials.gov. Oral Metformin for Treatment of ABCA4 Retinopathy. 15 January 2025. Available online: https://clinicaltrials.gov/study/NCT04545736 (accessed on 15 July 2025).
- Kaufman, Y.; Ma, L.; Washington, I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J. Biol. Chem. 2011, 286, 7958–7965. [Google Scholar] [CrossRef] [PubMed]
- Kubota, R.; Gregory, J.; Henry, S.; Mata, N.L. Pharmacotherapy for metabolic and cellular stress in degenerative retinal diseases. Drug Discov. Today 2020, 25, 292–304. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Search Orphan Drug Designations and Approvals: Emixustat. 4 January 2017. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=548816&utm (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase 2a Multicenter, Randomized, Masked Study Evaluating the Pharmacodynamics of Emixustat Hydrochloride in Subjects With Macular Atrophy Secondary to Stargardt Disease. 19 May 2021. Available online: https://clinicaltrials.gov/study/NCT03033108 (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase 3 Multicenter, Randomized, Double-Masked Study Comparing the Efficacy and Safety of Emixustat Hydrochloride with Placebo for the Treatment of Macular Atrophy Secondary to Stargardt Disease. 30 May 2024. Available online: https://clinicaltrials.gov/study/NCT03772665 (accessed on 15 July 2025).
- Business Wire. Kubota Vision Announces Positive Post Hoc Analysis from Phase 3 Clinical Trial of Emixustat in Patients with Stargardt Disease. 3 October 2022. Available online: https://www.businesswire.com/news/home/20221003005323/en/Kubota-Vision-Announces-Positive-Post-Hoc-Analysis-from-Phase-3-Clinical-Trial-of-Emixustat-in-Patients-with-Stargardt-Disease (accessed on 15 July 2025).
- Julien-Schraermeyer, S.; Illing, B.; Tschulakow, A.; Taubitz, T.; Guezguez, J.; Burnet, M.; Schraermeyer, U. Penetration, distribution, and elimination of remofuscin/soraprazan in Stargardt mouse eyes following a single intravitreal injection using pharmacokinetics and transmission electron microscopic autoradiography: Implication for the local treatment of Stargardt’s disease and dry age-related macular degeneration. Pharmacol. Res. Perspect. 2020, 8, e00683. [Google Scholar]
- Julien, S.; Schraermeyer, U. Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol. Aging 2012, 33, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tschulakow, A.; Tikhonovich, M.; Taubitz, T.; Illing, B.; Schultheiss, S.; Schraermeyer, U.; Julien-Schraermeyer, S. Preclinical results of a new pharmacological therapy approach for Stargardt disease and dry age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 256. [Google Scholar]
- U.S. Food and Drug Administration. Search Orphan Drug Designations and Approvals: Soraprazan. 22 December 2017. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=617617&utm (accessed on 15 July 2025).
- Central Committee on Research Involving Human Subjects, T.N. A Multi-National, Multi-Centre, Double-Masked, Placebo-Controlled Proof of Concept Trial to Evaluate the Safety and Efficacy of Oral Soraprazan in Stargardt Disease. 12 April 2024. Available online: https://onderzoekmetmensen.nl/en/trial/48130 (accessed on 15 July 2025).
- Prokopiou, E.; Kolovos, P.; Kalogerou, M.; Neokleous, A.; Nicolaou, O.; Sokratous, K.; Kyriacou, K.; Georgiou, T. Omega-3 Fatty Acids Supplementation: Therapeutic Potential in a Mouse Model of Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Prokopiou, K.; Kolovos, P.; Tsangari, H.; Bandello, F.; Rossetti, L.M.; Mastropasqua, L.; Mohand-Said, S.; Georgiou, T. A prospective, multicentre, randomised, double-blind study designed to assess the potential effects of omega-3 fatty acids supplementation in dry age-related macular degeneration or Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2022, 63, 377-F0208. [Google Scholar]
- ClinicalTrials.gov. A Phase 2b Randomized, Double-Masked, Controlled Trial to Establish the Safety and Efficacy of Zimura™ (Complement C5 Inhibitor) Compared to Sham in Subjects With Autosomal Recessive Stargardt Disease. 4 December 2024. Available online: https://clinicaltrials.gov/study/NCT03364153 (accessed on 15 July 2025).
- Karen, O.H.C. FDA Clears First Clinical Trial of RNA Exon Editor Developed to Treat Stargardt Disease. 31 January 2024. Available online: https://crisprmedicinenews.com/news/fda-clears-first-clinical-trial-of-rna-exon-editor-developed-to-treat-stargardt-disease/ (accessed on 15 July 2025).
