

Effects of Targeted Radionuclide Therapy on Cancer Cells Beyond the Ablative Radiation Dose

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Current General Aspects of Targeted Radionuclide Therapy
2.1. Radiopharmaceuticals as the Basis of Targeted Radionuclide Therapy
2.2. Theranostics in Nuclear Medicine
2.3. Recent Clinical Outcomes of Targeted Radionuclide Therapy
3. Immediate Molecular and Cellular Responses to Ablative Radiation Dose
3.1. DNA Direct and Indirect Damage Induced by Radiation
3.2. DNA Damage Mediated by ROS and RNS
3.3. Molecular Pathways Involved in DNA Damage as a Response to Ablative Radiation Doses in Tumors
3.4. Cellular Signaling and Apoptosis
3.5. Apoptosis and Cell Death
3.6. Cell-Cycle Arrest and Checkpoint Activation
4. Tumor Microenvironment Changes: Immune-Mediated Cell Death Induced by Targeted Radionuclide Therapy
5. Overall Efficacy of Targeted Radionuclide Therapy Mediated by Long-Term and Non-Targeted Effects
6. Conclusions and Future Directions
7. Summarizing Limitations and Challenges of Targeted Radionuclide Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AEs | Abscopal effects |
| APCs | Antigen-presenting cells |
| ATM | Ataxia telangiectasia mutated |
| ATR | Rad3-related kinases |
| BAK | BCL2 homologous antagonist killer |
| BAX | BCL2-associated X |
| BER | Base excision repair |
| BRCA2 | Breast Cancer gene 2 |
| CAFs | Cancer-associated fibroblasts |
| cGAS | Cyclic GMP–AMP synthase |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| DDR | DNA damage response |
| DNA-PK | DNA-dependent protein kinase complex |
| DOTA | 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DSBs | Double-strand breaks |
| EGFR | Epidermal growth factor receptor |
| FAP | Fibroblast activation protein |
| FEN1 | Flap endonuclease 1 |
| GLUT1 | Glucose transporter 1 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HMGB1 | High-mobility group box 1 protein |
| HR | Homologous recombination |
| ICs | Immune checkpoints |
| iPD-L1 | Programmed death ligand 1 inhibitor |
| LDHR | Low-dose hyper-radiosensitivity |
| LDRT | Low-dose radiotherapy |
| MAPK | Mitogen-activated protein kinasa |
| mCRPC | Metastatic castration-resistance prostate cancer |
| MHC | Major histocompatibility complex |
| MIBG | Meta-iodobenzylguanidine |
| MMR | Non-targeted effects |
| NETs | Neuroendocrine tumors |
| NF-κB | Nuclear Factor kappa B |
| NHEJ | Non-homologous end joining |
| NK | Natural killer cells |
| NTEs | Non-targeted effects |
| OGG1 | 8-oxoguanina |
| PD-1 | Programmed cell death protein 1 |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| PD-L1 | Programmed death ligand 1 |
| PCNA | Proliferating cell nuclear antigen |
| PSMA | Prostate-specific membrane antigen |
| RIARs | Radiation-induced adaptive responses |
| RIBEs | Radiation-induced bystander effects |
| RICEs | Radiation-induced cohort effects |
| RIF | Radiation-induced foci |
| RIGI | Radiation-induced genomic instability |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SSBs | Single-strand breaks |
| SSTR | Somatostatin receptors |
| STING | Stimulator of interferon genes |
| TAMs | Tumor-associated macrophages |
| TATE | Tyr3-octreotate |
| TGF-β | Transforming Growth Factor-beta |
| TOC | Tyr3-octreotide |
| TP53 | Tumor Protein 53 |
| TRT | Targeted radionuclide therapy |
| TTAMs | Tumor-associated macrophages |
| VLA-4 | Very late antigen-4 |
| XLF | XRCC4-like factor |
| XRCC1 | X-ray repair cross-complementing protein 1 |
References
- Gow, M.; Seymour, C.; Boyd, M.; Mairs, R.; Prestiwch, W.; Mothersill, C. Dose Calculations for [131I] Meta-Iodobenzylguanidine-Induced Bystander Effects. Dose-Response 2014, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Terashima, S.; Tatemura, R.; Saito, W.; Hosokawa, Y. Evaluation of the influence of radiation-induced cohort effect in cell populations receiving different doses. Int. J. Radiat. Biol. 2025, 101, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.; Shields, L.; Hynes, C.; Howe, O.; McCleanc, B.; Lynga, F. DNA damage and cytokine production in non-target irradiated lymphocytes. Radiat. Res. 2019, 191, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.; Mairs, R.J.; Keith, W.N.; Ross, S.C.; Welsh, P.; Akabani, G.; Owens, J.; Vaidyanathan, G.; Carruthers, R.; Dorrens, J. An efficient targeted radiotherapy/gene therapy strategy utilising human telomerase promoters and radioastatine and harnessing radiation-mediated bystander effects. J. Gene Med. A Cross-Discip. J. Res. Sci. Gene Transf. Its Clin. Appl. 2004, 6, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, N.E.; Mairs, R.J.; Kirk, D.; Keith, W.N.; Carruthers, R.; McCluskey, A.G.; Brown, M.; Wilson, L.; Boyd, M. Application of targeted radiotherapy/gene therapy to bladder cancer cell lines. Eur. Urol. 2005, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, R.; Chan, C.; Lu, Y.; Winnik, M.A.; Cescon, D.W.; Reilly, R.M. 90Y-labeled gold nanoparticle depot (NPD) combined with anti-PD-L1 antibodies strongly inhibits the growth of 4T1 tumors in immunocompetent mice and induces an abscopal effect on a distant non-irradiated tumor. Mol. Pharm. 2022, 19, 4199–4211. [Google Scholar] [CrossRef] [PubMed]
- Nabrinsky, E.; Macklis, J.; Bitran, J. A review of the abscopal effect in the era of immunotherapy. Cureus 2022, 14, e29620. [Google Scholar] [CrossRef] [PubMed]
- Zboralski, D.; Osterkamp, F.; Christensen, E.; Bredenbeck, A.; Schumann, A.; Hoehne, A.; Schneider, E.; Paschke, M.; Ungewiss, J.; Haase, C.; et al. Fibroblast activation protein targeted radiotherapy induces an immunogenic tumor microenvironment and enhances the efficacy of PD-1 immune checkpoint inhibition. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 2621–2635. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhao, L.; Fu, K.; Lin, Q.; Wen, X.; Jacobson, O.; Sun, L.; Wu, H.; Zhang, X.; Guo, Z.; et al. Integrin αvβ3-targeted radionuclide therapy combined with immune checkpoint blockade immunotherapy synergistically enhances anti-tumor efficacy. Theranostics 2019, 9, 7948. