
New Advances in Small Molecules Targeted at Viral Capsid–Genome Binding



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structural Features of Cp–Genome Binding
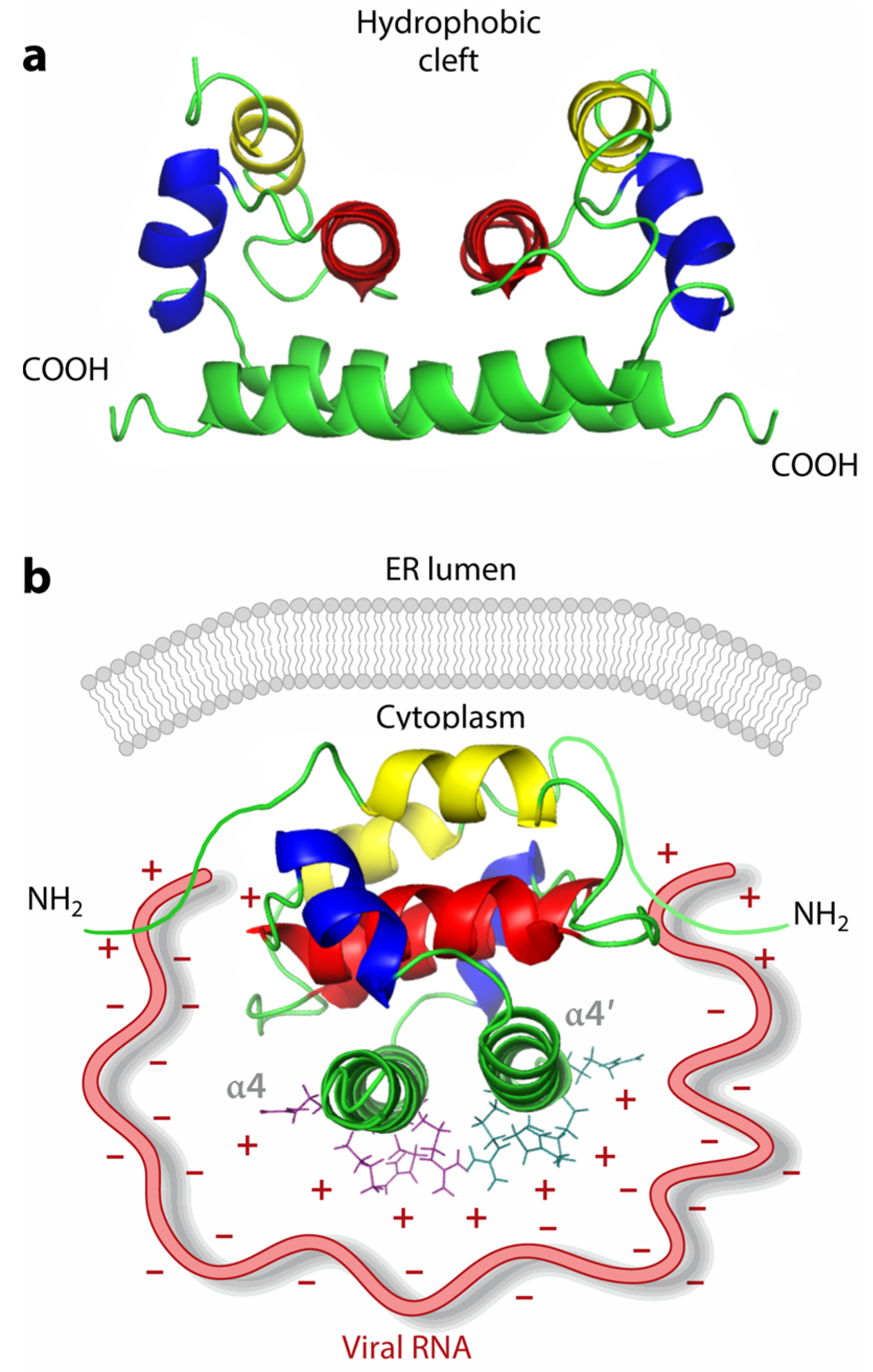
2.1. General Structure and Virus Composition
2.2. Genome-Binding Sites on Coat Proteins
2.3. Protein-Binding Sites on Genome
3. Small Molecules Targeting Capsid Protein–Genome Binding
3.1. Small Molecules Disrupting Capsid Subunit Interactions
3.2. Small-Molecule Inhibitors for Dengue Viruses
3.3. Example of CpAMs Targeting on Hepatitis B Virus
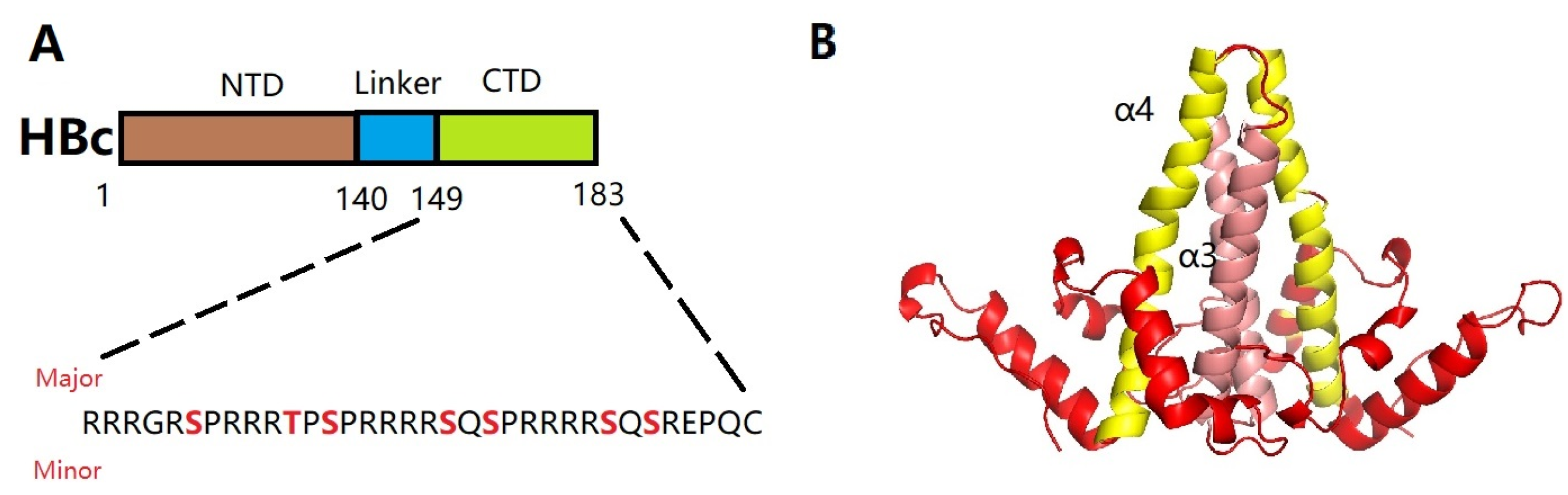
3.3.1. Hepatitis B Virus Nucleocapsid Assembly
3.3.2. A Brief Introduction to Research on CpAM
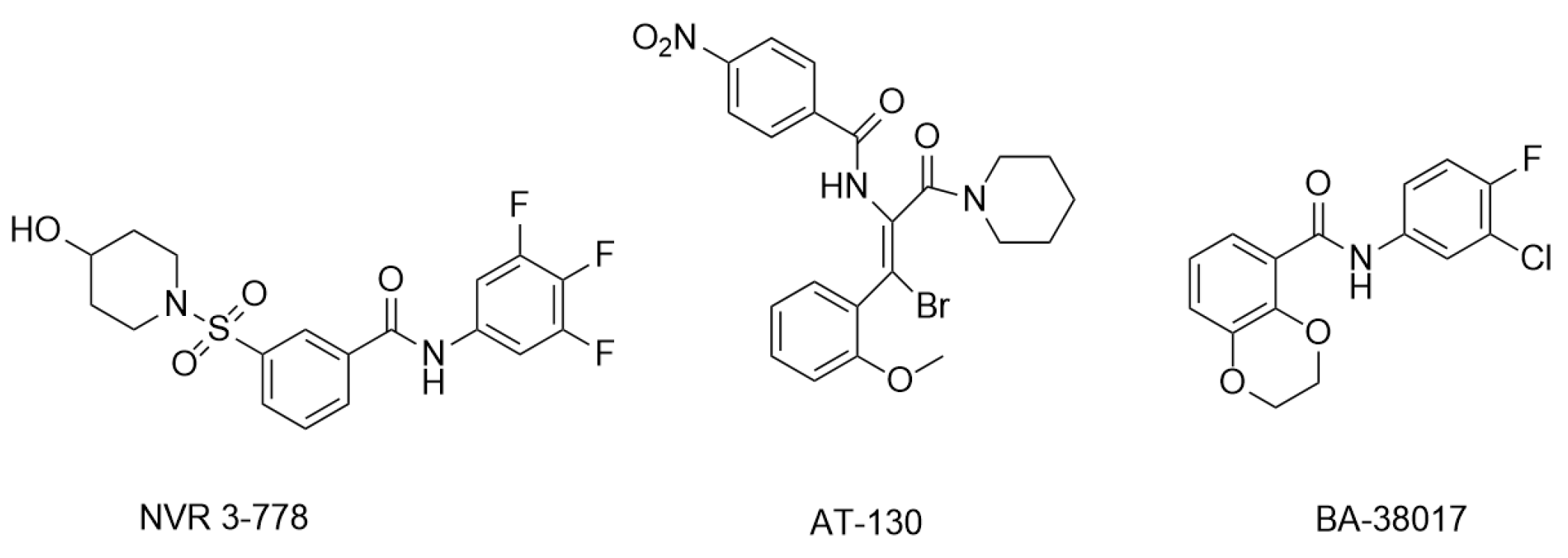
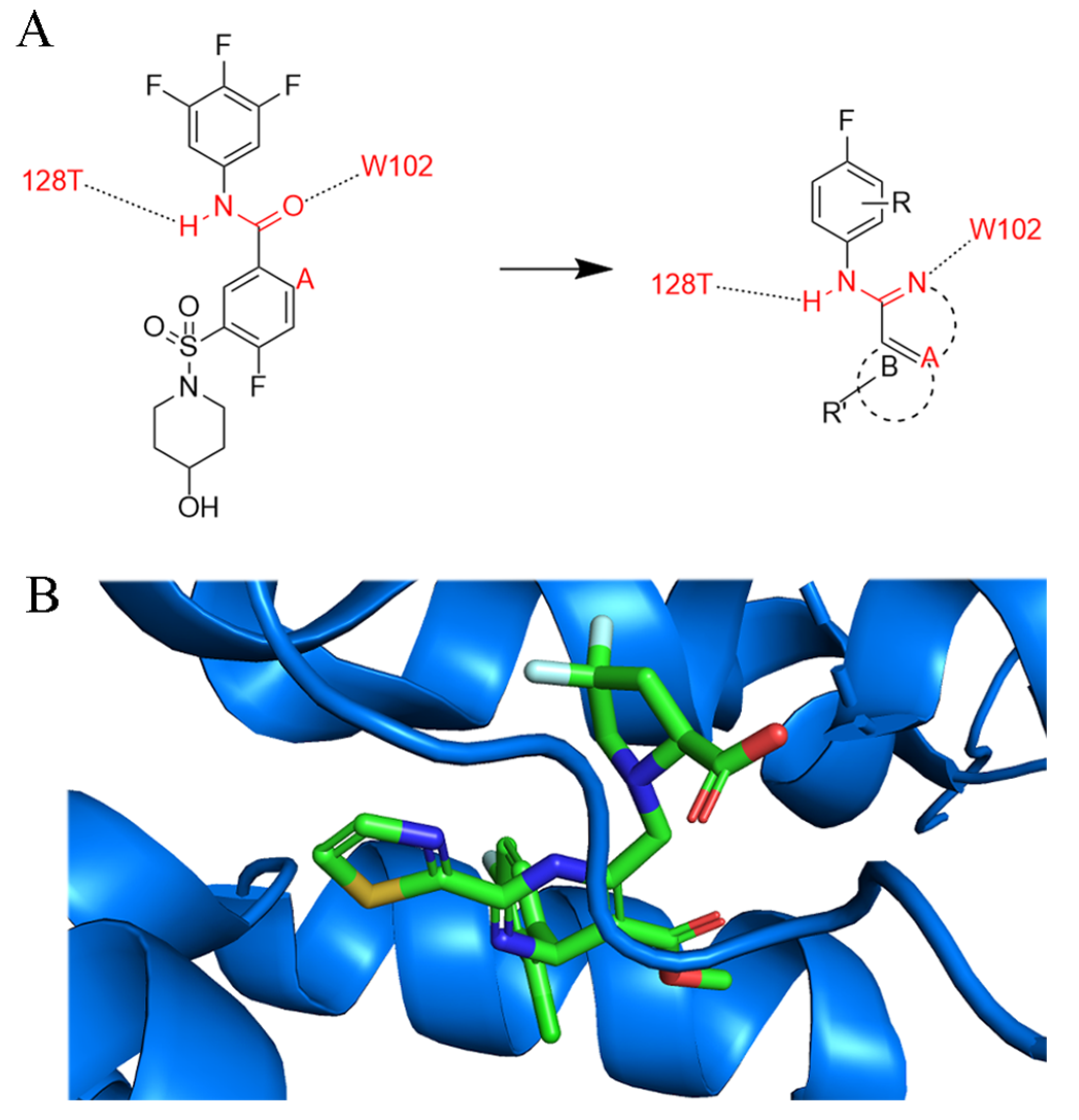
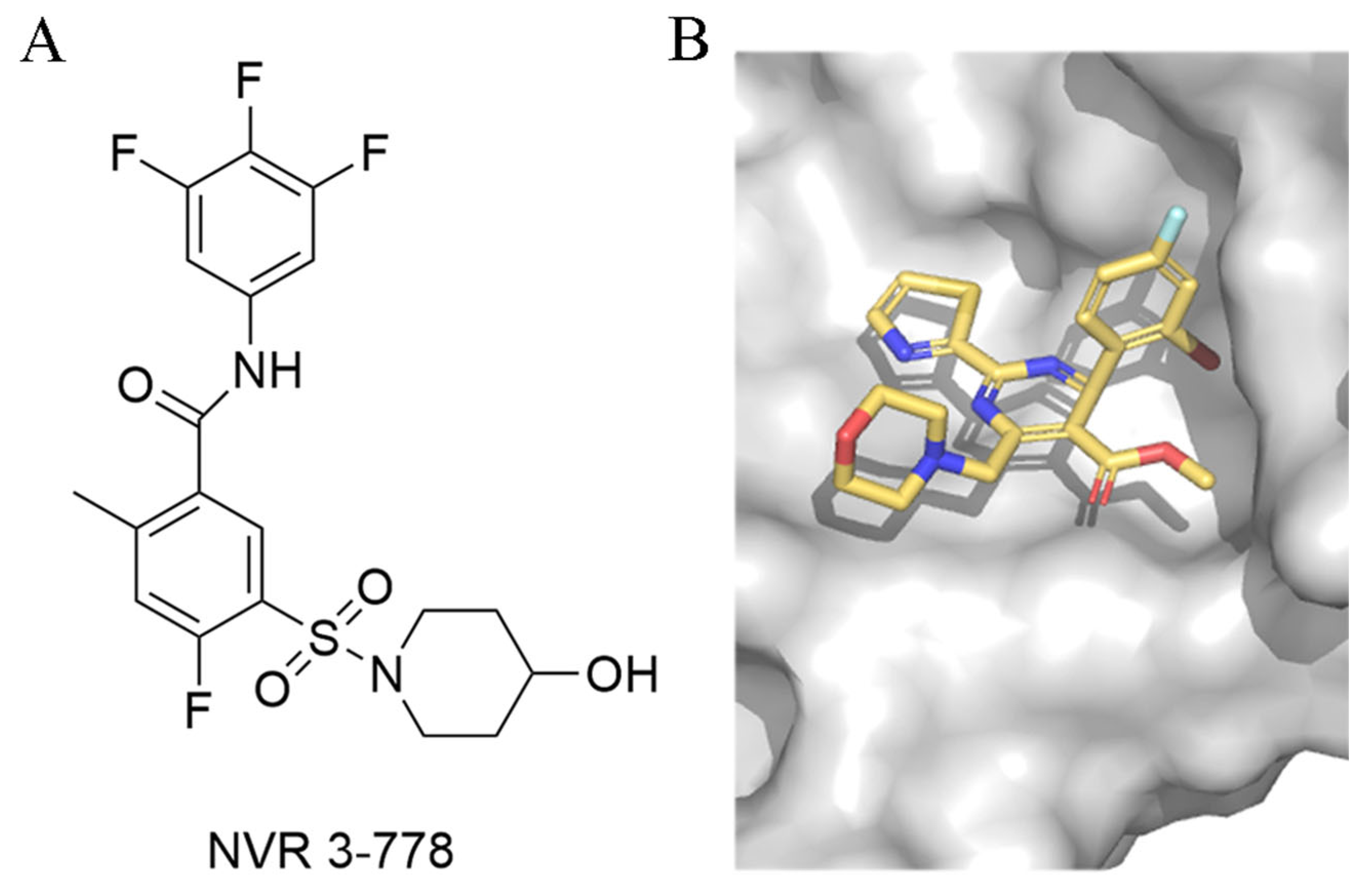
3.3.3. Sulfamoylbenzamide and Its Derivatives
3.3.4. Other Newly Developed Type-II CpAM
3.4. Small-Molecule Inhibitors for SARS-CoV-2
3.5. Summary on the Discovery of Potential Small Molecules
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallace, V.J.; Sakowski, E.G.; Preheim, S.P.; Prasse, C. Bacteria exposed to antiviral drugs develop antibiotic cross-resistance and unique resistance profiles. Commun. Biol. 2023, 6, 837. [Google Scholar] [CrossRef] [PubMed]
- Dinata, R.; Baindara, P.; Mandal, S.M. Evolution of Antiviral Drug Resistance in SARS-CoV-2. Viruses 2025, 17, 722. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Taylor, A. Arthritogenic Alphavirus Capsid Protein. Life 2021, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Airo, A.M.; Felix-Lopez, A.; Mancinelli, V.; Evseev, D.; Lopez-Orozco, J.; Shire, K.; Paszkowski, P.; Frappier, L.; Magor, K.E.; Hobman, T.C. Flavivirus Capsid Proteins Inhibit the Interferon Response. Viruses 2022, 14, 968. [Google Scholar] [CrossRef] [PubMed]
