
A Proton Magnetic Resonance Spectroscopy (1H MRS) Pilot Study Revealing Altered Glutamatergic and Gamma-Aminobutyric Acid (GABA)ergic Neurotransmission in Social Anxiety Disorder (SAD)

 , and
, and 
Abstract
1. Introduction
2. Results
2.1. Demographic and Clinical Participant Characteristics
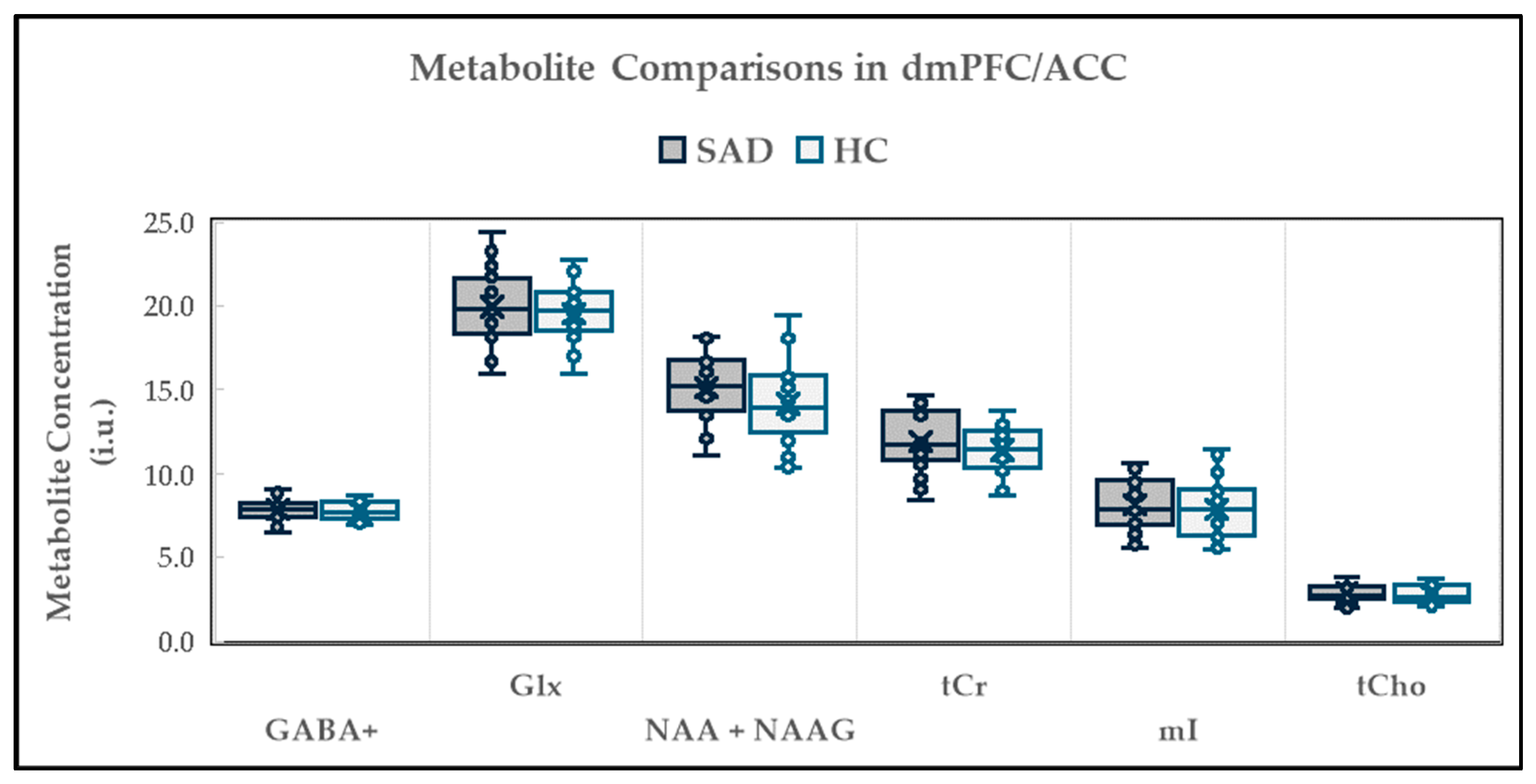
2.2. Neurochemical Alterations in dmPFC/ACC
2.2.1. Between-Group Neurochemical Comparison
2.2.2. Relationship Between Neurochemical Levels and Demographic Variables
2.2.3. Relationship Between Neurochemical Levels and Clinical Variables
2.2.4. Relationship Between Neurochemicals
2.3. Neurochemical Alterations in dlPFC
2.3.1. Between-Group Neurochemical Comparison
2.3.2. Relationship Between Neurochemical Levels and Demographic Variables
2.3.3. Relationship Between Neurochemical Levels and Clinical Variables
2.3.4. Relationship Between Neurochemicals
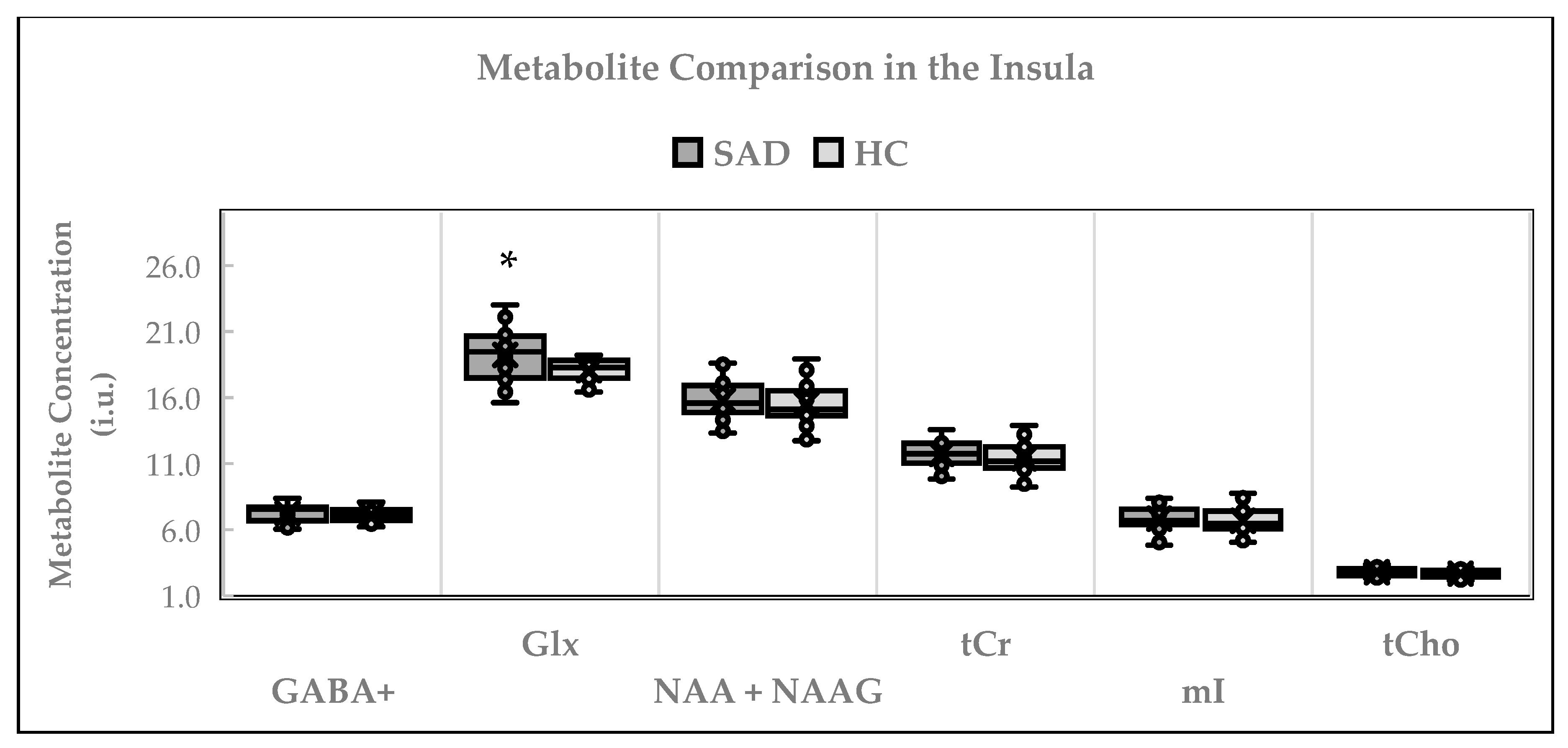
2.4. Neurochemical Alterations in Insula
2.4.1. Between-Group Neurochemical Comparison
2.4.2. Relationship Between Neurochemical Levels and Demographic Variables
2.4.3. Relationship Between Neurochemical Levels and Clinical Variables
2.4.4. Relationship Between Neurochemicals
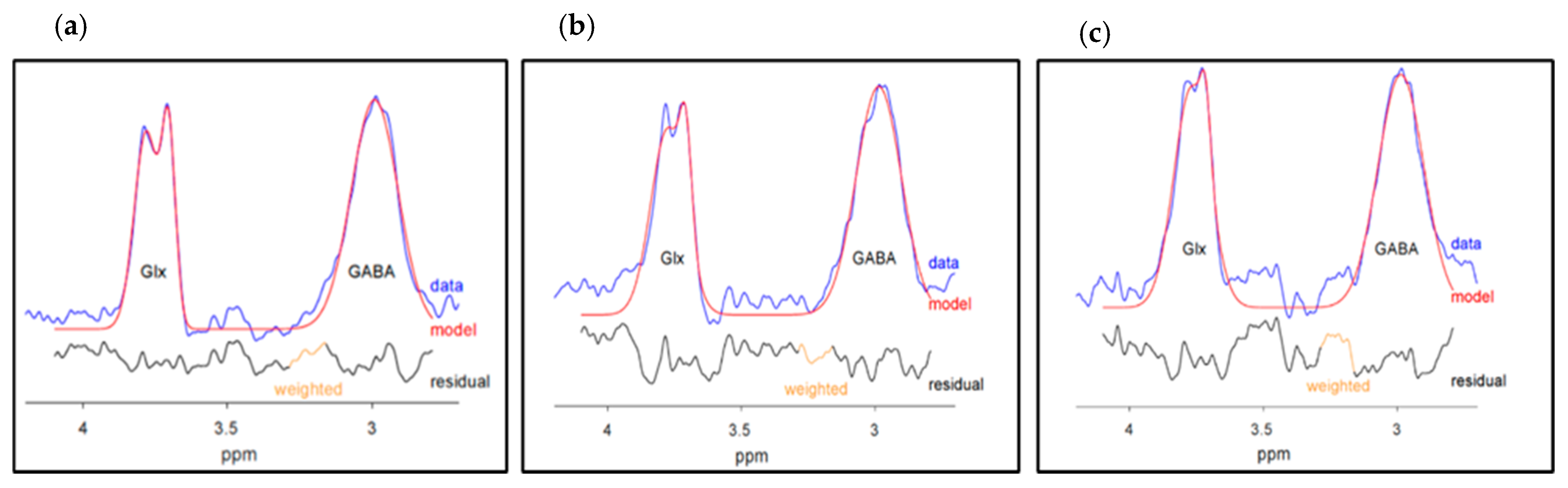
2.5. Spectroscopy Quality Measures
2.5.1. GABA+ and Glx Quality Measures
2.5.2. NAA + NAAG, tCr, mI, and tCho Quality Measures
3. Discussion
3.1. dmPFC/ACC
3.2. dlPFC
3.3. Insula
3.4. Study Limitations
4. Materials and Methods
4.1. Participants
4.2. Study Procedures
4.3. Outcome Measures
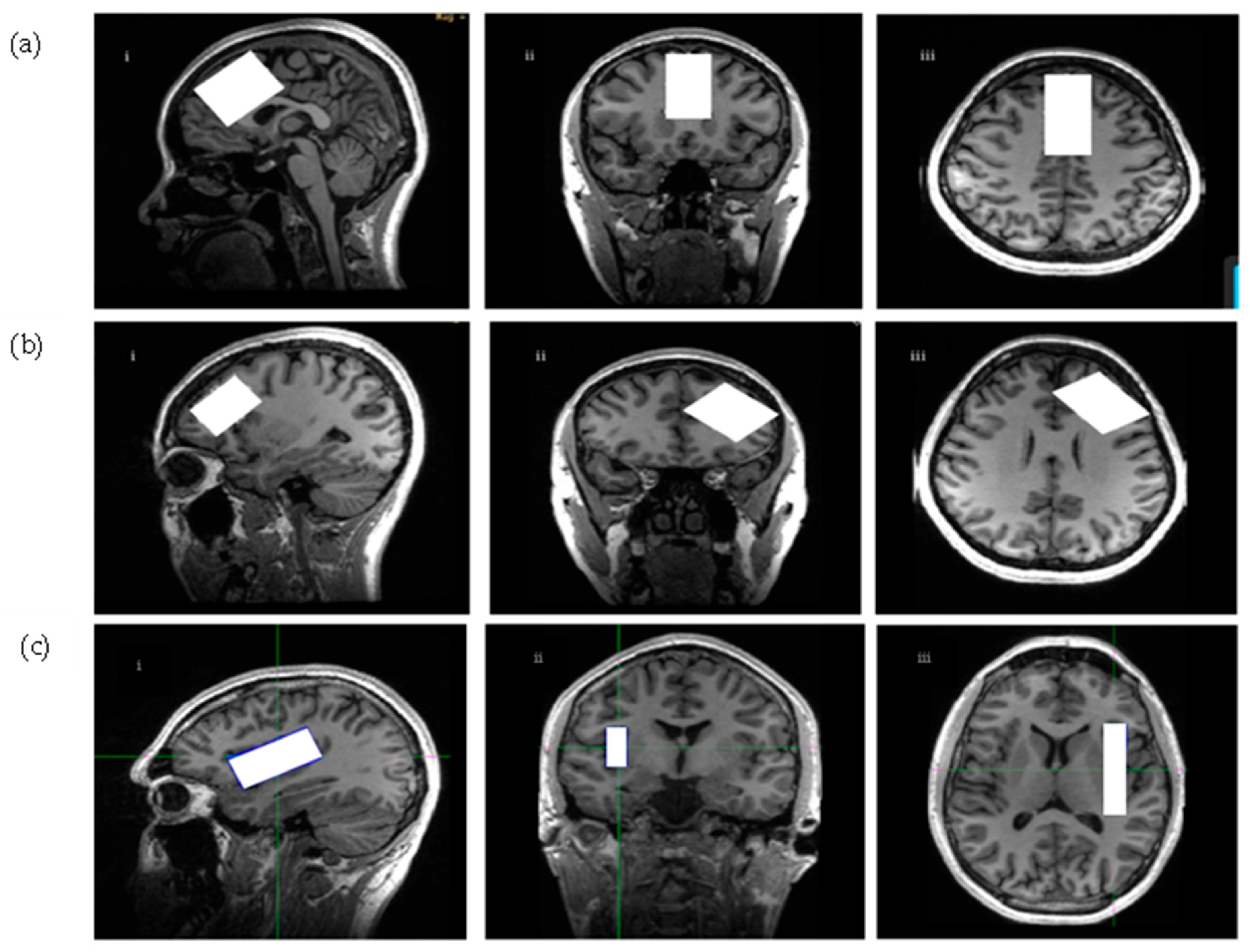
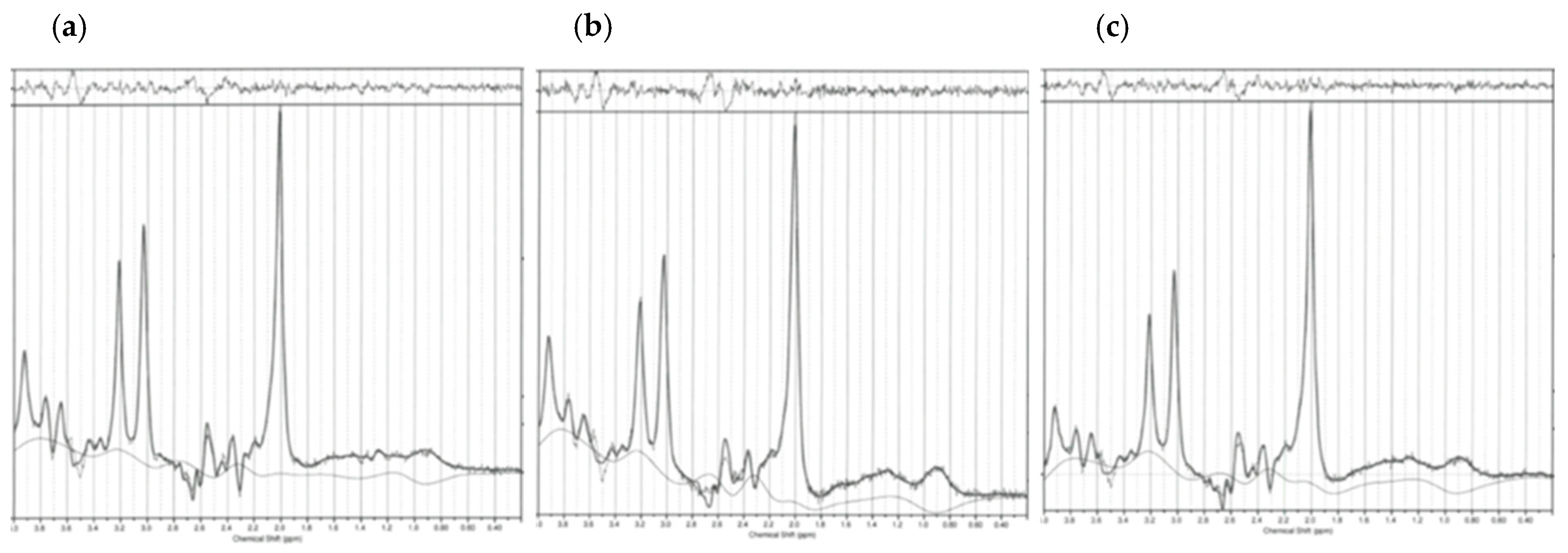
4.4. Scanning Procedures and Spectroscopic Parameters
4.4.1. Scanning Procedures
4.4.2. Spectroscopic Parameters
4.5. Spectroscopic Data Processing
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Merikangas, K.R.; Walters, E.E. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005, 62, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.J.; Lim, C.C.W.; Roest, A.M.; de Jonge, P.; Aguilar-Gaxiola, S.; Al-Hamzawi, A.; Alonso, J.; Benjet, C.; Bromet, E.J.; Bruffaerts, R.; et al. The cross-national epidemiology of social anxiety disorder: Data from the World Mental Health Survey Initiative. BMC Med. 2017, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Leichsenring, F.; Leweke, F. Social Anxiety Disorder. N. Engl. J. Med. 2017, 376, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E. Mental Disorders and Access to Mental Health Care; Statistics Canada: Ottawa, ON, Canada, 2023. [Google Scholar]
- Bruffaerts, R.; Harris, M.G.; Kazdin, A.E.; Vigo, D.V.; Sampson, N.A.; Chiu, W.T.; Al-Hamzawi, A.; Alonso, J.; Altwaijri, Y.A.; Andrade, L.; et al. Perceived helpfulness of treatment for social anxiety disorder: Findings from the WHO World Mental Health Surveys. Soc. Psychiatry Psychiatr. Epidemiol. 2022, 57, 2079–2095. [Google Scholar] [CrossRef] [PubMed]
- Fonzo, G.A.; Ramsawh, H.J.; Flagan, T.M.; Sullivan, S.G.; Letamendi, A.; Simmons, A.N.; Paulus, M.P.; Stein, M.B. Common and disorder-specific neural responses to emotional faces in generalized anxiety, social anxiety, and panic disorders. Br. J. Psychiatry 2015, 206, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Brehl, A.-K.; Kohn, N.; Fernandez, G.; Schene, A.H. A mechanistic model for individualized treatment of anxiety disorders based on predictive neural biomarkers. Psychol. Med. 2020, 50, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, S.; Rubin-Kahana, D.S.; Kloiber, S.; Kennedy, S.H.; Chavez, S.; Le Foll, B. Neurochemical Alterations in Social Anxiety Disorder (SAD): A Systematic Review of Proton Magnetic Resonance Spectroscopic Studies. Int. J. Mol. Sci. 2022, 23, 4754. [Google Scholar] [CrossRef] [PubMed]