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, M.C.; Patel, R.B.; Anderson, C.J. Combined Targeted Radiopharmaceutical Therapy and Immune Checkpoint Blockade: From Preclinical Advances to the Clinic. J. Nucl. Med. 2022, 63, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandus, J.; Fendler, W.P.; Lueckerath, K.; Berliner, C.; Kurzidem, S.; Hadaschik, E.; Klode, J.; Zimmer, L.; Livingstone, E.; Schadendorf, D.; et al. Response to Combined Peptide Receptor Radionuclide Therapy and Checkpoint Immunotherapy with Ipilimumab Plus Nivolumab in Metastatic Merkel Cell Carcinoma. J. Nucl. Med. 2022, 63, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Liu, S.V.; Subramaniam, D.S.; Torres, T.; Loda, M.; Esposito, G.; Giaccone, G. Phase I study of the (177)Lu-DOTA(0)-Tyr(3)-Octreotate (lutathera) in combination with nivolumab in patients with neuroendocrine tumors of the lung. J. Immunother. Cancer. 2020, 8, e000980. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Starzinski, S.; de Kouchkovsky, I.; Koshkin, V.; Bose, R.; Chou, J.; Desai, A.; Kwon, D.; Kaushal, S.; Trihy, L.; et al. Single-dose (177)Lu-PSMA-617 followed by maintenance pembrolizumab in patients with metastatic castration-resistant prostate cancer: An open-label, dose-expansion, phase 1 trial. Lancet Oncol. 2023, 24, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Chan, T.A.; Galluzzi, L.; Constanzo, J. Radiopharmaceuticals as combinatorial partners for immune checkpoint inhibitors. Trends Cancer 2023, 9, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, P.; Cruciani, V.; Berg-Larsen, A.; Schlicker, A.; Mobergslien, A.; Bartnitzky, L.; Berndt, S.; Zitzmann-Kolbe, S.; Kamfenkel, C.; Stargard, S.; et al. Immunostimulatory effects of targeted thorium-227 conjugates as single agent and in combination with anti-PD-L1 therapy. J. Immunother. Cancer 2021, 9, e002387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, X.; Gao, X.; Chen, X.; Li, L.; Li, G.; Liu, C.; Miao, Y.; Wang, R.; Hu, K. Radiopharmaceuticals and their applications in medicine. Signal Transduct. Target. Ther. 2025, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Ailawadhi, S.; Pafundi, D.; Peterson, J. Advances and future directions in radiopharmaceutical delivery for cancer treatment. Expert Rev. Anticancer Ther. 2025, 25, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Lawal, I.O.; Abubakar, S.O.; Ndlovu, H.; Mokoala, K.M.G.; More, S.S.; Sathekge, M.M. Advances in Radioligand Theranostics in Oncology. Mol. Diagn. Ther. 2024, 28, 265–289. [Google Scholar] [CrossRef] [PubMed]
- Barca, C.; Griessinger, C.M.; Faust, A.; Depke, D.; Essler, M.; Windhorst, A.D.; Devoogdt, N.; Brindle, K.M.; Schäfers, M.; Zinnhardt, B.; et al. Expanding Theranostic Radiopharmaceuticals for Tumor Diagnosis and Therapy. Pharmaceuticals 2021, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Suman, S.K.; Mukherjee, A. Antibody-based radiopharmaceuticals as theranostic agents: An overview. Curr. Med. Chem. 2022, 29, 5979–6005. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Wang, H.; Yu, B.; Li, Q.; Gan, L.; Chen, W. Astatine-211 and actinium-225: Two promising nuclides in targeted alpha therapy. Acta Biochim. Biophys. Sin. 2024, 57, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Ocampo-García, B.; Cruz-Nova, P.; Jiménez-Mancilla, N.; Luna-Gutiérrez, M.; Oros-Pantoja, R.; Lara-Almazán, N.; Pérez-Velasco, D.; Santos-Cuevas, C.; Ferro-Flores, G. (225)Ac-iPSMA-RGD for Alpha-Therapy Dual Targeting of Stromal/Tumor Cell PSMA and Integrins. Int. J. Mol. Sci. 2023, 24, 16553. [Google Scholar] [CrossRef] [PubMed]
- Solnes, L.B.; Shokeen, M.; Pandit-Taskar, N. Novel Agents and Future Perspectives on Theranostics. Semin. Radiat. Oncol. 2021, 31, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Mirshahvalad, S.A.; Beheshti, M.; Metser, U.; Jiang, D.M.; Wong, R.; Alrekhais, I.; Veit-Haibach, P. Theranostic: A Primer for Radiologists. Can. Assoc. Radiol. J. 2025, 8465371251338032. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu, S.; Has-Şimşek, D. Evaluating the efficacy of [177Lu] Lu-FAP-2286 in combination therapy for metastatic breast cancer. Eur. J. Nucl. Med. Mol. Imaging 2024, 51, 4188–4189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Su, X.; Xiang, B.; Zhang, S.; Zhou, X. Application of (68)Ga- and (177)Lu-Labeled FAP Inhibitor in Evaluation and Treatment of Cardiac Fibrosis After Myocardial Infarction. MedComm 2025, 6, e70198. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Benítez, D.; Luna-Gutiérrez, M.; Aguirre-De Paz, J.G.; Cruz-Nova, P.; Bravo-Villegas, G.; Vargas-Ahumada, J.E.; Vallejo-Armenta, P.; Morales-Avila, E.; Jiménez-Mancilla, N.; Oros-Pantoja, R.; et al. (68)Ga-DOTA-D-Alanine-BoroPro Radiotracer for Imaging of the Fibroblast Activation Protein in Malignant and Non-Malignant Diseases. Pharmaceutics 2024, 16, 532. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Yao, J.; Zhao, Z.; Zhang, X.; Lu, J. Application of 99mTc-Labeled WL12 peptides as a tumor PD-L1-Targeted SPECT Imaging Agent: Kit formulation, preclinical evaluation, and study on the influence of Coligands. Pharmaceuticals 2024, 17, 906. [Google Scholar] [CrossRef] [PubMed]
- Boschi, A.; Urso, L.; Uccelli, L.; Martini, P.; Filippi, L. 99mTc-labeled FAPI compounds for cancer and inflammation: From radiochemistry to the first clinical applications. EJNMMI Radiopharm. Chem. 2024, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Beaino, W.; Fecek, R.J.; Fabian, K.P.L.; Laymon, C.M.; Kurland, B.F.; Storkus, W.J.; Anderson, C.J. Combined VLA-4-Targeted Radionuclide Therapy and Immunotherapy in a Mouse Model of Melanoma. J. Nucl. Med. 2018, 59, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Rouanet, J.; Benboubker, V.; Akil, H.; Hennino, A.; Auzeloux, P.; Besse, S.; Pereira, B.; Delorme, S.; Mansard, S.; D’Incan, M.; et al. Immune checkpoint inhibitors reverse tolerogenic mechanisms induced by melanoma targeted radionuclide therapy. Cancer Immunol. Immunother. 2020, 69, 2075–2088. [Google Scholar] [CrossRef] [PubMed]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Rickles, D.; Dadachova, E. Evaluating the Combination of Radioimmunotherapy and Immunotherapy in a Melanoma Mouse Model. Int. J. Mol. Sci. 2020, 21, 773. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.E.; Allen, K.J.H.; Jiao, R.B.; Frank, C.; Rickles, D.; Dadachova, E. Mechanistic Insights into Synergy between Melanin-Targeting Radioimmunotherapy and Immunotherapy in Experimental Melanoma. Int. J. Mol. Sci. 2020, 21, 8721. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.; Nigam, S.; Tatum, D.S.; Raphael, I.; Xu, J.D.; Kumar, R.; Plakseychuk, E.; Latoche, J.D.; Vincze, S.; Li, B.; et al. Novel theranostic agent for PET imaging and targeted radiopharmaceutical therapy of tumour-infiltrating immune cells in glioma. EBioMedicine 2021, 71, 10. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.Y.; Xu, M.X.; Chen, J.Y.; Ding, J.; Wang, P.P.; Huo, L.; Li, F.; Liu, Z.B. PET imaging facilitates antibody screening for synergistic radioimmunotherapy with a 177Lu-labeled αPD-L1 antibody. Theranostics 2021, 11, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Vito, A.; Rathmann, S.; Mercanti, N.; El-Sayes, N.; Mossman, K.; Valliant, J. Combined Radionuclide Therapy and Immunotherapy for Treatment of Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4843. [Google Scholar] [CrossRef] [PubMed]
