

Amphiregulin in Fibrotic Diseases and Cancer


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanism of Action of AREG
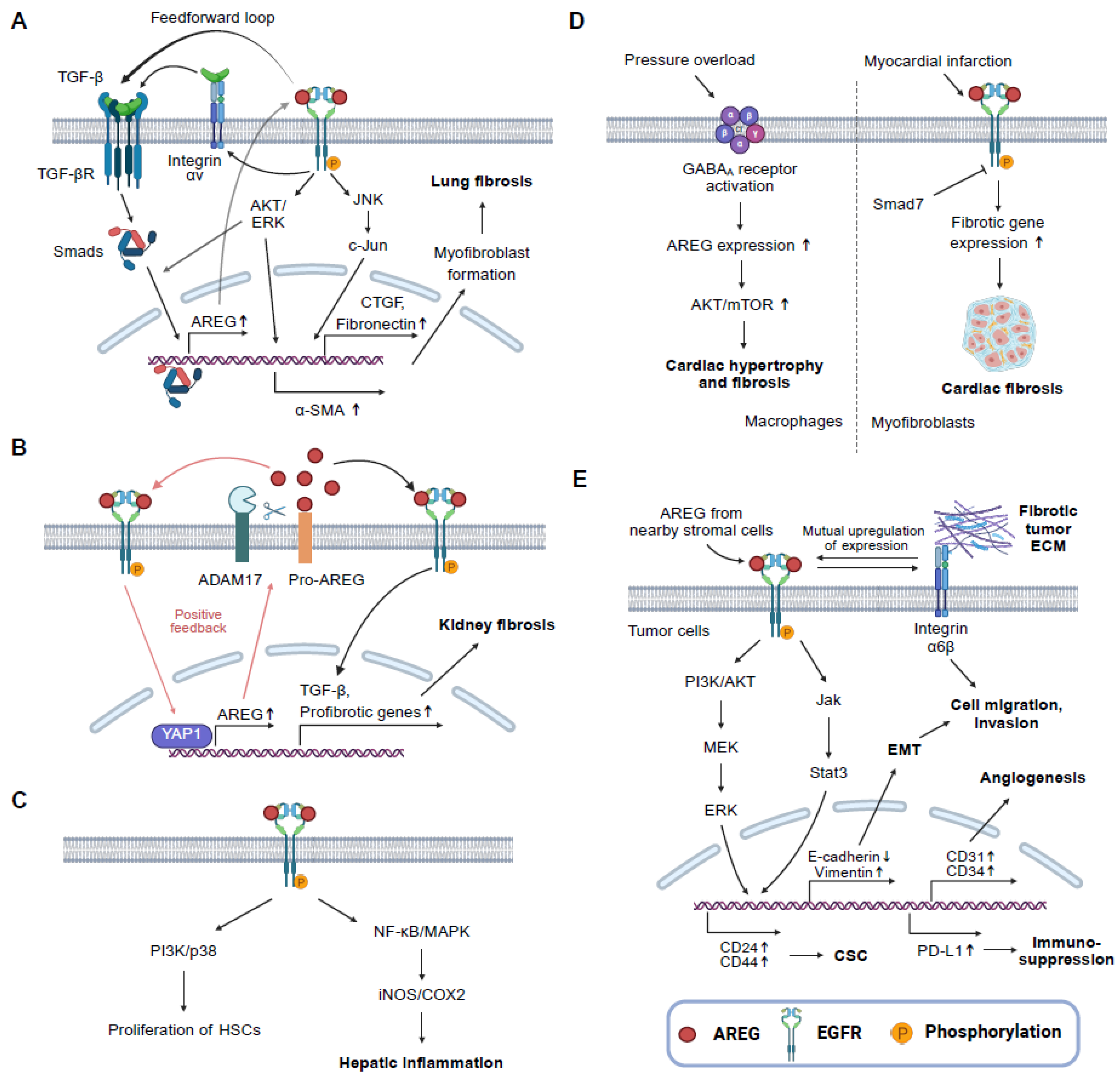
3. AREG and Fibrotic Diseases
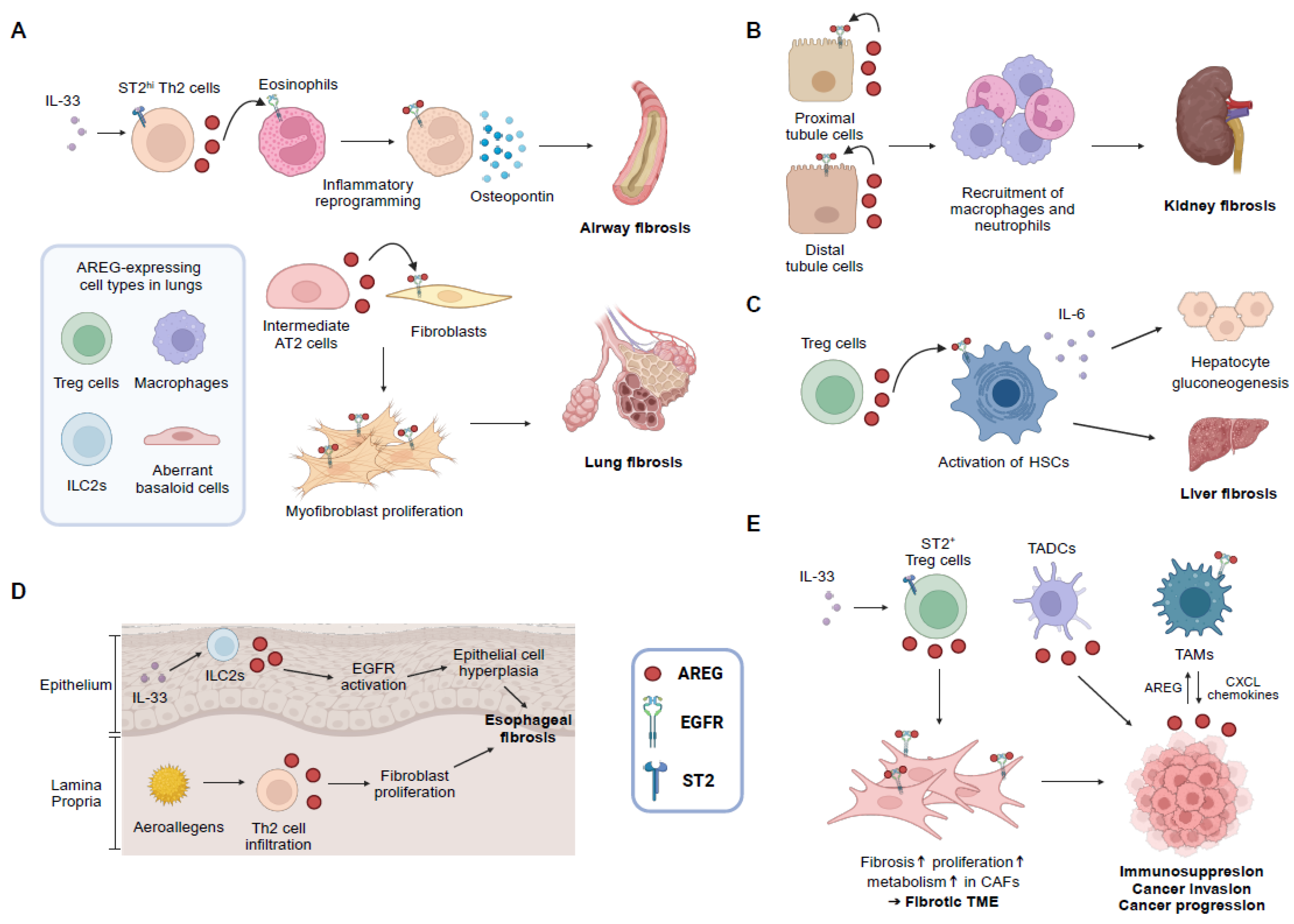
3.1. Lung Fibrosis
3.2. Kidney Fibrosis
3.3. Liver Fibrosis
3.4. Cardiac Fibrosis
3.5. Intestinal Fibrosis
3.6. Radiation-Induced Fibrosis
3.7. Other Types of Fibrosis
4. AREG and Cancer
5. Therapeutic Targeting of AREG: Preclinical and Clinical Trials for Human Application
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | A disintegrin and metalloprotease |
| AKI | Acute kidney injury |
| AREG | Amphiregulin |
| α-SMA | α-smooth muscle actin |
| CCl4 | Carbon tetrachloride |
| CKD | Chronic kidney disease |
| DKD | Diabetic kidney disease |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| EoE | Eosinophilic esophagitis |
| ERK | Extracellular signal-regulated kinase |
| FPC | Fructose, palmitate, and cholesterol-rich |
| HB-EGF | Heparin-binding EGF-like growth factor |
| HSCs | Hepatic stellate cells |
| IBD | Inflammatory bowel disease |
| IIM | Inflammatory myopathy |
| IL | Interleukin |
| ILC2s | Type 2 innate lymphoid cells |
| IPF | Idiopathic pulmonary fibrosis |
| KO | Knockout |
| MAIT | Mucosal-associated invariant T |
| MAPK | Mitogen-activated protein kinase |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MNPs | Mononuclear phagocytes |
| PD-L1 | Programmed death-ligand 1 |
| PI3K | Phosphoinositide 3-kinase |
| PLCγ | Phospholipase C-γ |
| SAMiRNA | Self-Assembled-Micelle inhibitory RNA |
| SBRT | Stereotactic body radiotherapy |
| SSc | Systemic sclerosis |
| SSc-ILD | Systemic sclerosis-associated interstitial lung disease |
| TBI | Total-body irradiation |
| TGF | Transforming growth factor |
| Th | T helper |
| TKI | Tyrosine kinase inhibitor |
| TME | Tumor microenvironment |
References
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Fibrogenesis of parenchymal organs. Proc. Am. Thorac. Soc. 2008, 5, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Guo, Y.; Li, P.; Xiong, Q.; Zhao, Y.Y. Deciphering the cellular mechanisms underlying fibrosis-associated diseases and therapeutic avenues. Pharmacol. Res. 2021, 163, 105316. [Google Scholar] [CrossRef] [PubMed]
- Lurje, I.; Gaisa, N.T.; Weiskirchen, R.; Tacke, F. Mechanisms of organ fibrosis: Emerging concepts and implications for novel treatment strategies. Mol. Asp. Med. 2023, 92, 101191. [Google Scholar] [CrossRef] [PubMed]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 2011, 13, e27. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Chakraborty, D.; Distler, J.H.W. Cellular and molecular mechanisms in fibrosis. Exp. Dermatol. 2021, 30, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Umbarkar, P.; Ejantkar, S.; Tousif, S.; Lal, H. Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models. Cells 2021, 10, 2412. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr161. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Feghali-Bostwick, C.; Soare, A.; Asano, Y.; Distler, O.; Abraham, D.J. Review: Frontiers of Antifibrotic Therapy in Systemic Sclerosis. Arthritis Rheumatol. 2017, 69, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct. Target. Ther. 2022, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Chauhan, S.B.; Kumar, A.; Kumar, S.; Engwerda, C.R.; Sundar, S.; Kumar, R. Amphiregulin in cellular physiology, health, and disease: Potential use as a biomarker and therapeutic target. J. Cell Physiol. 2022, 237, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 2019, 50, 645–654 e646. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.W. Amphiregulin as a driver of tissue fibrosis. Am. J. Transplant. 2020, 20, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Shoyab, M.; McDonald, V.L.; Bradley, J.G.; Todaro, G.J. Amphiregulin: A bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc. Natl. Acad. Sci. USA 1988, 85, 6528–6532. [Google Scholar] [CrossRef] [PubMed]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta 2011, 1816, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, J.L.; Scott, J.A.; Bouizar, Z.; Robling, A.; Pitfield, S.E.; Riese, D.J., 2nd; Foley, J. Amphiregulin-EGFR signaling regulates PTHrP gene expression in breast cancer cells. Breast Cancer Res. Treat. 2008, 110, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.W.; Johnson, J.L.; Bhasin, A.; Johnston, A.; Gudjonsson, J.E.; Rittie, L.; Elder, J.T. Metalloproteinase-mediated, context-dependent function of amphiregulin and HB-EGF in human keratinocytes and skin. J. Investig. Dermatol. 2010, 130, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Kefaloyianni, E.; Muthu, M.L.; Kaeppler, J.; Sun, X.; Sabbisetti, V.; Chalaris, A.; Rose-John, S.; Wong, E.; Sagi, I.; Waikar, S.S.; et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 2016, 1, e87023. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.W.; Stuart, P.E.; Lambert, S.; Gandarillas, A.; Rittie, L.; Johnston, A.; Elder, J.T. Membrane-Tethered Intracellular Domain of Amphiregulin Promotes Keratinocyte Proliferation. J. Investig. Dermatol. 2016, 136, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.; Minutti, C.M.; Knipper, J.A. Immune- and non-immune-mediated roles of regulatory T-cells during wound healing. Immunology 2019, 157, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Garcia-Trevijano, E.R.; Castillo, J.; Erroba, E.; Lee, D.C.; Prieto, J.; Avila, M.A. Amphiregulin: An early trigger of liver regeneration in mice. Gastroenterology 2005, 128, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Schelfhout, V.R.; Coene, E.D.; Delaey, B.; Waeytens, A.A.; De Rycke, L.; Deleu, M.; De Potter, C.R. The role of heregulin-alpha as a motility factor and amphiregulin as a growth factor in wound healing. J. Pathol. 2002, 198, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Wang, S.; Pan, Y.; Yang, J.; Yin, L.; Dou, H.; Hou, Y. Amphiregulin secreted by umbilical cord multipotent stromal cells protects against ferroptosis of macrophages via the activating transcription factor 3-CD36 axis to alleviate endometrial fibrosis. Stem Cells 2024, 42, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Santos, F.; Ding, L.; Wu, Z.; Phan, S. Amphiregulin Promotes Fibroblast Activation in Pulmonary Fibrosis. FASEB J. 2016, 30, 50–56. [Google Scholar] [CrossRef]