- Pitts, M.W.; Todorovic, C.; Blank, T.; Takahashi, L.K. The central nucleus of the amygdala and corticotropin-releasing factor: Insights into contextual fear memory. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 7379–7388. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.-Z. From Structure to Behavior in Basolateral Amygdala-Hippocampus Circuits. Front. Neural Circuits 2017, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Q.; Mu, W.; Rodriguez, M.; Qian, M.; Berger, T. The effect of shame on patients with social anxiety disorder in internet-based cognitive behavioral therapy: Randomized controlled trial. JMIR Ment. Health 2020, 7, e15797. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, J.; Benson, B.; Farber, M.; Pine, D.; Ernst, M. Altered striatal intrinsic functional connectivity in pediatric anxiety. Neuropsychologia 2016, 85, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.P.I.; Simon, D.; Miltner, W.H.R.; Straube, T. Altered activation of the ventral striatum under performance-related observation in social anxiety disorder. Psychol. Med. 2017, 47, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Bas-Hoogendam, J.M.; van Steenbergen, H.; Nienke Pannekoek, J.; Fouche, J.-P.; Lochner, C.; Hattingh, C.J.; Cremers, H.R.; Furmark, T.; Månsson, K.N.T.; Frick, A.; et al. Voxel-based morphometry multi-center mega-analysis of brain structure in social anxiety disorder. NeuroImage Clin. 2017, 16, 678–688. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, R.A. In Vivo NMR Spectroscopy: Principles and Techniques; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Epperson, C.N.; Gueorguieva, R.; Czarkowski, K.A.; Stiklus, S.; Sellers, E.; Krystal, J.H.; Rothman, D.L.; Mason, G.F. Preliminary evidence of reduced occipital GABA concentrations in puerperal women: A 1H-MRS study. Psychopharmacology 2006, 186, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine-glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Gardier, A.M. Fast-acting antidepressant activity of ketamine: Highlights on brain serotonin, glutamate, and GABA neurotransmission in preclinical studies. Pharmacol. Ther. 2019, 199, 58–90. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Xu, A.; Cui, S.; Sun, M.R.; Xue, Y.C.; Wang, J.H. Impaired GABA synthesis, uptake, and release are associated with depression-like behaviors induced by chronic mild stress. Transl. Psychiatry 2016, 6, e910. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.M. Glutamate: The Master Neurotransmitter and Its Implications in Chronic Stress and Mood Disorders. Front. Hum. Neurosci. 2021, 15, 722323. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate-Glutamine Cycle. Adv. Neurobiol. 2016, 13, 1–7. [Google Scholar] [PubMed]
- Xu, H.; Liu, L.; Tian, Y.; Wang, J.; Li, J.; Zheng, J.; Zhao, H.; He, M.; Xu, T.-L.; Duan, S.; et al. A Disinhibitory Microcircuit Mediates Conditioned Social Fear in the Prefrontal Cortex. Neuron 2019, 102, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, S.; Lin, A.; Stanwell, P. Glutamate and glutamine: A review of in vivo MRS in the human brain. NMR Biomed. 2013, 26, 1630–1646. [Google Scholar] [CrossRef] [PubMed]
- Pollack, M.H.; Jensen, J.E.; Simon, N.M.; Kaufman, R.E.; Renshaw, P.F. High-field MRS study of GABA, glutamate and glutamine in social anxiety disorder: Response to treatment with levetiracetam. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Grills, R. Brain Neurochemicals in Generalized Social Anxiety Disorder: A Proton Magnetic Resonance Spectroscopy Study. Ph.D. Thesis, University of Alberta, Edmonton, AB, Canada, 2011. [Google Scholar]
- Howells, F.M.; Hattingh, C.J.; Syal, S.; Breet, E.; Stein, D.J.; Lochner, C. (1)H-magnetic resonance spectroscopy in social anxiety disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 58, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.G.; Rowe, J.B. Neurotransmitter deficits from frontotemporal lobar degeneration. Brain 2018, 141, 1263–1285. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.G.; Rouse, M.A.; Jones, P.S.; Ye, R.; Hezemans, F.H.; O’Callaghan, C.; Frangou, P.; Kourtzi, Z.; Rua, C.; Carpenter, T.A.; et al. GABA and glutamate deficits from frontotemporal lobar degeneration are associated with disinhibition. Brain 2020, 143, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Plitman, E.; de la Fuente-Sandoval, C.; Reyes-Madrigal, F.; Chavez, S.; Gómez-Cruz, G.; León-Ortiz, P.; Graff-Guerrero, A. Elevated Myo-Inositol, Choline, and Glutamate Levels in the Associative Striatum of Antipsychotic-Naive Patients with First-Episode Psychosis: A Proton Magnetic Resonance Spectroscopy Study with Implications for Glial Dysfunction. Schizophr. Bull. 2016, 42, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Chhetri, D.R. Myo-Inositol and Its Derivatives: Their Emerging Role in the Treatment of Human Diseases. Front. Pharmacol. 2019, 10, 1172. [Google Scholar] [CrossRef] [PubMed]