- Dabagian, H.; Taghvaee, T.; Martorano, P.; Martinez, D.; Samanta, M.; Watkins, C.M.; Chai, R.; Mansfield, A.; Graham, T.J.; Maris, J.M.; et al. PARP Targeted Alpha-Particle Therapy Enhances Response to PD-1 Immune-Checkpoint Blockade in a Syngeneic Mouse Model of Glioblastoma. ACS Pharmacol. Transl. Sci. 2021, 4, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Czernin, J.; Current, K.; Mona, C.E.; Nyiranshuti, L.; Hikmat, F.; Radu, C.G.; Lückerath, K. Immune-Checkpoint Blockade Enhances (225)Ac-PSMA617 Efficacy in a Mouse Model of Prostate Cancer. J. Nucl. Med. 2021, 62, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Jagodinsky, J.C.; Jin, W.J.; Bates, A.M.; Hernandez, R.; Grudzinski, J.J.; Marsh, I.R.; Chakravarty, I.; Arthur, I.S.; Zangl, L.M.; Brown, R.J.; et al. Temporal analysis of type 1 interferon activation in tumor cells following external beam radiotherapy or targeted radionuclide therapy. Theranostics 2021, 11, 6120–6137. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, D.; Lee, D.; Cheng, Y.; Baumhover, N.J.; Marks, B.M.; Sagastume, E.A.; Ballas, Z.K.; Johnson, F.L.; Morris, Z.S.; et al. Targeted Alpha-Particle Radiotherapy and Immune Checkpoint Inhibitors Induces Cooperative Inhibition on Tumor Growth of Malignant Melanoma. Cancers 2021, 13, 3676. [Google Scholar] [CrossRef] [PubMed]
- Guzik, P.; Siwowska, K.; Fang, H.Y.; Cohrs, S.; Bernhardt, P.; Schibli, R.; Müller, C. Promising potential of [(177)Lu]Lu-DOTA-folate to enhance tumor response to immunotherapy-a preclinical study using a syngeneic breast cancer model. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.D.; Heidari, P.; Ataeinia, B.; Sinevici, N.; Granito, A.; Kumar, H.M.; Wehrenberg-Klee, E.; Mahmood, U. Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide. Pharmaceutics 2022, 14, 1460. [Google Scholar] [CrossRef]
- Wen, X.; Zeng, X.; Cheng, X.; Zeng, X.; Liu, J.; Zhang, Y.; Li, Y.; Chen, H.; Huang, J.; Guo, Z.; et al. PD-L1-Targeted Radionuclide Therapy Combined with αPD-L1 Antibody Immunotherapy Synergistically Improves the Antitumor Effect. Mol. Pharm. 2022, 19, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
- Potluri, H.K.; Ferreira, C.A.; Grudzinski, J.; Massey, C.; Aluicio-Sarduy, E.; Engle, J.W.; Kwon, O.; Marsh, I.R.; Bednarz, B.P.; Hernandez, R.; et al. Antitumor efficacy of (90)Y-NM600 targeted radionuclide therapy and PD-1 blockade is limited by regulatory T cells in murine prostate tumors. J. Immunother. Cancer 2022, 10, e005060. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.J.; Zeng, X.Y.; Shi, C.R.; Liu, J.; Zhang, Y.R.; Shi, M.Q.; Li, J.C.; Chen, H.J.; Zhuang, R.Q.; Chen, X.Y.; et al. Optimum Combination of Radiopharmaceuticals-Based Targeting-Triggering-Therapy Effect and PD-L1 Blockade Immunotherapy. Adv. Therap. 2023, 6, 11. [Google Scholar] [CrossRef]
- Luna-Gutiérrez, M.; Azorín-Vega, E.; Oros-Pantoja, R.; Ocampo-García, B.; Cruz-Nova, P.; Jiménez-Mancilla, N.; Bravo-Villegas, G.; Santos-Cuevas, C.; Meléndez-Alafort, L.; Ferro-Flores, G. Lutetium-177 labeled iPD-L1 as a novel immunomodulator for cancer-targeted radiotherapy. EJNMMI Radiopharm. Chem. 2025, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pang, Y.; Zhou, Y.; Chen, J.; Fu, H.; Guo, W.; Xu, W.; Xue, X.; Su, G.; Sun, L.; et al. Antitumor efficacy and potential mechanism of FAP-targeted radioligand therapy combined with immune checkpoint blockade. Signal Transduct. Target. Ther. 2024, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, S.C.; Oosterwijk, E.; Molkenboer-Kuenen, J.; Frielink, C.; Franssen, G.M.; Boreel, D.F.; Tamborino, G.; Gloudemans, M.; Hendrikx, M.; Kroon, D.; et al. Towards effective CAIX-targeted radionuclide and checkpoint inhibition combination therapy for advanced clear cell renal cell carcinoma. Theranostics 2024, 14, 3693–3707. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, Y.; Pang, Y.; Fu, K.; Luo, Q.; Sun, L.; Wu, H.; Lin, Q.; Su, G.; Chen, X.; et al. FAP-targeted radioligand therapy with (68)Ga/(177)Lu-DOTA-2P(FAPI)(2) enhance immunogenicity and synergize with PD-L1 inhibitors for improved antitumor efficacy. J. Immunother. Cancer 2025, 13, e010212. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, H.; Wu, Y.; Luo, C.; Yang, G.; Luo, Q.; Jia, B.; Han, C.; Liu, Z.; Wang, F. Nuclear imaging of PD-L1 expression promotes the synergistic antitumor efficacy of targeted radionuclide therapy and immune checkpoint blockade. Eur. J. Nucl. Med. Mol. Imaging 2025, 52, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Ramonaheng, K.; Qebetu, M.; Ndlovu, H.; Swanepoel, C.; Smith, L.; Mdanda, S.; Mdlophane, A.; Sathekge, M. Activity quantification and dosimetry in radiopharmaceutical therapy with reference to (177)Lutetium. Front. Nucl. Med. 2024, 4, 1355912. [Google Scholar] [CrossRef] [PubMed]
- Tran-Gia, J.; Cicone, F.; Koole, M.; Giammarile, F.; Gear, J.; Deshayes, E.; Gabiña, P.M.; Cremonesi, M.; Wadsley, J.; Bernhardt, P.; et al. Rethinking Dosimetry: A European Perspective. J. Nucl. Med. 2025, 66, jnumed.124.269378. [Google Scholar] [CrossRef] [PubMed]
- Yusufaly, T.; Roncali, E.; Brosch-Lenz, J.; Uribe, C.; Jha, A.K.; Currie, G.; Dutta, J.; El-Fakhri, G.; McMeekin, H.; Pandit-Taskar, N.; et al. Computational Nuclear Oncology Toward Precision Radiopharmaceutical Therapies: Current Tools, Techniques, and Uncharted Territories. J. Nucl. Med. 2025, 66, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H. Direct Correlation of Tumor Absorbed Dose with Overall Survival in Metastatic Castration-Resistant Prostate Cancer Treated with (177)Lu Prostate-Specific Membrane Antigen. J. Nucl. Med. Technol. 2025, 53, jnmt.125.269658. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, H.; Yousefirizi, F.; Shiri, I.; Brosch-Lenz, J.; Mollaheydar, E.; Fele-Paranj, A.; Shi, K.; Zaidi, H.; Alberts, I.; Soltani, M.; et al. Theranostic digital twins: Concept, framework and roadmap towards personalized radiopharmaceutical therapies. Theranostics 2024, 14, 3404–3422. [Google Scholar] [CrossRef] [PubMed]