- Cheng, W.H.; Kao, S.Y.; Chen, C.L.; Yuliani, F.S.; Lin, L.Y.; Lin, C.H.; Chen, B.C. Amphiregulin induces CCN2 and fibronectin expression by TGF-beta through EGFR-dependent pathway in lung epithelial cells. Respir. Res. 2022, 23, 381. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, Y.; Li, Y.; Zhang, Q.; Zhong, L.; Pan, W.; Ji, K.; Zhang, S.; Chen, Z.; Liu, Y.; et al. Cancer-associated fibroblasts derived amphiregulin promotes HNSCC progression and drug resistance of EGFR inhibitor. Cancer Lett. 2025, 622, 217710. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, Z.; Wang, G.; Geng, J.; Wu, H.; Liu, X.; Bin, E.; Sui, J.; Dai, H.; Tang, N. Sustained amphiregulin expression in intermediate alveolar stem cells drives progressive fibrosis. Cell Stem Cell 2024, 31, 1344–1358.e1346. [Google Scholar] [CrossRef] [PubMed]
- Son, S.S.; Hwang, S.; Park, J.H.; Ko, Y.; Yun, S.I.; Lee, J.H.; Son, B.; Kim, T.R.; Park, H.O.; Lee, E.Y. In vivo silencing of amphiregulin by a novel effective Self-Assembled-Micelle inhibitory RNA ameliorates renal fibrosis via inhibition of EGFR signals. Sci. Rep. 2021, 11, 2191. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Kim, T.R.; Park, J.H.; Yun, S.I.; Choi, H.; Choi, J.W.; Jeon, C.; Park, H.O. SAMiRNA Targeting Amphiregulin Alleviate Total-Body-Irradiation-Induced Renal Fibrosis. Radiat. Res. 2022, 197, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Yoon, P.O.; Park, J.W.; Lee, C.M.; Kim, S.H.; Kim, H.N.; Ko, Y.; Bae, S.J.; Yun, S.; Park, J.H.; Kwon, T.; et al. Self-assembled Micelle Interfering RNA for Effective and Safe Targeting of Dysregulated Genes in Pulmonary Fibrosis. J. Biol. Chem. 2016, 291, 6433–6446. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chiao, P.; Sun, Y. Amphiregulin in Cancer: New Insights for Translational Medicine. Trends Cancer 2016, 2, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Long, Q.; Zhu, D.; Fu, D.; Zhang, B.; Han, L.; Qian, M.; Guo, J.; Xu, J.; Cao, L.; et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 2019, 18, e13027. [Google Scholar] [CrossRef] [PubMed]
- Baldys, A.; Gooz, M.; Morinelli, T.A.; Lee, M.H.; Raymond, J.R., Jr.; Luttrell, L.M.; Raymond, J.R., Sr. Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry 2009, 48, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Krall, J.A.; Beyer, E.M.; MacBeath, G. High- and low-affinity epidermal growth factor receptor-ligand interactions activate distinct signaling pathways. PLoS ONE 2011, 6, e15945. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, C.; Baldessari, C.; Napolitano, M.; Orsi, G.; Grizzi, G.; Bertolini, F.; Barbieri, F.; Cascinu, S. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit. Rev. Oncol. Hematol. 2018, 123, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Nele Van Der, S.; Elisa, G.; Daniela, C.; Alessandro, L.; Christian, D.R.; Godefridus, J.P. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resist. 2018, 1, 230–249. [Google Scholar] [CrossRef]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., 2nd. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Busser, B.; Sancey, L.; Josserand, V.; Niang, C.; Favrot, M.C.; Coll, J.L.; Hurbin, A. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Mol. Ther. 2010, 18, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Busser, B.; Sancey, L.; Josserand, V.; Niang, C.; Khochbin, S.; Favrot, M.C.; Coll, J.L.; Hurbin, A. Amphiregulin promotes resistance to gefitinib in nonsmall cell lung cancer cells by regulating Ku70 acetylation. Mol. Ther. 2010, 18, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Obermann, J.L.; Pike, L.J. Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J. Biol. Chem. 2014, 289, 26178–26188. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, E.; Lin, S.; Hirayama, D.; Matsuda, K.; Tanave, A.; Sumiyama, K.; Tsukiji, S.; Otani, T.; Furuse, M.; Sorkin, A.; et al. Low-affinity ligands of the epidermal growth factor receptor are long-range signal transmitters in collective cell migration of epithelial cells. Cell Rep. 2024, 43, 114986. [Google Scholar] [CrossRef] [PubMed]
- Schramm, F.; Schaefer, L.; Wygrecka, M. EGFR Signaling in Lung Fibrosis. Cells 2022, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Liu, N. EGFR signaling in renal fibrosis. Kidney Int. Suppl. 2014, 4, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5, F1000-Faculty. [Google Scholar] [CrossRef] [PubMed]
- Kefaloyianni, E.; Keerthi Raja, M.R.; Schumacher, J.; Muthu, M.L.; Krishnadoss, V.; Waikar, S.S.; Herrlich, A. Proximal Tubule-Derived Amphiregulin Amplifies and Integrates Profibrotic EGF Receptor Signals in Kidney Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFbeta activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Engelman, J.A.; Arteaga, C.L. Overcoming resistance to tyrosine kinase inhibitors: Lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle 2009, 8, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Edgar, K.A.; Crocker, L.; Cheng, E.; Wagle, M.C.; Wongchenko, M.; Yan, Y.; Wilson, T.R.; Dompe, N.; Neve, R.M.; Belvin, M.; et al. Amphiregulin and PTEN evoke a multimodal mechanism of acquired resistance to PI3K inhibition. Genes Cancer 2014, 5, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Kindermann, M.; Knipfer, L.; Atreya, I.; Wirtz, S. ILC2s in infectious diseases and organ-specific fibrosis. Semin. Immunopathol. 2018, 40, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Aoki, A.; Morimoto, Y.; Kiuchi, M.; Okano, M.; Nakayama, T. The immunopathology of lung fibrosis: Amphiregulin-producing pathogenic memory T helper-2 cells control the airway fibrotic responses by inducing eosinophils to secrete osteopontin. Semin. Immunopathol. 2019, 41, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Neumark, N.; Cosme, C., Jr.; Rose, K.A.; Kaminski, N. The Idiopathic Pulmonary Fibrosis Cell Atlas. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L887–L893. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Koma, Y.I.; Miyako, S.; Torigoe, R.; Yokoo, H.; Omori, M.; Yamanaka, K.; Ishihara, N.; Tsukamoto, S.; Kodama, T.; et al. AREG Upregulation in Cancer Cells via Direct Interaction with Cancer-Associated Fibroblasts Promotes Esophageal Squamous Cell Carcinoma Progression Through EGFR-Erk/p38 MAPK Signaling. Cells 2024, 13, 1733. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Fu, W.; Turley, S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 2021, 54, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liu, T.; Wu, Z.; Hu, B.; Nakashima, T.; Ullenbruch, M.; Gonzalez De Los Santos, F.; Phan, S.H. Bone Marrow CD11c+ Cell-Derived Amphiregulin Promotes Pulmonary Fibrosis. J. Immunol. 2016, 197, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kurche, J.S.; Stancil, I.T.; Michalski, J.E.; Yang, I.V.; Schwartz, D.A. Dysregulated Cell-Cell Communication Characterizes Pulmonary Fibrosis. Cells 2022, 11, 3319. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.A.; Loffredo, L.F.; Santos-Alexis, K.L.; Ringham, O.R.; Arpaia, N. Regulation of the alveolar regenerative niche by amphiregulin-producing regulatory T cells. J. Exp. Med. 2023, 220, e20221462. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Fukami, T.; Yagi, H.; Kuroki, M.; Yotsumoto, F. Potential for molecularly targeted therapy against epidermal growth factor receptor ligands. Anticancer Res. 2009, 29, 823–830. [Google Scholar] [PubMed]