- Urenjak, J.; Williams, S.R.; Gadian, D.G.; Noble, M. Specific expression of N-acetyl aspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J. Neurochem. 1992, 59, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Schuff, N.; Meyerhoff, D.J.; Mueller, S.; Chao, L.; Sacrey, D.T.; Laxer, K.; Weiner, M.W. N-acetylaspartate as a marker of neuronal injury in neurodegenerative disease. Adv. Exp. Med. Biol. 2006, 576, 241–262; discussion 361–363. [Google Scholar] [PubMed]
- Bagnato, F.; Gauthier, S.A.; Laule, C.; Moore, G.R.W.; Bove, R.; Cai, Z.; Cohen-Adad, J.; Harrison, D.M.; Klawiter, E.C.; Morrow, S.A.; et al. Imaging Mechanisms of Disease Progression in Multiple Sclerosis: Beyond Brain Atrophy. J. Neuroimaging 2020, 30, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Blinkouskaya, Y.; Caçoilo, A.; Gollamudi, T.; Jalalian, S.; Weickenmeier, J. Brain aging mechanisms with mechanical manifestations. Mech. Ageing Dev. 2021, 200, 111575. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Richter-Landsberg, C.; Leibfritz, D. Multinuclear NMR Studies on the Energy Metabolism of Glial and Neuronal Cells. Dev. Neurosci. 1993, 15, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Cecil, K.M. Proton magnetic resonance spectroscopy: Technique for the neuroradiologist. Neuroimaging Clin. N. Am. 2013, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Tupler, L.A.; Davidson, J.R.T.; Smith, R.D.; Lazeyras, F.; Charles, H.C.; Krishnan, K.R.R. A Repeat Proton Magnetic Resonance Spectroscopy Study in Social Phobia. Biol. Psychiatry 1997, 42, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.; Renshaw, P.F. Mitochondrial dysfunction in bipolar disorder: Evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 2005, 10, 900–919. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.J.; Bell, C.J.; Malizia, A.L. Brain mechanisms of social anxiety disorder. J. Clin. Psychiatry 1998, 59, 4–11. [Google Scholar] [PubMed]
- Kim, H.; McGrath, B.M.; Silverstone, P.H. A review of the possible relevance of inositol and the phosphatidylinositol second messenger system (PI-cycle) to psychiatric disorders—Focus on magnetic resonance spectroscopy (MRS) studies. Hum. Psychopharmacol. 2005, 20, 309–326. [Google Scholar] [CrossRef]
- Filipović, D.; Todorović, N.; Bernardi, R.E.; Gass, P. Oxidative and nitrosative stress pathways in the brain of socially isolated adult male rats demonstrating depressive- and anxiety-like symptoms. Brain Struct. Funct. 2017, 222, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Fedoce, A.d.G.; Ferreira, F.; Bota, R.G.; Bonet-Costa, V.; Sun, P.Y.; Davies, K.J.A. The role of oxidative stress in anxiety disorder: Cause or consequence? Free Radic. Res. 2018, 52, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Hnilicová, P.; Strbak, O.; Kolisek, M.; Kurča, E.; Zeleňák, K.; Sivák, Š.; Kantorova, E. Molecular Sciences Current Methods of Magnetic Resonance for Noninvasive Assessment of Molecular Aspects of Pathoetiology in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 6117. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Kaur, G. Withania somnifera as a Potential Anxiolytic and Anti-inflammatory Candidate Against Systemic Lipopolysaccharide-Induced Neuroinflammation. Neuromol. Med. 2018, 20, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, N.; Cohen Kadosh, K. Excitatory and inhibitory neurochemical markers of anxiety in young females. Dev. Cogn. Neurosci. 2024, 66, 101363. [Google Scholar] [CrossRef] [PubMed]