- Brosch-Lenz, J.; Yousefirizi, F.; Zukotynski, K.; Beauregard, J.M.; Gaudet, V.; Saboury, B.; Rahmim, A.; Uribe, C. Role of Artificial Intelligence in Theranostics: Toward Routine Personalized Radiopharmaceutical Therapies. PET Clin. 2021, 16, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Filippi, L.; Chiaravalloti, A.; Schillaci, O.; Cianni, R.; Bagni, O. Theranostic approaches in nuclear medicine: Current status and future prospects. Expert Rev. Med. Devices 2020, 17, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Dabhade, A.; Bharadia, H.; Parekh, P.S.; Yadav, M.R.; Chorawala, M.R. Navigating the landscape of theranostics in nuclear medicine: Current practice and future prospects. Z. Naturforsch. C J. Biosci. 2024, 79, 235–266. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. (177)Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Sitani, K.; Parghane, R.V.; Talole, S.; Basu, S. Long-term outcome of indigenous (177)Lu-DOTATATE PRRT in patients with Metastatic Advanced Neuroendocrine Tumours: A single institutional observation in a large tertiary care setting. Br. J. Radiol. 2021, 94, 20201041. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.; Kluge, A.W.; Kulkarni, H.; Schorr-Neufing, U.; Niepsch, K.; Bitterlich, N.; van Echteld, C.J. [(177)Lu-DOTA](0)-D-Phe(1)-Tyr(3)-Octreotide ((177)Lu-DOTATOC) For Peptide Receptor Radiotherapy in Patients with Advanced Neuroendocrine Tumours: A Phase-II Study. Theranostics 2016, 6, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Luna-Gutiérrez, M.; Hernández-Ramírez, R.; Soto-Abundiz, A.; García-Pérez, O.; Ancira-Cortez, A.; López-Buenrostro, S.; Gibbens-Bandala, B.; Soldevilla-Gallardo, I.; Lara-Almazán, N.; Rojas-Pérez, M.; et al. Improving Overall Survival and Quality of Life in Patients with Prostate Cancer and Neuroendocrine Tumors Using (177)Lu-iPSMA and (177)Lu-DOTATOC: Experience after 905 Treatment Doses. Pharmaceutics 2023, 15, 1988. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Herrmann, K.; Krause, B.J.; Rahbar, K.; Chi, K.N.; Morris, M.J.; Sartor, O.; Tagawa, S.T.; Kendi, A.T.; Vogelzang, N.; et al. Health-related quality of life and pain outcomes with [(177)Lu]Lu-PSMA-617 plus standard of care versus standard of care in patients with metastatic castration-resistant prostate cancer (VISION): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023, 24, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Castellano, D.; Herrmann, K.; de Bono, J.S.; Shore, N.D.; Chi, K.N.; Crosby, M.; Piulats, J.M.; Fléchon, A.; Wei, X.X.; et al. (177)Lu-PSMA-617 versus a change of androgen receptor pathway inhibitor therapy for taxane-naive patients with progressive metastatic castration-resistant prostate cancer (PSMAfore): A phase 3, randomised, controlled trial. Lancet 2024, 404, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Emmett, L.; Subramaniam, S.; Crumbaker, M.; Joshua, A.M.; Sandhu, S.; Nguyen, A.; Weickhardt, A.; Lee, S.T.; Ng, S.; Francis, R.J.; et al. Overall survival and quality of life with [(177)Lu]Lu-PSMA-617 plus enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer (ENZA-p): Secondary outcomes from a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2025, 26, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Bülbül, O.; Ünek, İ.T.; Kefi, A.; Tuna, E.B.; Bekiş, R. Factors affecting overall survival and progression-free survival in patients with metastatic castration resistant prostate cancer received (177)Lu PSMA I&T therapy. Hell. J. Nucl. Med. 2020, 23, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; and Ghosh, A. Activation of DNA damage response signaling in mammalian cells by ionizing radiation. Free. Radic. Res. 2021, 55, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, M.; Cremonesi, M.; Indovina, L.; Chianelli, M.; Pacilio, M.; Danieli, R.; Chiesa, C.; Zanzonico, P. Radiobiology and radiation dosimetry in nuclear medicine. In Nuclear Oncology: From Pathophysiology to Clinical Applications; Springer: Berlin/Heidelberg, Germany, 2022; pp. 345–410. [Google Scholar]
- Khazaei Monfared, Y.; Heidari, P.; Klempner, S.J.; Mahmood, U.; Parikh, A.R.; Hong, T.S.; Strickland, M.R.; Esfahani, S.A. DNA Damage by Radiopharmaceuticals and Mechanisms of Cellular Repair. Pharmaceutics 2023, 15, 2761. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A. DNA Damage, Repair Mechanisms, and Chromosomal Aberrations. In Proton Beam Radiotherapy: Physics and Biology; Tsuboi, K., Sakae, T., Gerelchuluun, A., Eds.; Springer: Singapore, 2020; pp. 183–208. [Google Scholar]
- Salvatori, M.; Cremonesi, M.; Indovina, L.; Chianelli, M.; McEwan, A.J.B.; Zanzonico, P. Radiobiology and Radiation Dosimetry in Nuclear Medicine: Therapy, Diagnosis, and Considerations for Sensitive Populations. In Nuclear Oncology: Pathophysiology and Clinical Applications; Strauss, H.W., Mariani, G., Volterrani, D., Larson, S.M., Eds.; Springer: New York, NY, USA, 2013; pp. 121–149. [Google Scholar]
- Fabbrizi, M.R.; Doggett, T.J.; Hughes, J.R.; Melia, E.; Dufficy, E.R.; Hill, R.M.; Goula, A.; Phoenix, B.; Parsons, J.L. Inhibition of key DNA double strand break repair protein kinases enhances radiosensitivity of head and neck cancer cells to X-ray and proton irradiation. Cell Death Discov. 2024, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand–break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Bussink, J.; Span, P.N. γ-H2AX foci in peripheral blood lymphocytes to quantify radiation-induced DNA damage after 177Lu-DOTA-Octreotate peptide receptor radionuclide therapy. J. Nucl. Med. 2015, 56, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Tamborino, G.; Nonnekens, J.; De Saint-Hubert, M.; Struelens, L.; Feijtel, D.; de Jong, M.; Konijnenberg, M.W. Dosimetric evaluation of the effect of receptor heterogeneity on the therapeutic efficacy of peptide receptor radionuclide therapy: Correlation with DNA damage induction and in vivo survival. J. Nucl. Med. 2022, 63, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Schumann, S.; Scherthan, H.; Lapa, C.; Serfling, S.; Muhtadi, R.; Lassmann, M.; Eberlein, U. DNA damage in blood leucocytes of prostate cancer patients during therapy with 177 Lu-PSMA. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, U.; Nowak, C.; Bluemel, C.; Buck, A.K.; Werner, R.A.; Scherthan, H.; Lassmann, M. DNA damage in blood lymphocytes in patients after 177 Lu peptide receptor radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Ranzani, M.; Hindupur, S.; Cicconi, A.; Sastre-Moreno, G.; Kristian, A.; Engstler, B.S.; Pinch, B.; Hartnagel, C.; Gorses, D.; Simon, E. Abstract LB222: Elucidating the cellular responses and mechanism of action of 177Lu-based radioligand therapy. Cancer Res. 2024, 84, LB222. [Google Scholar] [CrossRef]