- Ascidian. Ascidian Therapeutics Announces First-Ever IND for an RNA Exon Editor as FDA Approves Trial Plan and Fast Tracks ACDN-01 in Stargardt Disease and Other ABCA4 Retinopathies. 29 January 2024. Available online: https://ascidian-tx.com/wp-content/uploads/2024/01/Ascidian-IND-Acceptance-Release_FINAL.1.29.24.pdf (accessed on 15 July 2025).
- ClinicalTrials.gov. ACDN-01-001: Open-Label, Single Ascending Dose Study to Evaluate the Safety, Tolerability, and Preliminary Efficacy of Subretinal ACDN-01 in Participants With ABCA4-related Retinopathy. 22 January 2025. Available online: https://clinicaltrials.gov/study/NCT06467344 (accessed on 15 July 2025).
- ClinicalTrials.gov. Safety and Efficacy of a Single Subretinal Injection of JWK006 Gene Therapy in Subjects with Stargardt Disease (STGD1). 8 March 2024. Available online: https://clinicaltrials.gov/study/NCT06300476 (accessed on 15 July 2025).
- Bank, D. Sonpiretigene Isteparvovec. 6 March 2025. Available online: https://go.drugbank.com/drugs/DB17844?utm (accessed on 15 July 2025).
- Allen, C.H. MCO-010 optogenetic gene therapy for severe vision loss in Stargardt disease. Opthalmol. Times, 17 November 2023; p. 48. [Google Scholar]
- Akula, M.; McNamee, S.M.; Love, Z.; Nasraty, N.; Chan, N.P.M.; Whalen, M.; Avola, M.O.; Olivares, A.M.; Leehy, B.D.; Jelcick, A.S.; et al. Retinoic acid related orphan receptor alpha is a genetic modifier that rescues retinal degeneration in a mouse model of Stargardt disease and Dry AMD. Gene Ther. 2024, 31, 413–421. [Google Scholar] [CrossRef] [PubMed]
- San, W.; Zhou, Q.; Shen, D.; Cao, D.; Chen, Y.; Meng, G. Roles of retinoic acid-related orphan receptor alpha in high glucose-induced cardiac fibroblasts proliferation. Front. Pharmacol. 2025, 16, 1539690. [Google Scholar] [CrossRef] [PubMed]
- Boukhtouche, F.; Vodjdani, G.; Jarvis, C.I.; Bakouche, J.; Staels, B.; Mallet, J.; Mariani, J.; Lemaigre-Dubreuil, Y.; Brugg, B. Human retinoic acid receptor-related orphan receptor alpha1 overexpression protects neurones against oxidative stress-induced apoptosis. J. Neurochem. 2006, 96, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Phase 1/2 Study to Assess the Safety and Efficacy of OCU410ST for STARGARDT DISEASE. 12 March 2024. Available online: https://clinicaltrials.gov/study/NCT05956626 (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase Ib, Open-Label Study to Evaluate the Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Tinlarebant in Japanese Patients with Stargardt Disease and a Phase II/III, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety, Tolerability, and Efficacy of Tinlarebant in Japanese Patients with Stargardt Disease. 28 November 2024. Available online: https://clinicaltrials.gov/study/NCT06388083 (accessed on 15 July 2025).
- ClinicalTrials.gov. Phase 3, Multicenter, Randomized, Double-Masked, Placebo-Controlled Study to Evaluate the Safety and Efficacy of Tinlarebant in the Treatment of Stargardt Disease in Adolescent Subjects. 7 June 2024. Available online: https://clinicaltrials.gov/study/NCT05244304 (accessed on 15 July 2025).
- ClinicalTrials.gov. Phase 1/2, Open-Label, Dose-Finding Followed by 2-Year Extension Study to Evaluate Safety and Tolerability of Tinlarebant in Adolescent Subjects With Stargardt Disease. 2024. Available online: https://clinicaltrials.gov/study/NCT05266014#study-plan (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase 2 Multicenter, Double-Masked, Randomized, Placebo-Controlled Study to Investigate the Long Term Safety, Tolerability, Pharmacokinetics and Effects of ALK-001 on the Progression of Stargardt Disease. 27 April 2025. Available online: https://clinicaltrials.gov/study/NCT02402660 (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase 2 Multicenter, Double-Masked, Randomized, Placebo-Controlled Study to Investigate the Long Term Safety, Tolerability, Pharmacokinetics and Effects of ALK-001 on the Progression of Stargardt Disease. 21 June 2024. Available online: https://clinicaltrials.gov/study/NCT04239625 (accessed on 15 July 2025).