- Michels, L.; Schulte-Vels, T.; Schick, M.; O’Gorman, R.L.; Zeffiro, T.; Hasler, G.; Mueller-Pfeiffer, C. Prefrontal GABA and glutathione imbalance in posttraumatic stress disorder: Preliminary findings. Psychiatry Res. 2014, 224, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Brühl, A.B.; Delsignore, A.; Komossa, K.; Weidt, S. Neuroimaging in social anxiety disorder—A meta-analytic review resulting in a new neurofunctional model. Neurosci. Biobehav. Rev. 2014, 47, 260–280. [Google Scholar] [CrossRef] [PubMed]
- Bas-Hoogendam, J.M.; Blackford, J.U.; Brühl, A.B.; Blair, K.S.; van der Wee, N.J.A.; Westenberg, P.M. Neurobiological candidate endophenotypes of social anxiety disorder. Neurosci. Biobehav. Rev. 2016, 71, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Bas-Hoogendam, J.M.; van Steenbergen, H.; Tissier, R.L.M.; van der Wee, N.J.A.; Westenberg, P.M. Altered Neurobiological Processing of Unintentional Social Norm Violations: A Multiplex, Multigenerational Functional Magnetic Resonance Imaging Study on Social Anxiety Endophenotypes. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2020, 5, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Mizzi, S.; Pedersen, M.; Lorenzetti, V.; Heinrichs, M.; Labuschagne, I. Resting-state neuroimaging in social anxiety disorder: A systematic review. Mol. Psychiatry 2022, 27, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Zilverstand, A.; Sorger, B.; Sarkheil, P.; Goebel, R. fMRI neurofeedback facilitates anxiety regulation in females with spider phobia. Front. Behav. Neurosci. 2015, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ke, J.; Qi, R.; Xu, Q.; Zhong, Y.; Liu, T.; Li, J.; Zhang, L.; Lu, G. Increased Inhibition of the Amygdala by the mPFC may Reflect a Resilience Factor in Post-traumatic Stress Disorder: A Resting-State fMRI Granger Causality Analysis. Front. Psychiatry 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Fernández, G.; Hermans, E.; Vogel, S.; Zhang, Y.; Li, H.; Klumpers, F. How acute stress may enhance subsequent memory for threat stimuli outside the focus of attention: DLPFC-amygdala decoupling. Neuroimage 2018, 171, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.C.; Simon, N.M.; Dougherty, D.D.; Hoge, E.A.; Worthington, J.J.; Chow, C.; Kaufman, R.E.; Gold, A.L.; Fischman, A.J.; Pollack, M.H.; et al. A PET Study of Tiagabine Treatment Implicates Ventral Medial Prefrontal Cortex in Generalized Social Anxiety Disorder. Neuropsychopharmacology 2009, 34, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Thoeringer, C.K.; Ripke, S.; Unschuld, P.G.; Lucae, S.; Ising, M.; Bettecken, T.; Uhr, M.; Keck, M.E.; Mueller-Myhsok, B.; Holsboer, F.; et al. The GABA transporter 1 (SLC6A1): A novel candidate gene for anxiety disorders. J. Neural Transm. 2009, 116, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Cloak, C.C.; Ernst, T. Magnetic resonance spectroscopy studies of GABA in neuropsychiatric disorders. J. Clin. Psychiatry 2003, 64, 7–14. [Google Scholar] [PubMed]
- Ratai, E.M.; Annamalai, L.; Burdo, T.; Joo, C.G.; Bombardier, J.P.; Fell, R.; Hakimelahi, R.; He, J.; Lentz, M.R.; Campbell, J.; et al. Brain creatine elevation and N-Acetylaspartate reduction indicates neuronal dysfunction in the setting of enhanced glial energy metabolism in a macaque model of neuroAIDS. Magn. Reson. Med. 2011, 66, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Meftahi, G.H.; Hatef, B.; Pirzad Jahromi, G. Creatine Activity as a Neuromodulator in the Central Nervous System. Arch. Razi Inst. 2023, 78, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Ducray, A.D.; Schlepp, J.A.; Qualls, R.; Andres, R.H.; Seiler, R.W.; Schlattner, U.; Wallimann, T.; Widmer, H.R. Creatine treatment promotes differentiation of GABA-ergic neuronal precursors in cultured fetal rat spinal cord. J. Neurosci. Res. 2007, 85, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Gerbatin, R.R.; Silva, L.F.A.; Hoffmann, M.S.; Della-Pace, I.D.; do Nascimento, P.S.; Kegler, A.; de