- Wołek, K.; Cieplak, M. Self-assembly of model proteins into virus capsids. J. Phys. Condens. Matter 2017, 29, 474003. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Song, H.; Shi, Y.; Qi, J.; Gao, G.F. Crystal Structure of the Capsid Protein from Zika Virus. J. Mol. Biol. 2018, 430, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Valiente, L.; Riomoros-Barahona, V.; Gil-Redondo, J.C.; Castón, J.R.; Valbuena, A.; Mateu, M.G. A RNA Dodecahedral Cage Inside a Human Virus Plays a Dual Biological Role in Virion Assembly and Genome Release Control. J. Mol. Biol. 2025, 437, 168922. [Google Scholar] [CrossRef] [PubMed]
- Sabir, A.J.; Le, N.P.K.; Singh, P.P.; Karniychuk, U. Endogenous ZAP affects Zika virus RNA interactome. RNA Biol. 2024, 21, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.M.; Brown, D.T.; Rothnagel, R.; Chiu, W.; Schoepp, R.J.; Johnston, R.E.; Prasad, B.V. Three-dimensional structure of a membrane-containing virus. Proc. Natl. Acad. Sci. USA 1993, 90, 9095–9099. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.I.; Ren, J.; Wang, X.; Rao, Z.; Fry, E.E. Hepatitis A Virus Capsid Structure. Cold Spring Harb. Perspect. Med. 2019, 9, a031807. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, L.; Carey, B.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Capsid—The Clever Caper. Viruses 2017, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sun, Z.; Cui, C.; Wang, S.; Yang, D.; Shi, Z.; Wei, X.; Wang, P.; Sun, W.; Zhu, J.; et al. Structural Insights into Alphavirus Assembly Revealed by the Cryo-EM Structure of Getah Virus. Viruses 2022, 14, 327. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Caetano-Anollés, G. Identification of Capsid/Coat Related Protein Folds and Their Utility for Virus Classification. Front. Microbiol. 2017, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Neves-Martins, T.C.; Mebus-Antunes, N.C.; Caruso, I.P.; Almeida, F.C.L.; Da Poian, A.T. Unique structural features of flaviviruses’ capsid proteins: New insights on structure-function relationship. Curr. Opin. Virol. 2021, 47, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Morando, M.A.; Barbosa, G.M.; Cruz-Oliveira, C.; Da Poian, A.T.; Almeida, F.C.L. Dynamics of Zika Virus Capsid Protein in Solution: The Properties and Exposure of the Hydrophobic Cleft Are Controlled by the α-Helix 1 Sequence. Biochemistry 2019, 58, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhao, Q.; Yang, X.; Chen, C.; Yang, K.; Wu, C.; Zhang, T.; Duan, Y.; Xue, X.; Mi, K.; et al. Structural insight into the Zika virus capsid encapsulating the viral genome. Cell Res. 2018, 28, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Requião, R.D.; Carneiro, R.L.; Moreira, M.H.; Ribeiro-Alves, M.; Rossetto, S.; Palhano, F.L.; Domitrovic, T. Viruses with different genome types adopt a similar strategy to pack nucleic acids based on positively charged protein domains. Sci. Rep. 2020, 10, 5470. [Google Scholar] [CrossRef] [PubMed]
- Sotcheff, S.; Routh, A. Understanding Flavivirus Capsid Protein Functions: The Tip of the Iceberg. Pathogens 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.; Slon Campos, J.L.; Poggianella, M.; Burrone, O.R. Dengue virus capsid anchor modulates the efficiency of polyprotein processing and assembly of viral particles. J. Gen. Virol. 