- Lofgren, K.A.; Sreekumar, S.; Jenkins, E.C., Jr.; Ernzen, K.J.; Kenny, P.A. Anti-tumor efficacy of an MMAE-conjugated antibody targeting cell surface TACE/ADAM17-cleaved Amphiregulin in breast cancer. Antib. Ther. 2021, 4, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Hosur, V.; Farley, M.L.; Burzenski, L.M.; Shultz, L.D.; Wiles, M.V. ADAM17 is essential for ectodomain shedding of the EGF-receptor ligand amphiregulin. FEBS Open Bio 2018, 8, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Tape, C.J.; Jodrell, D.I.; Murphy, G. Anti-tumour effects of a specific anti-ADAM17 antibody in an ovarian cancer model in vivo. PLoS ONE 2012, 7, e40597. [Google Scholar] [CrossRef] [PubMed]
- Dosch, J.; Ziemke, E.; Wan, S.; Luker, K.; Welling, T.; Hardiman, K.; Fearon, E.; Thomas, S.; Flynn, M.; Rios-Doria, J.; et al. Targeting ADAM17 inhibits human colorectal adenocarcinoma progression and tumor-initiating cell frequency. Oncotarget 2017, 8, 65090–65099. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Park, J.W.; Cho, W.K.; Zhou, Y.; Han, B.; Yoon, P.O.; Chae, J.; Elias, J.A.; Lee, C.G. Modifiers of TGF-beta1 effector function as novel therapeutic targets of pulmonary fibrosis. Korean J. Intern. Med. 2014, 29, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Trachalaki, A.; Sultana, N.; Wells, A.U. An update on current and emerging drug treatments for idiopathic pulmonary fibrosis. Expert. Opin. Pharmacother. 2023, 24, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Suri, G.S.; Kaur, G.; Jha, C.K.; Tiwari, M. Understanding idiopathic pulmonary fibrosis—Clinical features, molecular mechanism and therapies. Exp. Gerontol. 2021, 153, 111473. [Google Scholar] [CrossRef] [PubMed]
- Zaman, T.; Lee, J.S. Risk factors for the development of idiopathic pulmonary fibrosis: A review. Curr. Pulmonol. Rep. 2018, 7, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis. Adv. Respir. Med. 2023, 91, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Bonella, F.; Spagnolo, P.; Ryerson, C. Current and Future Treatment Landscape for Idiopathic Pulmonary Fibrosis. Drugs 2023, 83, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.; Athar, Z.M.; Hiba, T. Current and Novel Treatment Modalities of Idiopathic Pulmonary Fibrosis. Cureus 2024, 16, e56140. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Refini, R.M.; Bergantini, L.; d’Alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-Term Follow-Up of Patients with Idiopathic Pulmonary Fibrosis Treated with Pirfenidone or Nintedanib: A Real-Life Comparison Study. Front. Mol. Biosci. 2020, 7, 581828. [Google Scholar] [CrossRef] [PubMed]
- Marijic, P.; Schwarzkopf, L.; Schwettmann, L.; Ruhnke, T.; Trudzinski, F.; Kreuter, M. Pirfenidone vs. nintedanib in patients with idiopathic pulmonary fibrosis: A retrospective cohort study. Respir. Res. 2021, 22, 268. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, M. Palliative care in advanced pulmonary fibrosis. Curr. Opin. Pulm. Med. 2024, 30, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Hanata, N.; Nagafuchi, Y.; Sugimori, Y.; Kobayashi, S.; Tsuchida, Y.; Iwasaki, Y.; Shoda, H.; Fujio, K. Serum Amphiregulin and Heparin-Binding Epidermal Growth Factor as Biomarkers in Patients with Idiopathic Inflammatory Myopathy. J. Clin. Med. 2021, 10, 3730. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Fan, X.; Mao, Y.; Jiang, J. Amphiregulin in lung diseases: A review. Medicine 2024, 103, e37292. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Luo, Z.; Zhou, Y. Regeneration-Associated Transitional State Cells in Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 6757. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ding, L.; Wu, Z.; Gonzalez De Los Santos, F.; Phan, S. Role of dendritic cell-derived amphiregulin in pulmonary fibrosis (CCR5P.203). J. Immunol. 2015, 194, 186.5. [Google Scholar] [CrossRef]
- Yao, H.C.; Zhu, Y.; Lu, H.Y.; Ju, H.M.; Xu, S.Q.; Qiao, Y.; Wei, S.J. Type 2 innate lymphoid cell-derived amphiregulin regulates type II alveolar epithelial cell transdifferentiation in a mouse model of bronchopulmonary dysplasia. Int. Immunopharmacol. 2023, 122, 110672. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Hirahara, K.; Kiuchi, M.; Wada, T.; Ichikawa, T.; Kanno, T.; Okano, M.; Kokubo, K.; Onodera, A.; Sakurai, D.; et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity 2018, 49, 134–150.e136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lee, J.Y.; Lee, C.M.; Cho, W.K.; Kang, M.J.; Koff, J.L.; Yoon, P.O.; Chae, J.; Park, H.O.; Elias, J.A.; et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J. Biol. Chem. 2012, 287, 41991–42000. [Google Scholar] [CrossRef] [PubMed]
- Conroy, K.P.; Kitto, L.J.; Henderson, N.C. alphav integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Sakao, S.; Tatsumi, K. Molecular mechanisms of lung-specific toxicity induced by epidermal growth factor receptor tyrosine kinase inhibitors. Oncol. Lett. 2012, 4, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoshiba, K.; Yokohori, N.; Nagai, A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res. 2003, 63, 5054–5059. [Google Scholar] [PubMed]
- Ma, H.; Wu, X.; Li, Y.; Xia, Y. Research Progress in the Molecular Mechanisms, Therapeutic Targets, and Drug Development of Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 963054. [Google Scholar] [CrossRef] [PubMed]