- ClinicalTrials.gov. Prospective, Randomised, Double-Blind Study to Assess the Therapeutic Potential of Omega-3 Fatty Acids Supplementation in Dry Macular Degeneration and Stargardt Disease (Macular Degeneration Omega-3 Study—MADEOS. 23 February 2021. Available online: https://clinicaltrials.gov/study/NCT03297515 (accessed on 15 July 2025).
- ClinicalTrials.gov. An Open Label Study to Determine the Long Term Safety, Tolerability and Biological Activity of SAR422459 in Patients With Stargardt’s Macular Degeneration. 1 October 2024. Available online: https://clinicaltrials.gov/study/NCT01736592 (accessed on 15 July 2025).
- ClinicalTrials.gov. A Phase 2a, Open Label Multicenter Clinical Trial to Evaluate the Safety and Effects of a Single Intravitreal Injection of vMCO-010 Optogenetic Therapy in Subjects With Stargardt Disease. 11 September 2023. Available online: https://clinicaltrials.gov/study/NCT05417126 (accessed on 15 July 2025).
- ClinicalTrials.gov. Stem Cells Therapy in Degenerative Diseases of the Retina. 17 December 2018. Available online: https://clinicaltrials.gov/study/NCT03772938 (accessed on 15 July 2025).
- U.S. Food and Drug Administration. Stem Cell Ophthalmology Treatment Study (SCOTS). 23 October 2019. Available online: https://clinicaltrials.gov/study/NCT01920867 (accessed on 15 July 2025).
- ClinicalTrials.gov. Bone Marrow Derived Stem Cell Ophthalmology Treatment Study II. 16 April 2024. Available online: https://clinicaltrials.gov/study/NCT03011541 (accessed on 15 July 2025).
- Weiss, J.N.; Levy, S. Stem Cell Ophthalmology Treatment Study (SCOTS): Bone Marrow-Derived Stem Cells in the Treatment of Stargardt Disease. Medicines 2020, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Ocugen. Ocugen Announces Positive Opinion of EMA’s Committee for Advanced Therapies for ATMP Classification for Novel Modifier Gene Therapy Candidate OCU410 for Geographic Atrophy and OCU410ST for Stargardt Disease. Available online: https://ir.ocugen.com/news-releases/news-release-details/ocugen-announces-positive-opinion-emas-committee-advanced (accessed on 15 July 2025).