- Ritt, P.; Jobic, C.; Beck, M.; Schmidkonz, C.; Kuwert, T.; Uder, M.; Brand, M. Dissimilar DNA damage to blood lymphocytes after 177Lu-labeled DOTATOC or prostate-specific membrane antigen therapy. J. Nucl. Med. 2021, 62, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Bertolet, A.; Ramos-Méndez, J.; Paganetti, H.; Schuemann, J. The relation between microdosimetry and induction of direct damage to DNA by alpha particles. Phys. Med. Biol. 2021, 66, 155016. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.J. Radiation biology. Alpha-particle after effects. Nature 1992, 355, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Göring, L.; Schumann, S.; Müller, J.; Buck, A.K.; Port, M.; Lassmann, M.; Scherthan, H.; Eberlein, U. Repair of α-particle-induced DNA damage in peripheral blood mononuclear cells after internal ex vivo irradiation with 223Ra. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Bannik, K.; Madas, B.; Jarzombek, M.; Sutter, A.; Siemeister, G.; Mumberg, D.; Zitzmann-Kolbe, S. Radiobiological effects of the alpha emitter Ra-223 on tumor cells. Sci. Rep. 2019, 9, 18489. [Google Scholar] [CrossRef] [PubMed]
- Howell, R.W. Advancements in the use of Auger electrons in science and medicine during the period 2015–2019. Int. J. Radiat. Biol. 2023, 99, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Lyu, X.; Yuan, S.; Wang, S.; Li, W.; Chen, Z.; Yu, H.; Li, f.; Jiang, Q. Oxidative stress: A critical hint in ionizing radiation induced pyroptosis. Radiat. Med. Prot. 2020, 1, 179–185. [Google Scholar] [CrossRef]
- Zheng, Z.; Su, J.; Bao, X.; Wang, H.; Bian, C.; Zhao, Q.; Jiang, X. Mechanisms and applications of radiation-induced oxidative stress in regulating cancer immunotherapy. Front. Immunol. 2023, 14, 1247268. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yu, L.; Wang, J.; Gao, X.; Chen, Y.; Pan, W.; Tang, B. A mitochondria-targeted nanoradiosensitizer activating reactive oxygen species burst for enhanced radiation therapy. Chem. Sci. 2018, 9, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Sun, X.; Zhao, H.; Guan, H.; Gao, S.; Zhou, P.K. Double-strand DNA break repair: Molecular mechanisms and therapeutic targets. MedComm 2023, 4, e388. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, F.P.; Safwat, A.; Kassem, M.; Gautier, L.; Overgaard, J.; Horsman, M.R. Cancer stem cell overexpression of nicotinamide N-methyltransferase enhances cellular radiation resistance. Radiother. Oncol. 2011, 99, 373–378. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, F.P. Intrinsic radiation resistance of mesenchymal cancer stem cells and implications for treatment response in a murine sarcoma model. Dan. Med. J. 2012, 59, B4388. [Google Scholar] [PubMed]
- Van Haren, M.J.; Zhang, Y.; Thijssen, V.; Buijs, N.; Gao, Y.; Mateuszuk, L.; Fedak, F.A.; Kij, A.; Campagna, R.; Sartini, D. Macrocyclic peptides as allosteric inhibitors of nicotinamide N-methyltransferase (NNMT). RSC Chem. Biol. 2021, 2, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Van Haren, M.J.; Buijs, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W. Potent inhibition of nicotinamide N-methyltransferase by alkene-linked bisubstrate mimics bearing electron deficient aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef] [PubMed]
- van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; Escudé Martinez de Castilla, P. Esterase-sensitive prodrugs of a potent bisubstrate inhibitor of nicotinamide N-methyltransferase (NNMT) display cellular activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Yacoub, A.; Contessa, J.; Caron, R.; Amorino, G.; Valerie, K.; Hagan, M.P.; Grant, S.; Schmidt-Ullrich, R. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat. Res. 2003, 159, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, H.; Liu, Y.; Zhang, H.; Tian, M. Radionuclide imaging of apoptosis for clinical application. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Lundsten, S.; Spiegelberg, D.; Stenerlöw, B.; Nestor, M. The HSP90 inhibitor onalespib potentiates 177Lu-DOTATATE therapy in neuroendocrine tumor cells. Int. J. Oncol. 2019, 55, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Lundsten, S.; Berglund, H.; Jha, P.; Krona, C.; Hariri, M.; Nelander, S.; Lane, D.P.; Nestor, M. p53-Mediated Radiosensitization of 177Lu-DOTATATE in Neuroblastoma Tumor Spheroids. Biomolecules 2021, 11, 1695. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.C.; Tse, B.W.C.; Fletcher, N.L.; Houston, Z.H.; Lund, M.; Volpert, M.; Stewart, C.; Sokolowski, K.A.; Jeet, V.; Thurecht, K.J.; et al. Targeted beta therapy of prostate cancer with (177)Lu-labelled Miltuximab® antibody against glypican-1 (GPC-1). EJNMMI Res. 2020, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. Mechanisms of cell killing response from low linear energy transfer (LET) radiation originating from 177Lu radioimmunotherapy targeting disseminated intraperitoneal tumor xenografts. Int. J. Mol. Sci. 2016, 17, 736. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.C.; Knox, S.J. Radiobiology of radioimmunotherapy with 90Y ibritumomab tiuxetan (Zevalin). In Seminars in Oncology; W.B. Saunders: Philadelphia, PA, USA, 2003; pp. 6–10. [Google Scholar]
- Suominen, M.I.; Fagerlund, K.M.; Rissanen, J.P.; Konkol, Y.M.; Morko, J.P.; Peng, Z.; Alhoniemi, E.J.; Laine, S.K.; Corey, E.; Mumberg, D. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin. Cancer Res. 2017, 23, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Singh Jaggi, J.; Henke, E.; Seshan, S.V.; Kappel, B.J.; Chattopadhyay, D.; May, C.; McDevitt, M.R.; Nolan, D.; Mittal, V.; Benezra, R.; et al. Selective alpha-particle mediated depletion of tumor vasculature with vascular normalization. PLoS ONE 2007, 2, e267. [Google Scholar] [CrossRef]