- Koya, D. Diabetic kidney disease: Its current trends and future therapeutic perspectives. J. Diabetes Investig. 2019, 10, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Boor, P. Kidney fibrosis: Emerging diagnostic and therapeutic strategies. Mol. Asp. Med. 2023, 93, 101206. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Fu, P.; Ma, L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal Transduct. Target. Ther. 2023, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Melderis, S.; Hagenstein, J.; Warkotsch, M.T.; Dang, J.; Herrnstadt, G.R.; Niehus, C.B.; Neumann, K.; Panzer, U.; Berasain, C.; Avila, M.A.; et al. Amphiregulin Aggravates Glomerulonephritis via Recruitment and Activation of Myeloid Cells. J. Am. Soc. Nephrol. 2020, 31, 1996–2012. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Rodrigues-Diez, R.; Morgado-Pascual, J.L.; Valentijn, F.; Valdivielso, J.M.; Goldschmeding, R.; Ruiz-Ortega, M. Role of Epidermal Growth Factor Receptor (EGFR) and Its Ligands in Kidney Inflammation and Damage. Mediat. Inflamm. 2018, 2018, 8739473. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Taniguchi, Y.; Hamada Ode, K.; Shimamura, Y.; Inotani, S.; Nishikawa, H.; Matsumoto, T.; Horino, T.; Fujimoto, S.; Terada, Y. Clinical significance of amphiregulin in patients with chronic kidney disease. Clin. Exp. Nephrol. 2024, 28, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, I.M.; Kefalogianni, E.; Zhao, R.; Verma, A.; Sabbisetti, V.; Rahman, M.; Pradhan, N.; Srivastava, A.; He, J.; Chen, J.; et al. Associations of Serum Amphiregulin Levels with Kidney Failure and Mortality: The Chronic Renal Insufficiency Cohort (CRIC). Kidney Med. 2025, 7, 100958. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Harris, D.C.; Wang, Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology 2015, 30, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-edged Sword. J. Histochem. Cytochem. 2019, 67, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Palau, V.; Pascual, J.; Soler, M.J.; Riera, M. Role of ADAM17 in kidney disease. Am. J. Physiol. Ren. Physiol. 2019, 317, F333–F342. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Y.; Liu, X.S.; Huang, X.R.; Yu, X.Q.; Lan, H.Y. Diverse Role of TGF-beta in Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-beta1/p53 signaling in renal fibrogenesis. Cell Signal 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, X.M.; Huang, X.R.; Lan, H.Y. The preventive and therapeutic implication for renal fibrosis by targetting TGF-beta/Smad3 signaling. Clin. Sci. 2018, 132, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Harris, R.C. Interaction of the EGF Receptor and the Hippo Pathway in the Diabetic Kidney. J. Am. Soc. Nephrol. 2016, 27, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Melderis, S.; Warkotsch, M.T.; Dang, J.; Hagenstein, J.; Ehnold, L.I.; Herrnstadt, G.R.; Niehus, C.B.; Feindt, F.C.; Kylies, D.; Puelles, V.G.; et al. The Amphiregulin/EGFR axis protects from lupus nephritis via downregulation of pathogenic CD4+ T helper cell responses. J. Autoimmun. 2022, 129, 102829. [Google Scholar] [CrossRef] [PubMed]
- Buvall, L.; Menzies, R.I.; Williams, J.; Woollard, K.J.; Kumar, C.; Granqvist, A.B.; Fritsch, M.; Feliers, D.; Reznichenko, A.; Gianni, D.; et al. Selecting the right therapeutic target for kidney disease. Front. Pharmacol. 2022, 13, 971065. [Google Scholar] [CrossRef] [PubMed]
- Tawengi, M.; Al-Dali, Y.; Tawengi, A.; Benter, I.F.; Akhtar, S. Targeting the epidermal growth factor receptor (EGFR/ErbB) for the potential treatment of renal pathologies. Front. Pharmacol. 2024, 15, 1394997. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, S.I.; Choi, M.E. Therapeutic targets for treating fibrotic kidney diseases. Transl. Res. 2015, 165, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, F.; Li, X.; Ma, Z.; Jiang, D. Quercetin inhibits the amphiregulin/EGFR signaling-mediated renal tubular epithelial-mesenchymal transition and renal fibrosis in obstructive nephropathy. Phytother. Res. 2023, 37, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Unalp-Arida, A.; Ruhl, C.E. Liver fibrosis scores predict liver disease mortality in the United States population. Hepatology 2017, 66, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Lim, W.H.; Hui Lim, G.E.; Hao Tan, D.J.; Syn, N.; Muthiah, M.D.; Huang, D.Q.; Loomba, R. Mortality Outcomes by Fibrosis Stage in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2023, 21, 931–939.e935. [Google Scholar] [CrossRef] [PubMed]
- Savage, T.M.; Fortson, K.T.; de Los Santos-Alexis, K.; Oliveras-Alsina, A.; Rouanne, M.; Rae, S.S.; Gamarra, J.R.; Shayya, H.; Kornberg, A.; Cavero, R.; et al. Amphiregulin from regulatory T cells promotes liver fibrosis and insulin resistance in non-alcoholic steatohepatitis. Immunity 2024, 57, 303–318.e306. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.; Sigala, B.; Soeda, J.; Mouralidarane, A.; Morgan, M.; Mazzoccoli, G.; Rappa, F.; Cappello, F.; Cabibi, D.; Pazienza, V.; et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci. Rep. 2015, 5, 8812. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, A.; Takemura, K.; Tanaka, A.; Matsumoto, M.; Katsuyama, M.; Okanoue, T.; Yamaguchi, K.; Itoh, Y.; Iwata, K.; Amagase, K.; et al. Carfilzomib shows therapeutic potential for reduction of liver fibrosis by targeting hepatic stellate cell activation. Sci. Rep. 2024, 14, 19288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, W.M.; Sun, H.Y.; Peng, Y.; Huang, R.J.; Chen, C.Y.; Zhang, H.D.; Zhou, S.A.; Wu, H.P.; Tang, D.; et al. Hepatocyte-derived liver progenitor-like cells attenuate liver cirrhosis via induction of apoptosis in hepatic stellate cells. Hepatol. Commun. 2025, 9, e0614. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Lee, N.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Amphiregulin Induces iNOS and COX-2 Expression through NF-kappaB and MAPK Signaling in Hepatic Inflammation. Mediat. Inflamm. 2023, 2023, 2364121. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Kita, M.; Torihashi, S.; Miyamoto, S.; Won, K.J.; Sato, K.; Ozaki, H.; Karaki, H. Upregulation of iNOS by COX-2 in muscularis resident macrophage of rat intestine stimulated with LPS. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G930–G938. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, M.K.; Lim, S.Y.; Sung, S.H.; Kim, Y.C. Inhibition of inducible NO synthase, cyclooxygenase-2 and interleukin-1beta by torilin is mediated by mitogen-activated protein kinases in microglial BV2 cells. Br. J. Pharmacol. 