2019, 100, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Barnard, T.R.; Abram, Q.H.; Lin, Q.F.; Wang, A.B.; Sagan, S.M. Molecular Determinants of Flavivirus Virion Assembly. Trends Biochem. Sci. 2021, 46, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Jablunovsky, A.; Jose, J. The Dynamic Landscape of Capsid Proteins and Viral RNA Interactions in Flavivirus Genome Packaging and Virus Assembly. Pathogens 2024, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Gil-Cantero, D.; Mata, C.P.; Valiente, L.; Rodríguez-Huete, A.; Valbuena, A.; Twarock, R.; Stockley, P.G.; Mateu, M.G.; Castón, J.R. Cryo-EM of human rhinovirus reveals capsid-RNA duplex interactions that provide insights into virus assembly and genome uncoating. Commun. Biol. 2024, 7, 1501. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, S.; Westerhuis, B.M.; Domanska, A.; Koning, R.I.; Matadeen, R.; Koster, A.J.; Bakker, A.Q.; Beaumont, T.; Wolthers, K.C.; Butcher, S.J. Multiple capsid-stabilizing interactions revealed in a high-resolution structure of an emerging picornavirus causing neonatal sepsis. Nat. Commun. 2016, 7, 11387. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2009, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Byk, L.A.; Gamarnik, A.V. Properties and Functions of the Dengue Virus Capsid Protein. Annu. Rev. Virol. 2016, 3, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Neves-Martins, T.C.; Mebus-Antunes, N.C.; Neto, C.H.G.; Barbosa, G.M.; Almeida, F.C.L.; Caruso, I.P.; Poian, A.T.D. Self-assembly of dengue virus empty capsid-like particles in solution. iScience 2023, 26, 106197. [Google Scholar] [CrossRef] [PubMed]