Zorzi, V.N.; Cunha, J.M.; Botelho, P.; Neto, J.B.T.; et al. Delayed creatine supplementation counteracts reduction of GABAergic function and protects against seizures susceptibility after traumatic brain injury in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Takahashi, H.; Oikawa, D.; Tachibana, T.; Denbow, D.M.; Furuse, M. Brain creatine functions to attenuate acute stress responses through GABAnergic system in chicks. Neuroscience 2005, 132, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.D.; Bröer, S. Creatine as a booster for human brain function. How might it work? Neurochem. Int. 2015, 89, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Nersesova, L.; Petrosyan, M.; Arutjunyan, A. Neuroprotective Potential of Creatine. Hidden Resources of Its Therapeutic and Preventive Use. Neurochem. J. 2022, 16, 14–30. [Google Scholar] [CrossRef]
- Turegano-Lopez, M.; de las Pozas, F.; Santuy, A.; Rodriguez, J.-R.; DeFelipe, J.; Merchan-Perez, A. Tracing nerve fibers with volume electron microscopy to quantitatively analyze brain connectivity. Commun. Biol. 2024, 7, 796. [Google Scholar] [CrossRef] [PubMed]
- Colla, M.; Schubert, F.; Bubner, M.; Heidenreich, J.O.; Bajbouj, M.; Seifert, F.; Luborzewski, A.; Heuser, I.; Kronenberg, G. Glutamate as a spectroscopic marker of hippocampal structural plasticity is elevated in long-term euthymic bipolar patients on chronic lithium therapy and correlates inversely with diurnal cortisol. Mol. Psychiatry 2009, 14, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. Int. J. Mol. Sci. 2019, 20, 5674. [Google Scholar] [CrossRef] [PubMed]
- Nasir, M.; Trujillo, D.; Levine, J.; Dwyer, J.B.; Rupp, Z.W.; Bloch, M.H. Glutamate Systems in DSM-5 Anxiety Disorders: Their Role and a Review of Glutamate and GABA Psychopharmacology. Front. Psychiatry 2020, 11, 548505. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A. Milestone Review: Metabolic dynamics of glutamate and GABA mediated neurotransmission—The essential roles of astrocytes. J. Neurochem. 2023, 166, 109–137. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Diniz, V.L.S.; Dodd, G.T.; Cruzat, V. Glutamine metabolism and optimal immune and CNS function. Proc. Nutr. Soc. 2023, 82, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Adamczyk, A.; Shugart, Y.Y.; Samuels, J.F.; Grados, M.A.; Greenberg, B.D.; Knowles, J.A.; McCracken, J.T.; Rauch, S.L.; Murphy, D.L.; et al. A screen of SLC1A1 for OCD-related alleles. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Chubar, V.; Van Leeuwen, K.; Bijttebier, P.; Van Assche, E.; Bosmans, G.; Van den Noortgate, W.; van Winkel, R.; Goossens, L.; Claes, S. Gene-environment interaction: New insights into perceived parenting and social anxiety among adolescents. Eur. Psychiatry 2020, 63, e64. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.D.; Jüngling, K.; Paulukat, L.; Vieler, M.; Gaburro, S.; Sosulina, L.; Blaesse, P.; Sreepathi, H.K.; Ferraguti, F.; Pape, H.-C. Glutamic Acid Decarboxylase 65: A Link Between GABAergic Synaptic Plasticity in the Lateral Amygdala and Conditioned Fear Generalization. Neuropsychopharmacology 2014, 39, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Hettema, J.M.; An, S.S.; Neale, M.C.; Bukszar, J.; van den Oord, E.J.C.G.; Kendler, K.S.; Chen, X. Association between glutamic acid decarboxylase genes and anxiety disorders, major depression, and neuroticism. Mol. Psychiatry 2006, 11, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Unschuld, P.G.; Ising, M.; Specht, M.; Erhardt, A.; Ripke, S.; Heck, A.; Kloiber, S.; Straub, V.; Brueckl, T.; Müller-Myhsok, B.; et al. Polymorphisms in the GAD2 gene-region are associated with susceptibility for unipolar depression and with a risk factor for anxiety disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Henry, H.; Loup, M.; Eilers, B.; Bachmann, C. Endogenous synthesis and transport of creatine in the rat brain: An in situ hybridization study. Brain Res. Mol. Brain Res. 2001, 86, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O. Ammonia toxicity to the brain: Effects on creatine metabolism and transport and protective roles of creatine. Mol. Genet. Metab. 2010, 100, S53–S58. [Google Scholar] [CrossRef] [PubMed]