2009, 156, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Dashek, R.J.; Cunningham, R.P.; Taylor, C.L.; Alessi, I.; Diaz, C.; Meers, G.M.; Wheeler, A.A.; Ibdah, J.A.; Parks, E.J.; Yoshida, T.; et al. Hepatocellular RECK as a Critical Regulator of Metabolic Dysfunction-associated Steatohepatitis Development. Cell Mol. Gastroenterol. Hepatol. 2024, 18, 101365. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, M.J.; Tieppo, J.; Marroni, N.P.; Tunon, M.J.; Gonzalez-Gallego, J. Suppression of amphiregulin/epidermal growth factor receptor signals contributes to the protective effects of quercetin in cirrhotic rats. J. Nutr. 2011, 141, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Garcia-Trevijano, E.R.; Castillo, J.; Erroba, E.; Santamaria, M.; Lee, D.C.; Prieto, J.; Avila, M.A. Novel role for amphiregulin in protection from liver injury. J. Biol. Chem. 2005, 280, 19012–19020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Rehman, H.; Krishnasamy, Y.; Haque, K.; Schnellmann, R.G.; Lemasters, J.J.; Zhong, Z. Amphiregulin stimulates liver regeneration after small-for-size mouse liver transplantation. Am. J. Transplant. 2012, 12, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Latasa, M.U.; Nicou, A.; Cartagena-Lirola, H.; Castillo, J.; Goni, S.; Vespasiani-Gentilucci, U.; Zagami, M.G.; Lotersztajn, S.; Prieto, J.; et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008, 48, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Fuchs, B.C.; Yamada, S.; Lauwers, G.Y.; Kulu, Y.; Goodwin, J.M.; Lanuti, M.; Tanabe, K.K. Mouse model of carbon tetrachloride induced liver fibrosis: Histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; San-Miguel, B.; Fernandez, A.; Ortiz de Urbina, J.; Gonzalez-Gallego, J.; Tunon, M.J. Melatonin limits the expression of profibrogenic genes and ameliorates the progression of hepatic fibrosis in mice. Transl. Res. 2015, 165, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, Y.; Ohara, D.; Takeuchi, Y.; Watanabe, H.; Kondoh, G.; Taura, K.; Uemoto, S.; Hirota, K. Foxp3+ Regulatory T Cells Inhibit CCl4-Induced Liver Inflammation and Fibrosis by Regulating Tissue Cellular Immunity. Front. Immunol. 2020, 11, 584048. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Fan, J.; Zhou, H. Bile acid-mediated signaling in cholestatic liver diseases. Cell Biosci. 2023, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, E.; Rodriguez-Ortigosa, C.M.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Alvarez-Sola, G.; Barcena-Varela, M.; Colyn, L.; Arcelus, S.; Jimenez, M.; et al. The Epidermal Growth Factor Receptor Ligand Amphiregulin Protects from Cholestatic Liver Injury and Regulates Bile Acids Synthesis. Hepatology 2019, 69, 1632–1647. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.H.; Wu, S.; Liang, P.; Ma, D.; Zhang, J.; Chen, H.; Zhong, Z.; Liu, J.; Jiang, H.; Feng, X.; et al. Mucosal-associated invariant T cells promote ductular reaction through amphiregulin in biliary atresia. eBioMedicine 2024, 103, 105138. [Google Scholar] [CrossRef] [PubMed]
- Mohagheghi, S.; Geramizadeh, B.; Nikeghbalian, S.; Khodadadi, I.; Karimi, J.; Khajehahmadi, Z.; Gharekhanloo, F.; Tavilani, H. Intricate role of yes-associated protein1 in human liver cirrhosis: TGF-β1 still is a giant player. IUBMB Life 2019, 71, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Yin, X.; Pan, X.; Zhang, J.; Fan, X.; Li, J.; Zhai, X.; Jiang, L.; Hao, P.; Wang, J.; et al. Post-myocardial infarction fibrosis: Pathophysiology, examination, and intervention. Front. Pharmacol. 2023, 14, 1070973. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Schlittler, M.; Pramstaller, P.P.; Rossini, A.; De Bortoli, M. Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Perspective from Fibroblasts. Int. J. Mol. Sci. 2023, 24, 14845. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Lopez, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; Gonzalez, A.; Colan, S.D.; Seidman, J.G.; et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.; Qu, Z.; Weiss, J.N. Cardiac fibrosis and arrhythmogenesis: The road to repair is paved with perils. J. Mol. Cell Cardiol. 2014, 70, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Fujiu, K.; Shibata, M.; Nakayama, Y.; Ogata, F.; Matsumoto, S.; Noshita, K.; Iwami, S.; Nakae, S.; Komuro, I.; Nagai, R.; et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat. Med. 2017, 23, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Sugita, J.; Fujiu, K.; Nakayama, Y.; Matsubara, T.; Matsuda, J.; Oshima, T.; Liu, Y.; Maru, Y.; Hasumi, E.; Kojima, T.; et al. Cardiac macrophages prevent sudden death during heart stress. Nat. Commun. 2021, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat. Commun. 2018, 9, 816. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Huang, S.; Wang, J.; Xia, T.; Liu, H.; You, Y.; Wang, Z.; Liu, K. The GABA(A) Receptor Influences Pressure Overload-Induced Heart Failure by Modulating Macrophages in Mice. Front. Immunol. 2021, 12, 670153. [Google Scholar] [CrossRef]
- Zuo, C.; Li, X.; Huang, J.; Chen, D.; Ji, K.; Yang, Y.; Xu, T.; Zhu, D.; Yan, C.; Gao, P. Osteoglycin attenuates cardiac fibrosis by suppressing cardiac myofibroblast proliferation and migration through antagonizing lysophosphatidic acid 3/matrix metalloproteinase 2/epidermal growth factor receptor signalling. Cardiovasc. Res. 2018, 114, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Shinde, A.V.; Hanna, A.; Alex, L.; Hernandez, S.C.; Li, R.; Chen, B.; Conway, S.J.; Frangogiannis, N.G. Smad7 effects on TGF-beta and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 2022, 132, e146926. [Google Scholar] [CrossRef] [PubMed]
- Grzebyk, E.; Pazgan-Simon, M.; Jagas, J.; Zuwala-Jagiello, J.; Gorka-Dynysiewicz, J. Left ventricular function is related with amphiregulin and fibrosis markers in cirrhotic cardiomyopathy. J. Physiol. Pharmacol. 2022, 73, 97–107. [Google Scholar] [CrossRef]
- Ji, M.; Liu, Y.; Zuo, Z.; Xu, C.; Lin, L.; Li, Y. Downregulation of amphiregulin improves cardiac hypertrophy via attenuating oxidative stress and apoptosis. Biol. Direct 2022, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Warunek, J.J.; Fan, L.; Zhang, X.; Wang, S.; Sanders, S.M.; Li, T.; Mathews, L.R.; Dwyer, G.K.; Wood-Trageser, M.A.; Traczek, S.; et al. Dysregulated Treg repair responses lead to chronic rejection after heart transplantation. J. Clin. Investig. 2024, 134, e173593. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Emoto, T.; Sato, S.; Yoshida, T.; Shoda, M.; Endoh, H.; Nagao, M.; Hamana, T.; Inoue, T.; Hayashi, T.; et al. Left atrial single-cell transcriptomics reveals amphiregulin as a surrogate marker for atrial fibrillation. Commun. Biol. 2024, 7, 1601. [Google Scholar] [CrossRef] [PubMed]