- Panday, H.; Jha, A.K.; Dwivedi, V.D. Investigation of small molecules disrupting dengue virus assembly by inhibiting capsid protein and blocking RNA encapsulation. Mol. Divers. 2024, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jones, C.T.; Groesch, T.D.; Kuhn, R.J.; Post, C.B. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. USA 2004, 101, 3414–3419. [Google Scholar] [CrossRef] [PubMed]
- Mebus-Antunes, N.C.; Ferreira, W.S.; Barbosa, G.M.; Neves-Martins, T.C.; Weissmuller, G.; Almeida, F.C.L.; Da Poian, A.T. The interaction of dengue virus capsid protein with negatively charged interfaces drives the in vitro assembly of nucleocapsid-like particles. PLoS ONE 2022, 17, e0264643. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Majumder, S.; Datta, J.; Giri, K. Computational Insights on the Assembly of the Dengue Virus Membrane–Capsid–RNA Complex. J. Membr. Biol. 2025, 258, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Dörnbrack, K.; Beck, J.; Nassal, M. Relaxing the restricted structural dynamics in the human hepatitis B virus RNA encapsidation signal enables replication initiation in vitro. PLoS Pathog. 2022, 18, e1010362. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Luckenbaugh, L.; Hu, J. Multiple roles of PP2A binding motif in hepatitis B virus core linker and PP2A in regulating core phosphorylation state and viral replication. PLoS Pathog. 2021, 17, e1009230. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Schneider, W.M.; Rice, C.M. Hepatitis B Virus Nucleocapsid Assembly. J. Mol. Biol. 2025, 30, 169182. [Google Scholar] [CrossRef] [PubMed]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Böttcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ban, H.; Zheng, H.; Liu, M.; Chang, J.; Guo, J.-T. Protein phosphatase 1 catalyzes HBV core protein dephosphorylation and is co-packaged with viral pregenomic RNA into nucleocapsids. PLoS Pathog. 2020, 16, e1008669. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.Y. Okuda lecture: Challenges of hepatitis B in the era of antiviral therapy. J. Gastroenterol. Hepatol. 2019, 34, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Gu, Z.; Sun, C.; Chen, Z.; Zhang, T.; Chen, R.; Liu, T.; Liao, H.; Zou, J.; Yang, D.; et al. A novel hepatitis B virus capsid assembly modulator QL-007 inhibits HBV replication and infection through altering capsid assembly. Antivir. Res. 2023, 218, 105715. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antivir. Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Yuen Chan, H.L.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pei, Y.; Wang, L.; Li, S.; Jiang, C.; Tan, X.; Dong, Y.; Xiang, Y.; Ma, Y.; Liu, G. Discovery of (1H-Pyrazolo[3,4-c]pyridin-5-yl)sulfonamide Analogues as Hepatitis B Virus Capsid Assembly Modulators by Conformation Constraint. J. Med. Chem. 2020, 63, 6066–6089. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Dou, Y.; Liang, M.; Xie, Y.; Xue, P.; Liu, L.; Li, C.; Wang, Y.; Tao, F.; et al. Design, Synthesis, and Biological Evaluation of Novel Thioureidobenzamide (TBA) Derivatives as HBV Capsid Assembly Modulators. J. Med. Chem. 2023, 66, 13968–13990. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.G.; Kultgen, S.G.; Mani, N.; Fan, K.Y.; Ardzinski, A.; Stever, K.; Dorsey, B.D.; Mesaros, E.F.; Thi, E.P.; Graves, I.; et al. Rational Design, Synthesis, and Structure–Activity Relationship of a Novel Isoquinolinone-Based Series of HBV Capsid Assembly Modulators Leading to the Identification of Clinical Candidate AB-836. J. Med. Chem. 2024, 67, 16773–16795. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Mani, N.; Ardzinski, A.; Stever, K.; Cuconati, A.; Micolochick Steuer, H.; Thi, E.P.; Graves, I.E.; Espiritu, C.L.; Mesaros, E.; et al. Preclinical and clinical antiviral characterization of AB-836, a potent capsid assembly modulator against hepatitis B virus. Antivir. Res. 2024, 231, 106010. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jia, H.; Guo, X.; Desta, S.; Zhang, S.; Zhang, J.; Ding, X.; Liang, X.; Liu, X.; Zhan, P. Design, synthesis, and evaluation of novel heteroaryldihydropyrimidine derivatives as non-nucleoside hepatitis B virus inhibitors by exploring the solvent-exposed region. Chem. Biol. Drug Des. 2019, 95, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T. The SARS coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Koetzner, C.A.; Masters, P.S. A key role for the carboxy-terminal tail of the murine coronavirus nucleocapsid protein in coordination of genome packaging. Virology 2016, 494, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Han, Y.; Yu, J.; Zhou, R.; Lei, J. Structural basis of the C-terminal domain of SARS-CoV-2 N protein in complex with GMP reveals critical residues for RNA interaction. Bioorg. Med. Chem. Lett. 2024, 114, 130014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Long, X.-Y.; Ding, X.; Fan, S.-R.; Cai, J.-Y.; Yang, B.-J.; Zhang, X.-F.; Luo, R.; Yang, L.; Ruan, T.; et al. Novel nucleocapsid protein-targeting phenanthridine inhibitors of SARS-CoV-2. Eur. J. Med. Chem. 2022, 227, 113966. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cui, Y.; Han, X.; Hu, W.; Sun, M.; Zhang, Y.; Wang, P.-H.; Song, G.; Chen, W.; Lou, J. Liquid–liquid phase separation by SARS-CoV-2 nucleocapsid protein and RNA. Cell Res. 2020, 30, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yu, Y.; Sun, L.-M.; Xing, J.