- Kumar, P.; Koach, J.; Nekritz, E.; Mukherjee, S.; Braun, B.S.; DuBois, S.G.; Nasholm, N.; Haas-Kogan, D.; Matthay, K.K.; Weiss, W.A.; et al. Aurora Kinase A inhibition enhances DNA damage and tumor cell death with (131)I-MIBG therapy in high-risk neuroblastoma. EJNMMI Res. 2024, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Norenberg, J.P.; Anderson, T.L.; Prossnitz, E.R.; Stabin, M.G.; Atcher, R.W. Somatostatin-receptor-targeted alpha-emitting 213Bi is therapeutically more effective than beta(-)-emitting 177Lu in human pancreatic adenocarcinoma cells. Nucl. Med. Biol. 2007, 34, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Kim, J.-Y.; Jun, Y.; Nam, J.-S. Strategies to tackle radiation resistance by penetrating cancer stem cell line of scrimmage. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 18–39. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, L.; Fu, D.; Liu, W.; Puri, A.; Kellis, M.; Yang, J. Antigen presenting cells in cancer immunity and mediation of immune checkpoint blockade. Clin. Exp. Metastasis 2024, 41, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Ascic, E.; Pereira, C.F. Transcription factor-mediated reprogramming to antigen-presenting cells. Curr. Opin. Genet. Dev. 2025, 90, 102300. [Google Scholar] [CrossRef] [PubMed]
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Zagorulya, M.; Duong, E.; Spranger, S. Impact of anatomic site on antigen-presenting cells in cancer. J. Immunother. Cancer 2020, 8, e001204. [Google Scholar] [CrossRef] [PubMed]
- Marciscano, A.E.; Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin. Immunol. 2021, 52, 101481. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A "Game Changer" in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pfeifer, A.K.; Myschetzky, R.; Garbyal, R.S.; Rasmussen, P.; Knigge, U.; Bzorek, M.; Kristensen, M.H.; Kjaer, A. Induction of Anti-Tumor Immune Responses by Peptide Receptor Radionuclide Therapy with (177)Lu-DOTATATE in a Murine Model of a Human Neuroendocrine Tumor. Diagnostics 2013, 3, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wöhrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Dar, T.B.; Henson, R.M.; Shiao, S.L. Targeting Innate Immunity to Enhance the Efficacy of Radiation Therapy. Front. Immunol. 2018, 9, 3077. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.G.; Ronet, C.; Ochoa de Olza, M.; Barras, D.; Crespo, I.; Andreatta, M.; Corria-Osorio, J.; Spill, A.; Benedetti, F.; Genolet, R.; et al. Low-Dose Radiotherapy Reverses Tumor Immune Desertification and Resistance to Immunotherapy. Cancer Discov. 2022, 12, 108–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, X.; Huang, Q.; Zhang, M.; Lei, T.; Wang, F.; Zou, W.; Huang, R.; Hu, X.; Wang, C.; et al. Single-cell RNA-sequencing reveals radiochemotherapy-induced innate immune activation and MHC-II upregulation in cervical cancer. Signal Transduct. Target. Ther. 2023, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Zagorulya, M.; Yim, L.; Morgan, D.M.; Edwards, A.; Torres-Mejia, E.; Momin, N.; McCreery, C.V.; Zamora, I.L.; Horton, B.L.; Fox, J.G.; et al. Tissue-specific abundance of interferon-gamma drives regulatory T cells to restrain DC1-mediated priming of cytotoxic T cells against lung cancer. Immunity 2023, 56, 386–405. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.; Walker, K.L.; Grudzinski, J.J.; Aluicio-Sarduy, E.; Patel, R.; Zahm, C.D.; Pinchuk, A.N.; Massey, C.F.; Bitton, A.N.; Brown, R.J.; et al. (90)Y-NM600 targeted radionuclide therapy induces immunologic memory in syngeneic models of T-cell Non-Hodgkin’s Lymphoma. Commun. Biol. 2019, 2, 79. [Google Scholar] [CrossRef] [PubMed]
- Malamas, A.S.; Gameiro, S.R.; Knudson, K.M.; Hodge, J.W. Sublethal exposure to alpha radiation (223Ra dichloride) enhances various carcinomas’ sensitivity to lysis by antigen-specific cytotoxic T lymphocytes through calreticulin-mediated immunogenic modulation. Oncotarget 2016, 7, 86937–86947. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hsueh, P.C.; Li, Z.; Ho, P.C. Microenvironment-driven metabolic adaptations guiding CD8(+) T cell anti-tumor immunity. Immunity 2023, 56, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Hu, C.; Zheng, X.; Sun, C.; Li, Q. Feedback loop between hypoxia and energy metabolic reprogramming aggravates the radioresistance of cancer cells. Exp. Hematol. Oncol. 2024, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Elsaidi, H.R.; Zorniak, B.; Laurens, E.; Yang, J.; Bacchu, V.; Wang, M.; Wiebe, L.I. Synthesis and Biological Evaluation of Iodoglucoazomycin (I-GAZ), an Azomycin-Glucose Adduct with Putative Applications in Diagnostic Imaging and Radiotherapy of Hypoxic Tumors. ChemMedChem 2016, 11, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer. 2020, 123, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Matarèse, B.F.E.; Rusin, A.; Seymour, C.; Mothersill, C. Quantum Biology and the Potential Role of Entanglement and Tunneling in Non-Targeted Effects of Ionizing Radiation: A Review and Proposed Model. Int. J. Mol. Sci. 2023, 24, 16464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhou, T.; He, F.; Rong, Y.; Lee, S.H.; Wu, S.; Zuo, L. Reactive oxygen species formation and bystander effects in gradient irradiation on human breast cancer cells. Oncotarget 2016, 7, 41622–41636. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Hill, H.Z.; Stein, D.; Rao, D.V.; Howell, R.W. Free radical-initiated and gap junction-mediated bystander effect due to nonuniform distribution of incorporated radioactivity in a three-dimensional tissue culture model. Radiat. Res. 2001, 155, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.Y.; Butler, N.J.; Makrigiorgos, G.M.; Adelstein, S.J.; Kassis, A.I. Bystander effect produced by radiolabeled tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.; Ross, S.C.; Dorrens, J.; Fullerton, N.E.; Tan, K.W.; Zalutsky, M.R.; Mairs, R.J. Radiation-induced biologic bystander effect elicited in vitro by targeted radiopharmaceuticals labeled with alpha-, beta-, and auger electron-emitting radionuclides. J. Nucl. Med. 2006, 47, 1007–1015. [Google Scholar] [PubMed]