- Porvari, K.; Horioka, K.; Kaija, H.; Pakanen, L. Amphiregulin is overexpressed in human cardiac tissue in hypothermia deaths; associations between the transcript and stress hormone levels in cardiac deaths. Ann. Med. 2024, 56, 2420862. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Irie, E.; Ishihara, R.; Mizushima, I.; Hatai, S.; Hagihara, Y.; Takada, Y.; Tsunoda, J.; Iwata, K.; Matsubara, Y.; Yoshimatsu, Y.; et al. Enrichment of type I interferon signaling in colonic group 2 innate lymphoid cells in experimental colitis. Front. Immunol. 2022, 13, 982827. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Sheng, H. Amphiregulin promotes intestinal epithelial regeneration: Roles of intestinal subepithelial myofibroblasts. Endocrinology 2010, 151, 3728–3737. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, B.; Pang, X.; Song, X.; Gu, Y.; Xie, R.; Liu, T.; Xu, X.; Wang, B.; Cao, H. Clostridium butyricum, a butyrate-producing potential probiotic, alleviates experimental colitis through epidermal growth factor receptor activation. Food Funct. 2022, 13, 7046–7061. [Google Scholar] [CrossRef] [PubMed]
- Xiu, W.; Chen, Q.; Wang, Z.; Wang, J.; Zhou, Z. Microbiota-derived short chain fatty acid promotion of Amphiregulin expression by dendritic cells is regulated by GPR43 and Blimp-1. Biochem. Biophys. Res. Commun. 2020, 533, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zeka, F.; Angori, S.; Rutishauser, D.; Moch, H.; Posovszky, C.; Amin, K.; Holtan, S.; Gungor, T.; Drozdov, D. High Amphiregulin Expression in Intestinal Biopsies of Pediatric Patients with Severe Acute Graft-Versus-Host Disease. Transplant. Cell Ther. 2025, 31, 323.e1–323.e9. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Ungaro, F.; Noviello, D.; Lovisa, S.; Peyrin-Biroulet, L.; Danese, S. Revisiting fibrosis in inflammatory bowel disease: The gut thickens. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson, J.; Wick, M.J. Mechanism of fibrosis and stricture formation in Crohn’s disease. Scand. J. Immunol. 2020, 92, e12990. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, G.; Lenti, M.V.; Di Sabatino, A. Therapeutic Targeting of Intestinal Fibrosis in Crohn’s Disease. Cells 2022, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13, 835005. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, W.; Yu, T.; Yu, Y.; Cui, X.; Zhou, Z.; Yang, H.; Yu, Y.; Bilotta, A.J.; Yao, S.; et al. Th17 Cell-Derived Amphiregulin Promotes Colitis-Associated Intestinal Fibrosis Through Activation of mTOR and MEK in Intestinal Myofibroblasts. Gastroenterology 2023, 164, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, M.; Giammarile, F.; Carrara, M.; Paez, D.; Hricak, H.; Ayati, N.; Li, J.J.; Mueller, M.; Aggarwal, A.; Al-Ibraheem, A.; et al. Radiotherapy and theranostics: A Lancet Oncology Commission. Lancet Oncol. 2024, 25, e545–e580. [Google Scholar] [CrossRef] [PubMed]
- Fijardo, M.; Kwan, J.Y.Y.; Bissey, P.A.; Citrin, D.E.; Yip, K.W.; Liu, F.F. The clinical manifestations and molecular pathogenesis of radiation fibrosis. EBioMedicine 2024, 103, 105089. [Google Scholar] [CrossRef] [PubMed]
- Meulenbroeks, C.; van Weelden, H.; Schwartz, C.; Voehringer, D.; Redegeld, F.A.M.; Rutten, V.; Willemse, T.; Sijts, A.; Zaiss, D.M.W. Basophil-derived amphiregulin is essential for UVB irradiation-induced immune suppression. J. Investig. Dermatol. 2015, 135, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nat. Rev. Dis. Primers 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Fang, S.; Gao, H.; Zhang, X.; Gu, D.; Liu, Y.; Wan, J.; Xie, J. A critical role of AREG for bleomycin-induced skin fibrosis. Cell Biosci. 2021, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Padilla, C.M.; Valenzi, E.; Tabib, T.; Nazari, B.; Sembrat, J.; Rojas, M.; Fuschiotti, P.; Lafyatis, R. Increased CD8+ tissue resident memory T cells, regulatory T cells and activated natural killer cells in systemic sclerosis lungs. Rheumatology 2024, 63, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Morina, G.; Sambataro, D.; Libra, A.; Palmucci, S.; Colaci, M.; La Rocca, G.; Ferro, F.; Carli, L.; Baldini, C.; Liuzzo, S.V.; et al. Recognition of Idiopathic Inflammatory Myopathies Underlying Interstitial Lung Diseases. Diagnostics 2025, 15, 275. [Google Scholar] [CrossRef] [PubMed]
- Ceribelli, A.; Tonutti, A.; Isailovic, N.; De Santis, M.; Selmi, C. Interstitial lung disease associated with inflammatory myositis: Autoantibodies, clinical phenotypes, and progressive fibrosis. Front. Med. 2023, 10, 1068402. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.; Patel, A.; Chatterjee, S.; Fernandez, A.P.; Farver, C.; Yadav, R.; Li, Y.; Danoff, S.K.; Saygin, D.; Huapaya, J.A.; et al. Idiopathic inflammatory myopathies related lung disease in adults. Lancet Respir. Med. 2025, 13, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Layoun, H.; Hajal, J.; Saliba, Y.; Smayra, V.; Habr, B.; Fares, N. Pirfenidone mitigates TGF-β1-mediated fibrosis in an idiopathic inflammatory myositis-associated interstitial lung disease model. Cytokine 2022, 154, 155899. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Kim, T.; Kim, H.; Jang, B.G.; Myung, J.K.; Kim, H.Y. Esophageal ILC2s mediate abnormal epithelial remodeling in eosinophilic esophagitis via Areg-EGFR signaling. Cell Mol. Immunol. 2025, 22, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Iwamura, C.; Kiuchi, M.; Kurosugi, A.; Onoue, M.; Matsumura, T.; Chiba, T.; Nakayama, T.; Kato, N.; Hirahara, K. Amphiregulin-producing T(H)2 cells facilitate esophageal fibrosis of eosinophilic esophagitis. J. Allergy Clin. Immunol. Glob. 2024, 3, 100287. [Google Scholar] [CrossRef] [PubMed]
- Ebott, J.; McAdams, J.; Kim, C.; Jansen, C.; Woodman, M.; De La Cruz, P.; Schrol, C.; Ribeiro, J.; James, N. Enhanced amphiregulin exposure promotes modulation of the high grade serous ovarian cancer tumor immune microenvironment. Front. Pharmacol. 2024, 15, 1375421. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Daigo, Y.; Takano, A.; Taniwaki, M.; Kato, T.; Hayama, S.; Murakami, H.; Takeshima, Y.; Inai, K.; Nishimura, H.; et al. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res. 2005, 65, 9176–9184. [Google Scholar] [CrossRef] [PubMed]