-Q.; Li, T.; Zhu, Y.; Wang, M.; Yu, Y.; Xue, W.; Xia, T.; et al. GCG inhibits SARS-CoV-2 replication by disrupting the liquid phase condensation of its nucleocapsid protein. Nat. Commun. 2021, 12, 2114. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Li, T.; Song, J. ATP and nucleic acids competitively modulate LLPS of the SARS-CoV2 nucleocapsid protein. Commun. Biol. 2023, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kang, J.; Lim, L.; Wei, Y.; Song, J. TDP-43 NTD can be induced while CTD is significantly enhanced by ssDNA to undergo liquid-liquid phase separation. Biochem. Biophys. Res. Commun. 2018, 499, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Ukmar-Godec, T.; Hutten, S.; Grieshop, M.P.; Rezaei-Ghaleh, N.; Cima-Omori, M.-S.; Biernat, J.; Mandelkow, E.; Söding, J.; Dormann, D.; Zweckstetter, M. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 2019, 10, 2909. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, C.; Li, B.; Wu, Y. New Advances in Small Molecules Targeted at Viral Capsid–Genome Binding. Int. J. Mol. Sci. 2025, 26, 6979. https://doi.org/10.3390/ijms26146979
Li J, Zhang C, Li B, Wu Y. New Advances in Small Molecules Targeted at Viral Capsid–Genome Binding. International Journal of Molecular Sciences. 2025; 26(14):6979. https://doi.org/10.3390/ijms26146979
Chicago/Turabian StyleLi, Jiamei, Chengfeng Zhang, Benteng Li, and Yuqing Wu. 2025. "New Advances in Small Molecules Targeted at Viral Capsid–Genome Binding" International Journal of Molecular Sciences 26, no. 14: 6979. https://doi.org/10.3390/ijms26146979
APA StyleLi, J., Zhang, C., Li, B., & Wu, Y. (2025). New Advances in Small Molecules Targeted at Viral Capsid–Genome Binding. International Journal of Molecular Sciences, 26(14), 6979. https://doi.org/10.3390/ijms26146979