- Motyka, S.; Moser, P.; Hingerl, L.; Hangel, G.; Heckova, E.; Strasser, B.; Eckstein, K.; Daniel Robinson, S.; Poser, B.A.; Gruber, S. The influence of spatial resolution on the spectral quality and quantification accuracy of whole-brain MRSI at 1.5 T, 3T, 7T, and 9.4 T. Magn. Reson. Med. 2019, 82, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Kreis, R.; Boer, V.; Choi, I.-Y.; Cudalbu, C.; de Graaf, R.A.; Gasparovic, C.; Heerschap, A.; Krššák, M.; Lanz, B.; Maudsley, A.A.; et al. Terminology and concepts for the characterization of in vivo MR spectroscopy methods and MR spectra: Background and experts’ consensus recommendations. NMR Biomed. 2021, 34, e4347. [Google Scholar] [CrossRef] [PubMed]
- Edden, R.A.E.; Puts, N.A.J.; Harris, A.D.; Barker, P.B.; Evans, C.J. Gannet: A batch-processing tool for the quantitative analysis of gamma-aminobutyric acid–edited MR spectroscopy spectra. J. Magn. Reson. Imaging 2014, 40, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Perneger, T.V. What’s wrong with Bonferroni adjustments? BMJ 1998, 316, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- First, M.; Williams, J.B.W.; Karg, R.S.; Spitzer, R.L. Structured Clinical Interview for DSM-5—Research Version (SCID-5 for DSM-5, Research Version; SCID-5-RV); American Psychiatric Association: Arlington, VA, USA, 2015. [Google Scholar]
- Liebowitz, M.R. Social phobia. Mod. Probl. Pharmacopsychiatry 1987, 22, 141–173. [Google Scholar] [PubMed]
- First, M.; Williams, J.B.W.; Smith Benjamin, L.; Spitzer, R.L. Structured Clinical Interview for DSM-5® Personality Disorders (SCID-5-PD); American Psychiatric Association: Arlington, VA, USA, 2016. [Google Scholar]
- Mattick, R.P.; Clarke, J.C. Development and validation of measures of social phobia scrutiny fear and social interaction anxiety. Behav. Res. Ther. 1998, 36, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.J.; Trivedi, M.H.; Ibrahim, H.M.; Carmody, T.J.; Arnow, B.; Klein, D.N.; Markowitz, J.C.; Ninan, P.T.; Kornstein, S.; Manber, R.; et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): A psychometric evaluation in patients with chronic major depression. Biol. Psychiatry 2003, 54, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.V.; Harnett-Sheehan, K.; Raj, B.A. The measurement of disability. Int. Clin. Psychopharmacol. 1996, 11, 89–95. [Google Scholar] [CrossRef] [PubMed]
- The Whoqol Group. Development of the World Health Organization WHOQOL-BREF Quality of Life Assessment. Psychol. Med. 1998, 28, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.L.; Heinrichs, N.; Kim, H.J.; Hofmann, S.G. The Liebowitz social anxiety scale as a self-report instrument: A preliminary psychometric analysis. Behav. Res. Ther. 2002, 40, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.R.; Hughes, D.L.; George, L.K.; Blazer, D.G. The epidemiology of social phobia: Findings from the Duke Epidemiological Catchment Area Study. Psychol. Med. 1993, 23, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Zhao, Y.J.; Zhang, Y.; He, F.; Pan, H.Q.; Cheung, T.; Ungvari, G.S.; Li, S.Y.; Xiang, Y.T. Psychometric Properties of the Quick Inventory of Depressive Symptomatology-Self-Report (QIDS-SR) in Depressed Adolescents. Front. Psychiatry 2020, 11, 598609. [Google Scholar] [CrossRef] [PubMed]
- Hambrick, J.P.; Turk, C.L.; Heimberg, R.G.; Schneier, F.R.; Liebowitz, M.R. Psychometric properties of disability measures among patients with social anxiety disorder. J. Anxiety Disord. 2004, 18, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Skevington, S.M.; Lotfy, M.; O’Connell, K.A. The World Health Organization’s WHOQOL-BREF quality of life assessment: Psychometric properties and results of the international field trial. A report from the WHOQOL group. Qual. Life Res. 2004, 13, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.W.; Listerud, J.; Kressel, H.Y. Combined chemical-shift and phase-selective imaging for fat suppression: Theory and initial clinical experience. Radiology 1991, 181, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Mescher, M.; Merkle, H.; Kirsch, J.; Garwood, M.; Gruetter, R. Simultaneous in vivo spectral editing and water suppression. NMR Biomed. 1998, 11, 266–272. [Google Scholar] [CrossRef]