- Ye, F.; Ning, J.; Liu, X.; Jin, X.; Wang, T.; Li, Q. The influence of non-DNA-targeted effects on carbon ion-induced low-dose hyper-radiosensitivity in MRC-5 cells. J. Radiat. Res. 2016, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.C.; Marples, B.; Lambin, P.; Short, S.C.; Turesson, I. Low-dose hypersensitivity: Current status and possible mechanisms. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Shetake, N.; Desai, S.; Kumar, A.; Samuel, G.; Pandey, B.N. Relevance of radiobiological concepts in radionuclide therapy of cancer. Int. J. Radiat. Biol. 2016, 92, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Morris, Z.S.; Wang, A.Z.; Knox, S.J. The Radiobiology of Radiopharmaceuticals. Semin. Radiat. Oncol. 2021, 31, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Janopaul-Naylor, J.R.; Shen, Y.; Qian, D.C.; Buchwald, Z.S. The Abscopal Effect: A Review of Pre-Clinical and Clinical Advances. Int. J. Mol. Sci. 2021, 22, 11061. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.; Correia, M.; Dias, A.G.; Pestana, A.; Soares, P.; Nunes, J.; Lima, J.; Máximo, V.; Boaventura, P. Evaluation of the role of mitochondria in the non-targeted effects of ionizing radiation using cybrid cellular models. Sci. Rep. 2020, 10, 6131. [Google Scholar] [CrossRef] [PubMed]
- Falahat, R.; Berglund, A.; Putney, R.M.; Perez-Villarroel, P.; Aoyama, S.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2013598118. [Google Scholar] [CrossRef] [PubMed]
- Amaresan, R.; Gopal, U. Cell surface GRP78: A potential mechanism of therapeutic resistant tumors. Cancer Cell Int. 2023, 23, 100. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jia, R.; Dai, Z.; Zhou, J.; Ruan, J.; Chng, W.; Cai, Z.; Zhang, X. Stress granules in cancer: Adaptive dynamics and therapeutic implications. iScience 2024, 27, 110359. [Google Scholar] [CrossRef] [PubMed]
- Swati; Chadha, V.D. Role of epigenetic mechanisms in propagating off-targeted effects following radiation based therapies—A review. Mutat. Res.-Rev. Mutat. Res. 2021, 787, 108370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Luh, F.; Ho, Y.S.; Yen, Y. Exosomes: A review of biologic function, diagnostic and targeted therapy applications, and clinical trials. J. Biomed. Sci. 2024, 31, 67. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.N.; Eke, I.; Makinde, A.Y.; Chopra, S.; Demaria, S.; Formenti, S.C.; Martello, S.; Bylicky, M.; Mitchell, J.B.; Aryankalayil, M.J. Radiation-induced Adaptive Response: New Potential for Cancer Treatment. Clin. Cancer Res. 2020, 26, 5781–5790. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Mladenov, E.; Mangelis, A.; Laskaratou, D.A.; Fragkoulis, G.I.; Hellweg, C.E.; Martin, O.A.; Emfietzoglou, D.; et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free Radic. Res. 2016, 50, S64–S78. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, C.; Acheva, A.; Salomaa, S. Clastogenic plasma factors: A short overview. Radiat. Env. Biophys. 2010, 49, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.; Du, Y. Systemic Radiotherapy of Bone Metastases With Radionuclides. Clin. Oncol. 2021, 33, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wu, X.; Yin, J.; Li, M.; Shen, J.; Li, J.; Zhao, Y.; Zhao, Q.; Wu, J.; Wen, Q.; et al. Identification of Genetic Mutations in Cancer: Challenge and Opportunity in the New Era of Targeted Therapy. Front. Oncol. 2019, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Teuter, M.; Hu, Y.; Ross, T.L.; Lolatte, K.; Ott, M.; Bengel, F.M.; Balakrishnan, A.; Bankstahl, J.P. Longitudinal multi-tracer imaging of hepatocellular carcinoma identifies novel stage- and oncogene-specific changes. Nucl. Med. Biol. 2025, 144–145, 109000. [Google Scholar] [CrossRef] [PubMed]
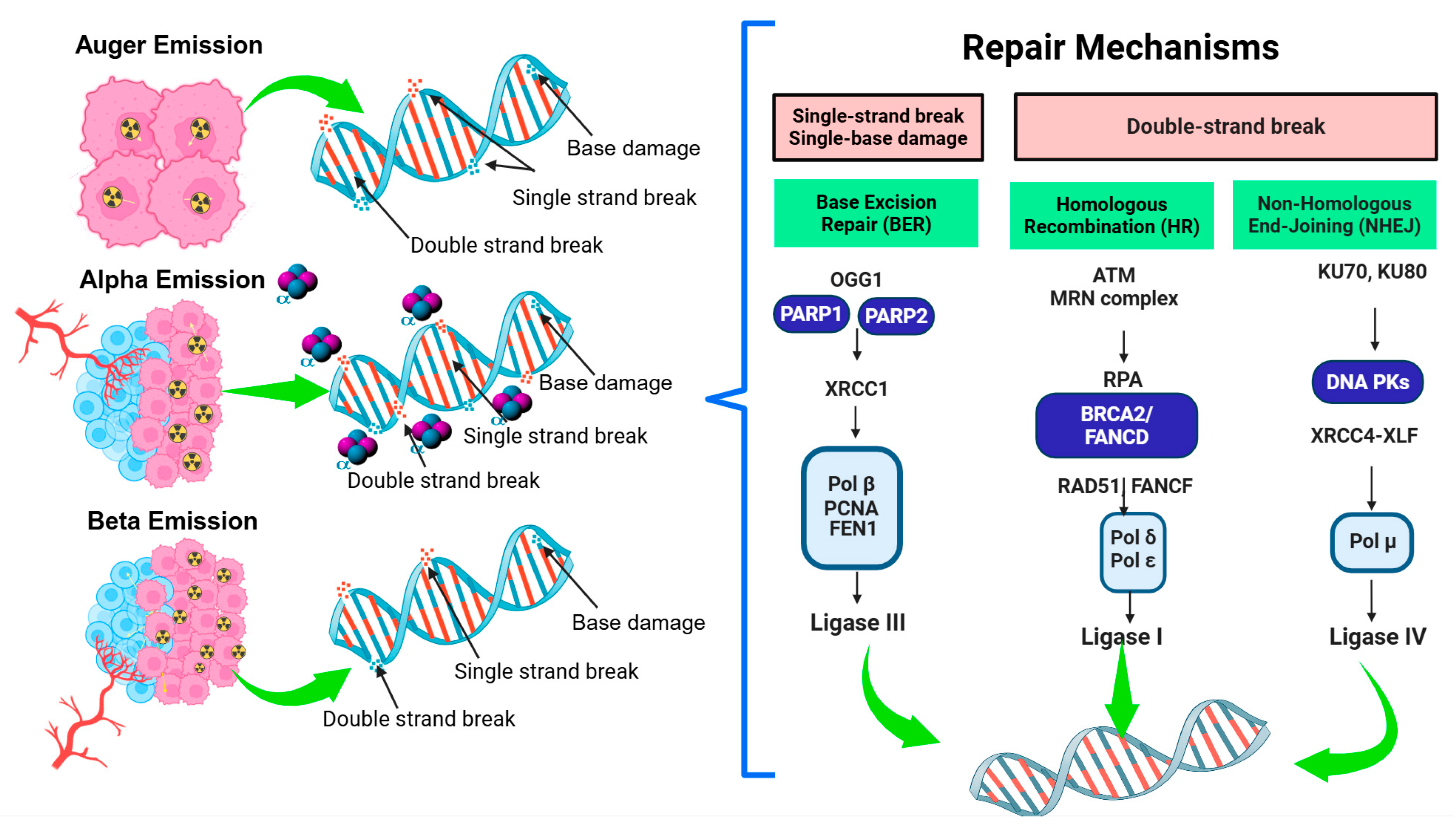


{kind=link}
{kind=link}
| Radiopharmaceutical | Target | IC Involved | Tumor Model | Results | References |
|---|---|---|---|---|---|
| 177Lu-LLP2A | Very late antigen-4 | PD-1/PD-L1, CTLA-4 | murine B16-F10 melanoma | Significant levels of apoptosis increases survival | [30] |
| 177Lu-EB-cRGDfK | integrin αvβ3 | PD-L1 | murine MC38 colon adenocarcinoma | Increases PD-L1 expression on T cells, reduces tumor volume, and increases overall survival | [9] |
| 131I-ICF01012 | melanin | PD-1/PD-L1, CTLA-4 | murine B16-F10 melanoma | Increases survival from 24 to 41 days | [31] |
| 213Bi-h8C3 | melanin | PD-1 | murine B16-F10 melanoma | Reduces tumor growth and increases survival without decreasing animal body weight | [32] |
| 177Lu-h8C3 | melanin | PD-1 | Cloudman S91 murine melanoma | Reduces tumor growth and increases survival | [33] |
| 225Ac -h8C3 | melanin | PD-1 | Cloudman S91 murine melanoma | No therapeutic efficacy | [33] |
| 227Th-TTC | mesothelin | PD-L1 | The murine cell line MC38 transfected with the human gene encoding for MSLN (hMSLN) | Increases the CD8 T-cell infiltration and the number of tumor-free surviving animals | [15] |
| 177Lu-Lumi804-αCD11 | CD11b+ cells | PD-1 CTLA-4 | GL261 glioma | Enhances the efficiency of the dual IC without altering the composition of immune cells within the TME | [34] |
| 177Lu-anti-PD-L1 | PD-L1 | PD-L1 | MC38 murine colon adenocarcinoma | Increases the infiltration of CD4+ and CD8+ T-cells | [35] |
| 177Lu-DNP-DOTA-BSA | No target | CTLA-4 PD-L1 | E0771 murine triple-negative breast cancer | Increases necrotic tissue within the tumor and decreases levels of F4/80+ macrophages | [36] |
| 211At-MM4 | PARP-1 | PD-1 | GL261 glioblastoma | Decreases tumor volume, disease-free mice were 100%. | [37] |
| 225Ac-PSMA | PSMA | PD-1 | RM1-PGLS prostate cancer | Enhances the efficiency of PD-1 | [38] |
| 90Y-NM600 | lipid rafts | PD-1 CTLA-4 | MOC2 head and neck squamous cell carcinoma | Reduces tumor growth and increases survival | [39] |
| 90Y-NM600 | lipid rafts | PD-1 CTLA-4 | B78 melanoma, B16 melanoma, 4T1 breast cancer NXS2 neuroblastoma | Low absorbed doses (2–5 Gy) activate the production of cytokines in TME, promoting tumor infiltration | [40] |
| 212Pb-VMT01 | melanocortin receptor | PD-1 CTLA-4 | B16F10 melanoma YUMM1.7 melanoma | Enhances infiltration of CD3+, CD4+ and CD8+ lymphocytes 43% of the mice showed complete tumor response | [41] |
| 177Lu-DOTA-folate | Folate receptor | CTLA-4 | NF9006 breast cancer | Reduces tumor growth and increases survival | [42] |
| 90Y-GZP | GranzymeB | PD-1 CTLA-4 | MC38 colon adenocarcinoma CT26 colon carcinoma | Promotes a dose-dependent response and increases survival | [43] |
| 131I-anti-PD-L1 | PD-L1 | PD-L1 | MC38 colon adenocarcinoma CT26 colon carcinoma | Delays significant tumor growth and prolongs survival | [44] |
| 90Y-NM600 | lipid rafts | PD-1 CTLA-4 | TRAMP-C1 prostate cancer, MycCAP Prostate cancer | Ineffective in the prostate cancer models studied | [45] |
| 177Lu-DOTA-EB-cRGDfK | integrin αvβ3 | PD-L1 | MC38 colon adenocarcinoma CT26 colon carcinoma | Inhibits tumor growth and protects against tumor recurrence | [46] |