- Masago, K.; Fujita, S.; Hatachi, Y.; Fukuhara, A.; Sakuma, K.; Ichikawa, M.; Kim, Y.H.; Mio, T.; Mishima, M. Clinical significance of pretreatment serum amphiregulin and transforming growth factor-alpha, and an epidermal growth factor receptor somatic mutation in patients with advanced non-squamous, non-small cell lung cancer. Cancer Sci. 2008, 99, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Hobor, S.; Van Emburgh, B.O.; Crowley, E.; Misale, S.; Di Nicolantonio, F.; Bardelli, A. TGFalpha and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin. Cancer Res. 2014, 20, 6429–6438. [Google Scholar] [CrossRef] [PubMed]
- Kappler, C.S.; Guest, S.T.; Irish, J.C.; Garrett-Mayer, E.; Kratche, Z.; Wilson, R.C.; Ethier, S.P. Oncogenic signaling in amphiregulin and EGFR-expressing PTEN-null human breast cancer. Mol. Oncol. 2015, 9, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, J.N.; Demory Beckler, M.; Gephart, J.D.; Franklin, J.L.; Bogatcheva, G.; Kremers, G.J.; Piston, D.W.; Ayers, G.D.; McConnell, R.E.; Tyska, M.J.; et al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 2011, 21, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Lindzen, M.; Lauriola, M.; Shirazi, N.; Sinha, S.; Abdul-Hai, A.; Levanon, K.; Korach, J.; Barshack, I.; Cohen, Y.; et al. An antibody to amphiregulin, an abundant growth factor in patients’ fluids, inhibits ovarian tumors. Oncogene 2016, 35, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Park, H.; Kim, K.J.; Kim, J.W.; Sung, J.H.; Nam, M.; Lee, J.H.; Jung, E.H.; Suh, K.J.; Lee, J.Y.; et al. Amphiregulin can predict treatment resistance to palliative first-line cetuximab plus FOLFIRI chemotherapy in patients with RAS wild-type metastatic colorectal cancer. Sci. Rep. 2021, 11, 23803. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, H.; Lu, J.; Zhang, Z.; Wu, H.; Liang, Z. AREG mediates the epithelial-mesenchymal transition in pancreatic cancer cells via the EGFR/ERK/NF-kappaB signalling pathway. Oncol. Rep. 2020, 43, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wang, S.; Pan, Y.; Zhi, W.; Gu, C.; Guo, T.; Zhai, J.; Li, C.; Chen, Y.Q.; Wang, R. Development, opportunities, and challenges of siRNA nucleic acid drugs. Mol. Ther. Nucleic Acids 2025, 36, 102437. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Landolt, L.; Spagnoli, G.C.; Hertig, A.; Brocheriou, I.; Marti, H.P. Fibrosis and cancer: Shared features and mechanisms suggest common targeted therapeutic approaches. Nephrol. Dial. Transplant. 2022, 37, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Benelli, R.; Vene, R.; Minghelli, S.; Carlone, S.; Gatteschi, B.; Ferrari, N. Celecoxib induces proliferation and Amphiregulin production in colon subepithelial myofibroblasts, activating erk1-2 signaling in synergy with EGFR. Cancer Lett. 2013, 328, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Mucciolo, G.; Araos Henriquez, J.; Jihad, M.; Pinto Teles, S.; Manansala, J.S.; Li, W.; Ashworth, S.; Lloyd, E.G.; Cheng, P.S.W.; Luo, W.; et al. EGFR-activated myofibroblasts promote metastasis of pancreatic cancer. Cancer Cell 2024, 42, 101–118.e111. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huo, R.; Kang, W.; Liu, Y.; Zhao, Z.; Fu, W.; Ma, R.; Zhang, X.; Tang, J.; Zhu, Z.; et al. Tumor-associated monocytes promote mesenchymal transformation through EGFR signaling in glioma. Cell Rep. Med. 2023, 4, 101177. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.Y.; Cho, K.H.; Jeong, K.J.; Cho, S.J.; Won, M.; Kim, S.H.; Cho, N.H.; Hur, G.M.; Yoon, S.H.; Park, H.W.; et al. Lysophosphatidic acid-induced amphiregulin secretion by cancer-associated fibroblasts augments cancer cell invasion. Cancer Lett. 2022, 551, 215946. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhao, H.; Gao, D.S.; Ni, A.; Li, H.; Chen, L.; Lu, X.; Chen, K.; Lu, B. Amphiregulin couples IL1RL1+ regulatory T cells and cancer-associated fibroblasts to impede antitumor immunity. Sci. Adv. 2023, 9, eadd7399. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Y.; Wang, Y.; Ye, P.; Li, J.; Li, H.; Ding, Q.; Xia, J. Amphiregulin Confers Regulatory T Cell Suppressive Function and Tumor Invasion via the EGFR/GSK-3beta/Foxp3 Axis. J. Biol. Chem. 2016, 291, 21085–21095. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Huang, M.S.; Cheng, D.E.; Hung, J.Y.; Yang, C.J.; Chou, S.H.; Kuo, P.L. Lung tumor-associated dendritic cell-derived amphiregulin increased cancer progression. J. Immunol. 2011, 187, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohba, M.; Ohmori, T. Molecular-Targeted Therapies for Epidermal Growth Factor Receptor and Its Resistance Mechanisms. Int. J. Mol. Sci. 2017, 18, 2420. [Google Scholar] [CrossRef] [PubMed]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Ten Dijke, P. Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors 2011, 29, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-beta Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.H.; Tham, C.L.; Harith, H.H.; Firdaus, N.; Israf, D.A. TGF-beta-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur. J. Pharmacol. 2021, 911, 174510. [Google Scholar] [CrossRef] [PubMed]
- Guernsey-Biddle, C.; High, P.; Carmon, K.S. Exploring the Potential of Epiregulin and Amphiregulin as Prognostic, Predictive, and Therapeutic Targets in Colorectal Cancer. Onco 2024, 4, 257–274. [Google Scholar] [CrossRef]
- Huang, W.S.; Wu, K.L.; Chen, C.N.; Chang, S.F.; Lee, D.Y.; Lee, K.C. Amphiregulin Upregulation in Visfatin-Stimulated Colorectal Cancer Cells Reduces Sensitivity to 5-Fluororacil Cytotoxicity. Biology 2024, 13, 821. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Beesetty, Y.; Lee, W.; Washington, M.K.; Chen, X.; Lockhart, A.C.; Merchant, N.B. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-alpha: Impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res. 2014, 74, 2062–2072. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fang, R.; Ma, R.; Long, Y.; He, R.; Lyu, H.; Chen, L.; Wen, Y. Amphiregulin promotes activated regulatory T cell-suppressive function via the AREG/EGFR pathway in laryngeal squamous cell carcinoma. Head. Face Med. 2024, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.J.; Ho, T.L.; Chao, C.C.; He, X.Y.; Chen, P.C.; Cheng, F.J.; Huang, W.C.; Huang, C.L.; Liu, P.I.; Tang, C.H. Particulate matter facilitates amphiregulin-dependent lung cancer proliferation through glutamine metabolism. Int. J. Biol. Sci. 2024, 20, 3126–3139. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.Y.; Wang, B.W.; Cheng, F.J.; Chen, C.H.; Hsia, T.C.; Wei, Y.L.; Chen, C.Y.; Hsieh, I.S.; Yeh, Y.L.; Wang, L.Y.; et al. Incense burning smoke sensitizes lung cancer cells to EGFR TKI by inducing AREG expression. Am. J. Cancer Res. 2018, 8, 2575–2589. [Google Scholar] [PubMed]