- Puts, N.A.; Edden, R.A. In vivo magnetic resonance spectroscopy of GABA: A methodological review. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.; Weismann, A.; Berndt, R. The Kondo resonance line shape in scanning tunneling spectroscopy: Instrumental aspects. J. Phys. Condens. Matter 2018, 30, 424001. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001, 14, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.; Devenyi, G.A.; Jezzard, P.; Hennessy, T.J.; Near, J. Advanced processing and simulation of MRS data using the FID appliance (FID-A)-An open source, MATLAB-based toolkit. Magn. Reson. Med. 2017, 77, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Gasparovic, C.; Song, T.; Devier, D.; Bockholt, H.J.; Caprihan, A.; Mullins, P.G.; Posse, S.; Jung, R.E.; Morrison, L.A. Use of tissue water as a concentration reference for proton spectroscopic imaging. Magn. Reson. Med. 2006, 55, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Woolrich, M.W.; Jbabdi, S.; Patenaude, B.; Chappell, M.; Makni, S.; Behrens, T.; Beckmann, C.; Jenkinson, M.; Smith, S.M. Bayesian analysis of neuroimaging data in FSL. NeuroImage 2009, 45, S173–S186. [Google Scholar] [CrossRef] [PubMed]
- Kurcyus, K.; Annac, E.; Hanning, N.M.; Harris, A.D.; Oeltzschner, G.; Edden, R.; Riedl, V. Opposite Dynamics of GABA and Glutamate Levels in the Occipital Cortex during Visual Processing. J. Neurosci. 2018, 38, 9967. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, S.; Truong, P.; Sailasuta, N.; Le Foll, B. Evaluating Back-to-Back and Day-to-Day Reproducibility of Cortical GABA+ Measurements Using Proton Magnetic Resonance Spectroscopy ((1)H MRS). Int. J. Mol. Sci. 2023, 24, 7713. [Google Scholar] [CrossRef] [PubMed]
- Bender, R.; Lange, S. Multiple test procedures other than Bonferroni’s deserve wider use. BMJ 1999, 318, 600–601. [Google Scholar] [CrossRef] [PubMed]
- Teresi, J.A.; Yu, X.; Stewart, A.L.; Hays, R.D. Guidelines for Designing and Evaluating Feasibility Pilot Studies. Med. Care 2022, 60, 95–103. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAD Group (n = 26) | Control Group (n = 26) | |||
| M (±SD) or % | M (±SD) or % | t or χ2 | d or Phi | |
| Demographic Variables | ||||
| Age (years) | 25.27 ± 5.01 | 25.15 ± 4.43 | 0.09 | 0.02 |
| Age of onset of SAD (years) | 12.67 ± 3.32 | |||
| Sex (% female) | 61.5% | 61.5% | 0.00 | 0.00 |
| Comorbidities (%) | ||||
| Clinical Features of AvPD | 88.5% | |||
| Lifetime MDD | 26.9% | |||
| Other (PDD, GAD) | 19.2% | |||
| Education (% university of higher) | 57.7% | 30.8% | 3.45 | −0.30 |
| Years of Education | 14.94 ± 2.71 | 15.90 ± 2.45 | −1.34 | −0.37 |
| Race (% Caucasian) | 42.3% | 23.1% | 1.40 | 0.21 |
| Race (% East Asian) | 30.8% | 30.8% | 0.00 | 0.00 |
| Employed or Student (%) | 92.0% | 92.3% | 0.00 | 0.00 |
| Last year’s income (% ≥$50,000) | 61.5% | 42.3% | 1.23 | 0.19 |
| Married/Common Law (%) | 30.8% | 11.5% | 1.85 | −0.24 |
| Clinical Variables | ||||
| LSAS Total | 98.08 ± 13.56 | 7.77 ± 6.31 | 30.78 ‡ | 8.54 |
| LSAS Anxiety | 50.69 ± 7.95 | 5.23 ± 4.40 | 25.11 ‡ | 7.08 |
| LSAS Avoidance | 47.38 ± 8.22 | 2.54 ± 3.73 | 26.38 ‡ | 7.32 |
| SIAS | 49.62 ± 11.71 | 4.88 ± 4.54 | 18.17 ‡ | 5.04 |
| QIDS-16 | 8.38 ± 5.12 | 2.23 ± 1.99 | 5.72 ‡ | 1.59 |
| SDS Total | 14.35 ± 7.47 | 0.85 ± 2.15 | 8.85 ‡ | 2.46 |
| SDS Work/School | 5.27 ± 3.01 | 0.35 ± 0.98 | 7.92 ‡ | 2.20 |
| SDS Social Activities | 6.31 ± 2.91 | 0.27 ± 0.88 | 10.14 ‡ | 2.81 |
| SDS Family Life/Home Responsibilities | 2.77 ± 2.71 | 0.27 ± 0.87 | 4.54 ‡ | 1.26 |
| WHOQoL BREF Physical | 50.92 ± 12.63 | 64.38 ± 7.66 | −4.68 ‡ | −1.29 |
| WHOQoL BREF Psychological | 54.38 ± 10.87 | 68.62 ± 8.49 | −5.27 ‡ | −1.46 |