- Ocugen. Ocugen, Inc. Announces Dosing Completion in the Phase 2 ArMaDa Clinical Trial for OCU410—A Multifunctional Modifier Gene Therapy for the Treatment of Geographic Atrophy Secondary to Dry Age-Related Macular Degeneration. 12 February 2025. Available online: https://ir.ocugen.com/node/13586/pdf (accessed on 15 July 2025).
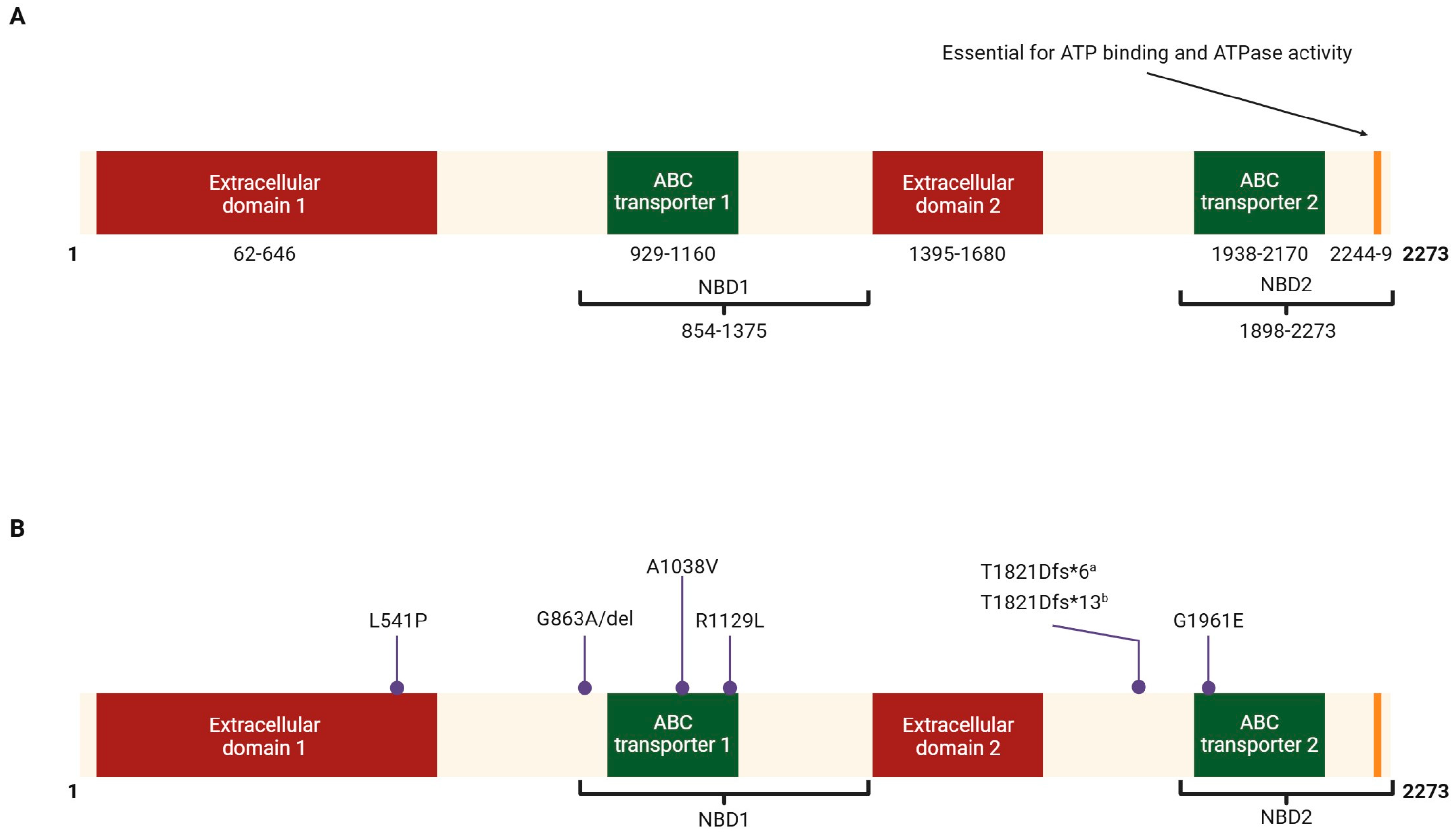

| Variant | Amino Acid Change | Prevalence | Country |
|---|---|---|---|
| 3386G>T | R1129L | 26% [38] | Spain |
| 10% [39] | Argentina | ||
| 5882G>A | G1961E | 20% [40] | Italy |
| 15% [41] | USA | ||
| 18% [34] | USA | ||
| 13% [37] | Germany | ||
| 10% [39] | Argentina | ||
| ~8% [38] | Spain | ||
| 3113C>T | A1038V | 9% [37] | Germany |
| 18% [34] | USA | ||
| 2894A>G | N965S | 10% [42] | China |
| 1622T>C | L541P | 9% [37] | Germany |
| 2588G>C | G863A, G863del | 7% [37] | Germany |
| 5461-10T>C | T1821Vfs*13, T1821Dfs*6 | 6% [37] | Germany |
| 5% [41] | USA |
| Disease | FAF Features |
|---|---|
| Stargardt disease | Central oval area of reduced AF, often surrounded by irregular AF |
| Best disease | Central round structure with regular or irregular intense AF |
| Rod-cone dystrophies | Central oval ring-shaped area of increased AF |
| Early-onset severe retinal dystrophy (with RPE65 mutations) | Complete absence of AF |
| Leber congenital amaurosis | AF is normal (except in EOSRD with RPE65 mutations) |
| X-linked retinoschisis | Central radial structures of AF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dayma, K.; Rajanala, K.; Upadhyay, A. Stargardt’s Disease: Molecular Pathogenesis and Current Therapeutic Landscape. Int. J. Mol. Sci. 2025, 26, 7006. https://doi.org/10.3390/ijms26147006
Dayma K, Rajanala K, Upadhyay A. Stargardt’s Disease: Molecular Pathogenesis and Current Therapeutic Landscape. International Journal of Molecular Sciences. 2025; 26(14):7006. https://doi.org/10.3390/ijms26147006
Chicago/Turabian StyleDayma, Kunal, Kalpana Rajanala, and Arun Upadhyay. 2025. "Stargardt’s Disease: Molecular Pathogenesis and Current Therapeutic Landscape" International Journal of Molecular Sciences 26, no. 14: 7006. https://doi.org/10.3390/ijms26147006
APA StyleDayma, K., Rajanala, K., & Upadhyay, A. (2025). Stargardt’s Disease: Molecular Pathogenesis and Current Therapeutic Landscape. International Journal of Molecular Sciences, 26(14), 7006. https://doi.org/10.3390/ijms26147006