- Piffko, A.; Yang, K.; Panda, A.; Heide, J.; Tesak, K.; Wen, C.; Zawieracz, K.; Wang, L.; Naccasha, E.Z.; Bugno, J.; et al. Radiation-induced amphiregulin drives tumour metastasis. Nature 2025. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Luetteke, N.C.; Qiu, T.H.; Fenton, S.E.; Troyer, K.L.; Riedel, R.F.; Chang, A.; Lee, D.C. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 1999, 126, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Jay, F.F.; Vaidya, M.; Porada, S.M.; Andrukhova, O.; Schneider, M.R.; Erben, R.G. Amphiregulin lacks an essential role for the bone anabolic action of parathyroid hormone. Mol. Cell Endocrinol. 2015, 417, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Ciarloni, L.; Mallepell, S.; Brisken, C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc. Natl. Acad. Sci. USA 2007, 104, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Veit, M.; Ahrens, B.; Seidel, J.; Sommer, A.; Bhakdi, S.; Reiss, K. Mutagenesis of the ADAM17-phosphatidylserine-binding motif leads to embryonic lethality in mice. Life Sci. Alliance 2019, 2, e201900430. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.R.; Kim, H.Y.; Kim, I.H.; Kim, K.C.; Ko, Y.; Park, J.H.; Yun, S.; Lee, I.C.; Kim, S.H.; Park, H.O. Safety pharmacology of self-assembled-micelle inhibitory RNA-targeting amphiregulin (SAMiRNA-AREG), a novel siRNA nanoparticle platform. Toxicol. Rep. 2021, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, T.R.; Kim, S.H.; Kim, I.H.; Lim, J.O.; Park, J.H.; Yun, S.; Lee, I.C.; Park, H.O.; Kim, J.C. Four-Week Repeated Intravenous Dose Toxicity of Self-Assembled-Micelle Inhibitory RNA-Targeting Amphiregulin in Mice. Int. J. Toxicol. 2021, 40, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, T.R.; Kim, S.H.; Kim, I.H.; Ko, Y.; Yun, S.; Lee, I.C.; Park, H.O.; Kim, J.C. Genotoxicity evaluation of self-assembled-micelle inhibitory RNA-targeting amphiregulin (SAMiRNA-AREG), a novel siRNA nanoparticle for the treatment of fibrotic disease. Drug Chem. Toxicol. 2022, 45, 2109–2115. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Kim, T.-R.; Kim, S.-H.; Kim, I.-H.; Kim, W.-I.; Park, J.-H.; Ko, Y.; Yun, S.; Park, H.-O.; Kim, J.-C. Systemic toxicity and toxicokinetics study of self-assembled-micelle inhibitory RNA-targeting amphiregulin in cynomolgus monkeys following intravenous injection. Toxicol. Res. 2025, 41, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, P.; Addison, M.L.; Dear, J.W.; Webb, D.J. Small interfering RNA: Discovery, pharmacology and clinical development-An introductory review. Br. J. Pharmacol. 2023, 180, 2697–2720. [Google Scholar] [CrossRef] [PubMed]
- Gavrilov, K.; Saltzman, W.M. Therapeutic siRNA: Principles, challenges, and strategies. Yale J. Biol. Med. 2012, 85, 187–200. [Google Scholar] [PubMed]
- Judge, A.; MacLachlan, I. Overcoming the innate immune response to small interfering RNA. Hum. Gene Ther. 2008, 19, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.; Judge, A.; MacLachlan, I. siRNA and innate immunity. Oligonucleotides 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Does the understanding of immune activation by RNA predict the design of safe siRNAs? Front. Biosci. 2008, 13, 4379–4392. [Google Scholar] [CrossRef] [PubMed]
- Bora, R.S.; Gupta, D.; Mukkur, T.K.; Saini, K.S. RNA interference therapeutics for cancer: Challenges and opportunities (review). Mol. Med. Rep. 2012, 6, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Yeseom Cho, K.; Tiwari, R.K. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Lee, S.K.; Goh, E.A.; Kwon, O.S.; Choi, W.; Kim, J.; Lee, M.S.; Choi, S.J.; Lim, S.S.; Moon, T.K.; et al. Weekly treatment with SAMiRNA targeting the androgen receptor ameliorates androgenetic alopecia. Sci. Rep. 2022, 12, 1607. [Google Scholar] [CrossRef] [PubMed]
- Ali Zaidi, S.S.; Fatima, F.; Ali Zaidi, S.A.; Zhou, D.; Deng, W.; Liu, S. Engineering siRNA therapeutics: Challenges and strategies. J. Nanobiotechnol. 2023, 21, 381. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, A. Innate immune regulations and various siRNA modalities. Drug Deliv. Transl. Res. 2023, 13, 2704–2718. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.L.; Chen, M.; Knifley, T.; Davis, K.A.; Harrison, S.M.W.; Stewart, R.L.; O’Connor, K.L. Integrin α6β4 Promotes Autocrine Epidermal Growth Factor Receptor (EGFR) Signaling to Stimulate Migration and Invasion toward Hepatocyte Growth Factor (HGF). J. Biol. Chem. 2015, 290, 27228–27238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Chen, Y.J.; Lin, C.Y.; Fong, Y.C.; Hsu, C.J.; Tsai, C.H.; Su, J.L.; Tang, C.H. Amphiregulin enhances α6β1 integrin expression and cell motility in human chondrosarcoma cells through Ras/Raf/MEK/ERK/AP-1 pathway. Oncotarget 2015, 6, 11434–11446. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.L.; Liu, J.; Qi, L.; Wang, C.; O’Connor, K.L. Integrin α6β4 Upregulates Amphiregulin and Epiregulin through Base Excision Repair-Mediated DNA Demethylation and Promotes Genome-wide DNA Hypomethylation. Sci. Rep. 2017, 7, 6174. [Google Scholar] [CrossRef] [PubMed]
- Gnosa, S.P.; Puig Blasco, L.; Piotrowski, K.B.; Freiberg, M.L.; Savickas, S.; Madsen, D.H.; Auf dem Keller, U.; Kronqvist, P.; Kveiborg, M. ADAM17-mediated EGFR ligand shedding directs macrophage-promoted cancer cell invasion. JCI Insight 2022, 7, e155296. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.R.; Son, B.; Lee, C.G.; Park, H.-O. Amphiregulin in Fibrotic Diseases and Cancer. Int. J. Mol. Sci. 2025, 26, 6945. https://doi.org/10.3390/ijms26146945
Kim TR, Son B, Lee CG, Park H-O. Amphiregulin in Fibrotic Diseases and Cancer. International Journal of Molecular Sciences. 2025; 26(14):6945. https://doi.org/10.3390/ijms26146945
Chicago/Turabian StyleKim, Tae Rim, Beomseok Son, Chun Geun Lee, and Han-Oh Park. 2025. "Amphiregulin in Fibrotic Diseases and Cancer" International Journal of Molecular Sciences 26, no. 14: 6945. https://doi.org/10.3390/ijms26146945
APA StyleKim, T. R., Son, B., Lee, C. G., & Park, H.-O. (2025). Amphiregulin in Fibrotic Diseases and Cancer. International Journal of Molecular Sciences, 26(14), 6945. https://doi.org/10.3390/ijms26146945