| 177Lu-iPD-L1 | PD-L1 | PD-1/ PD-L1 | 4T1 tumors Murine triple-negative breast cancer | Substantial increase in activated macrophages, IL-10, TGF beta, and PD-L1 expression levels | [47] |
| 177Lu-FAP-2286 | FAP | PD-1 | MCA205 mouse FAP-expressing tumors | Modulates the TME and increases CD8+ T-cell infiltration, significantly inhibiting tumor growth and improving survival | [8] |
| 177Lu-LNC1004 | FAP | PD-L1 | MC38/NIH3T3-FAP and CT26/NIH3T3-FAP tumor xenografts | suppression of malignant progression and increasing cell-to-cell communication, CD8+ T-cell activation, and expansion | [48] |
| 177Lu-DOTA-girentuximab | carbonic anhydrase IX (CAIX) | PD-1 CTLA-4 | Renca-CAIX or CT26-CAIX renal cell carcinoma | T-cell infiltration and modulated immune signaling pathways in the TME with complete tumor remission. | [49] |
| 177Lu-DOTA-2P(FAPI)2 | FAP | PD-L1 | CT26-FAP colorectal tumor | Tumor suppression, infiltrating CD8+ T-cells, and 100% tumor rejection after tumor cell re-inoculation | [50] |
| 177Lu-AB-3PRGD2 | integrin αvβ3 | PD-L1 | MC38 colon adenocarcinoma, B16-F10 melanoma | Tumor suppression, infiltrating CD8+ T-cells | [51] |
| Radiopharmaceutical (Cancer Type) | Median OS (Months) | PFS (Months) | Response Rates | References |
|---|---|---|---|---|
| 177Lu-DOTA-TATE (NETs) | >48 | 25.6 | Symptomatic improvement: 71.4% Partial response: 66.7% | [60,61] |
| 177Lu-DOTA-TOC (NETs) | >44.2 | 34.7 | Objective response: 33.9% Disease control: 66.1% | [62,63] |
| 177Lu-DOTA-iPSMA (mCRPC) | 21.7 | 10.6 | PSA decline: 73% Pain reduction: 88% | [63] |
| 177Lu-PSMA-617 (mCRPC) | 15.3 (34 months if applied before chemotherapy and combined with enzalutamide) | 8.7 (11.6 months if applied before chemotherapy) | PSA decline ≥50%: Disease control: 62% | [64,65,66] |
| 177Lu-PSMA I&T (mCRPC) | 17.1 | 7.4 | PSA decline of ≥50% | [67] |
| Radiopharmaceutical | Cancer Type | Therapeutic Target | Biomarker | Activity/Dose | Mechanism of Apoptosis Induction | References |
|---|---|---|---|---|---|---|
| 177Lu-DOTA-TATE + onalespib | Neuroendocrine tumors | Somatostatin receptors (SSTR2) | Increase activity in caspase 3/7, γH2AX, p53,p21 y BAX. Reduction EGFR | 5 kBq + | Binding to SSTR2 receptors, internalization, and release of radiation into the cell → DNA damage (level of DNA double-strand breaks) → apoptosis | [98,99] |
| 177Lu-DOTA-Miltuximab | Prostate cancer | Glypican-1 (GPC-1) | Cleaved caspase 3 | 6 MBq + | Miltuximab binds specifically to Glypican-1, internalization and release of radiation into the cell → DNA damage (level of DNA double-strand breaks)→, activation of caspases 3 and 9 (intrinsic pathway), and cell apoptosis. | [100] |
| 177Lu-PSMA-617 | Metastatic prostate cancer | Prostate-specific membrane antigen (PSMA) | Increase activity in γ-H2AX+53BP1 | 6.0 GBq (01 Gy to blood) +++ | DNA damage caused by beta emission generates free radicals, activating DNA damage pathways that lead to the activation of caspases (intrinsic pathway) and cell apoptosis. | [78] |
| 177Lu-trastuzumab | Metastatic breast cancer | HER2-positive tumors | Activate caspase 3, and PARP interferes with DNA-PK expression | 13.8 MBq ++ | HER2-specific binding induced cell death (DNA double-strand breaks), activation of p53, ATM, ATR, cytochrome c release, activation of caspases 9 and 3 (intrinsic pathway), and cell apoptosis. | [101] |
| 90Y-Ibritumomab | Non-Hodgkin’s lymphoma | CD20 antigen on B-cells | Arrest of cells in the G(2)/M phase of the cell cycle increases activity in caspase | 0.3 to 0.4 Gy ++ | Binding to CD20 → internalization → radiation damage (DNA double-strand breaks) → apoptosis via caspase activation and mitochondrial damage. | [102] |
| 223RaCl2 | Castration-resistant prostate cancer (CRPC) with bone metastases | selectively binds to hydroxyapatite in bone | Increase of γ-H2AX | 300 kBq/kg ++ | High LET → irreversible DNA damage (DNA double-strand breaks) → apoptosis | [103] |
| 225Ac-E4G1 | Tumor neovasculature in prostate cancer models. | Antibody E4G10 specifically binds the form of VE-cadherin | Increase in cleaved caspase-3 | 1.85 KBq ++ | High LET → irreversible DNA damage (DNA double-strand breaks) → apoptosis | [104] |
| 131I-MIBG | Neuroendocrine tumors, such as neuroblastoma and pheochromocytoma/paraganglioma. | Cells with norepinephrine uptake | Increase in cleaved caspase-3 and PARP, Cell-cycle arrest in G2/M | 37 MBq ++ | Accumulates in synaptic vesicles → intracellular damage by β- → activation of the mitochondrial apoptosis pathway. | [105] |
| 213Bi-DOTA-TOC | Neuroendocrine tumors | Somatostatin receptors (SSTR2) | release of apoptosis-specific mono-nucleosomes and oligonucleosomes | 37 KBq + | Binding to SSTR2 receptors, internalization, High LET → lethal DNA damage (DNA double-strand breaks) → immediate apoptosis in tumor cells | [106] |
| 225Ac-PSMA-RGD | Metastatic prostate cancer and tumor angiogenesis | PSMA, integrins | Increase in caspase-3/7 | 2–4 Gy + | Binding to PSMA and integrins, internalization, High LET → lethal DNA damage (DNA double-strand breaks) → immediate apoptosis in tumor cells | [22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferro-Flores, G.; Azorín-Vega, E.; Ocampo-García, B.; Luna-Gutiérrez, M.; Cruz-Nova, P.; Meléndez-Alafort, L. Effects of Targeted Radionuclide Therapy on Cancer Cells Beyond the Ablative Radiation Dose. Int. J. Mol. Sci. 2025, 26, 6968. https://doi.org/10.3390/ijms26146968
Ferro-Flores G, Azorín-Vega E, Ocampo-García B, Luna-Gutiérrez M, Cruz-Nova P, Meléndez-Alafort L. Effects of Targeted Radionuclide Therapy on Cancer Cells Beyond the Ablative Radiation Dose. International Journal of Molecular Sciences. 2025; 26(14):6968. https://doi.org/10.3390/ijms26146968
Chicago/Turabian StyleFerro-Flores, Guillermina, Erika Azorín-Vega, Blanca Ocampo-García, Myrna Luna-Gutiérrez, Pedro Cruz-Nova, and Laura Meléndez-Alafort. 2025. "Effects of Targeted Radionuclide Therapy on Cancer Cells Beyond the Ablative Radiation Dose" International Journal of Molecular Sciences 26, no. 14: 6968. https://doi.org/10.3390/ijms26146968
APA StyleFerro-Flores, G., Azorín-Vega, E., Ocampo-García, B., Luna-Gutiérrez, M., Cruz-Nova, P., & Meléndez-Alafort, L. (2025). Effects of Targeted Radionuclide Therapy on Cancer Cells Beyond the Ablative Radiation Dose. International Journal of Molecular Sciences, 26(14), 6968. https://doi.org/10.3390/ijms26146968