| WHOQoL BREF Social | 61.58 ± 22.13 | 86.50 ± 10.90 | −5.15 ‡ | −1.43 |
| WHOQoL BREF Environmental | 71.81 ± 14.73 | 85.62 ± 11.02 | −3.83 ‡ | −1.06 |
| GABA+ (i.u.) | Glx (i.u.) | GABA+/ Glx | NAA + NAAG (i.u.) | tCr (i.u.) | mI (i.u.) | tCho (i.u.) | |
|---|---|---|---|---|---|---|---|
| LSAS total | −1.68 | −0.84 | −0.13 | −1.80 | −0.83 | −1.69 | −0.33 |
| LSAS Anxiety | −2.40 * | −1.66 | 0.36 | −2.04 * | −1.47 | −1.79 | −0.93 |
| LSAS Avoidance | −0.66 | 0.17 | −0.72 | −1.18 | 0.13 | −1.21 | 0.27 |
| SIAS | −1.44 | −2.94 † | 1.73 | −1.37 | −0.81 | −1.24 | −1.15 |
| QIDS-16 | −0.03 | 0.14 | 0.07 | −1.54 | −1.30 | −1.56 | −1.46 |
| SDS total | −1.49 | −0.12 | −0.90 | −1.24 | −2.43 * | −2.54 * | −2.61 † |
| SDS Work/School | −1.89 | −0.53 | −0.70 | −1.70 | −2.86 ‡ | −2.82 † | −3.11 † |
| SDS Social Life | −1.44 | −0.84 | −0.60 | −1.38 | −2.03 * | −1.79 | −1.58 |
| SDS Family Life | −0.44 | 1.15 | −1.57 | −0.23 | −0.95 | −1.28 | −0.92 |
| WHOQoL BREF, Physical | −0.84 | −0.39 | 0.00 | 0.86 | 0.75 | 1.73 | 1.82 |
| WHOQoL BREF, Psychological | 0.21 | −0.43 | −0.73 | 0.70 | 0.00 | 1.23 | 0.54 |
| WHOQoL BREF, Social | 1.03 | −0.60 | 2.26 * | 0.37 | −0.69 | 0.14 | −0.44 |
| WHOQoL BREF, Environmental | −0.41 | −0.37 | 0.40 | 0.59 | 0.67 | 1.16 | 1.47 |
| GABA+ (i.u.) | Glx (i.u.) | GABA+ /Glx | NAA + NAAG (i.u.) | tCr (i.u.) | mI (i.u.) | tCho (i.u.) | |
|---|---|---|---|---|---|---|---|
| LSAS total | −0.98 | 1.32 | −1.37 | −1.06 | −0.83 | 0.10 | 0.40 |
| LSAS Anxiety | −1.66 | 0.65 | −1.46 | −1.23 | −0.45 | −0.13 | 0.50 |
| LSAS Avoidance | 0.27 | 1.74 | −0.80 | −0.45 | −1.23 | 0.54 | 0.03 |
| SIAS | −0.61 | 0.06 | −1.29 | −2.13 * | 0.03 | 0.26 | −0.24 |
| QIDS-16 | 0.30 | 0.37 | −0.31 | −1.56 | −0.40 | 1.00 | −0.92 |
| SDS total | 1.33 | 0.59 | 0.92 | −0.58 | −0.60 | 0.45 | −0.67 |
| SDS Work/School | 0.57 | 0.44 | 0.63 | −1.46 | −0.72 | 0.13 | −1.00 |
| SDS Social Life | 1.60 | 0.92 | 0.62 | −0.95 | −0.35 | 1.03 | −0.32 |
| SDS Family Life | 1.19 | 0.37 | 1.05 | −0.20 | −0.22 | 0.23 | −0.07 |
| WHOQoL BREF, Physical | −0.09 | −0.75 | 1.09 | 1.36 | 0.20 | −0.66 | 2.19 * |
| WHOQoL BREF, Psychological | −0.69 | −0.91 | 0.70 | 1.28 | −0.94 | −2.15 * | 0.25 |
| WHOQoL BREF, Social | −1.88 | −2.36 * | 1.08 | 0.27 | −2.15 * | −2.11 * | 0.97 |
| WHOQoL BREF, Environmental | −1.38 | −1.46 | 0.60 | 0.62 | −0.74 | −0.63 | 1.07 |
| GABA+ (i.u.) | Glx (i.u.) | GABA+/ Glx | NAA + NAAG (i.u.) | tCr (i.u.) | mI (i.u.) | tCho (i.u.) | |
|---|---|---|---|---|---|---|---|
| LSAS total | 0.40 | 0.55 | 0.49 | −0.79 | −1.17 | −1.23 | −1.15 |
| LSAS Anxiety | −0.23 | −0.13 | −0.33 | −1.54 | −2.23 * | −1.77 | −1.57 |
| LSAS Avoidance | −0.56 | 0.39 | 1.45 | 0.29 | 0.07 | −0.43 | −0.80 |
| SIAS | −1.85 | −1.01 | −0.72 | −0.92 | −1.83 | −1.25 | −0.94 |
| QIDS-16 | 1.21 | −0.30 | 0.76 | −0.20 | −1.43 | −1.78 | 0.28 |
| SDS total | −0.07 | −0.33 | 0.72 | −0.20 | −0.77 | −0.97 | −0.94 |
| SDS Work/School | −0.37 | −0.27 | 0.62 | −0.96 | −0.93 | −0.87 | −1.22 |
| SDS Social Life | −0.30 | −0.80 | 0.36 | −1.07 | −0.50 | −0.41 | −0.25 |
| SDS Family Life | 0.56 | −0.95 | 1.23 | 0.13 | −0.38 | −0.95 | −0.51 |
| WHOQoL BREF, Physical | −1.61 | −0.22 | −0.89 | 0.23 | 0.85 | 1.50 | 0.94 |
| WHOQoL BREF, Psychological | −0.29 | 0.27 | 0.21 | 0.76 | 0.55 | 0.77 | −0.08 |
| WHOQoLBREF, Social | −0.18 | 0.92 | −0.66 | 0.62 | −0.32 | 0.37 | −0.24 |
| WHOQoL BREF, Environmental | −0.69 | 0.24 | 0.18 | 1.74 | 1.69 | 1.96 * | 0.52 |
| Scan Parameters | dmPFC/ACC | dlPFC | Insula |
|---|---|---|---|
| Echo Time (TE) | 68 msec | 68 msec | 68 msec |
| Repetition Time (TR) | 1500 msec | 1500 msec | 1500 msec |
| Number of Acquisitions 1 | 384 | 384 | 512 |
| Number of Excitations (NEX) | 8 | 8 | 8 |
| Number of Points | 4096 | 4096 | 4096 |
| Spectral Width | 5000 Hz | 5000 Hz | 5000 Hz |
| Scan Time | 10.4 min | 10.4 min | 13.5 min |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsaid, S.; Wang, R.; Kloiber, S.; Desmond, K.L.; Le Foll, B. A Proton Magnetic Resonance Spectroscopy (1H MRS) Pilot Study Revealing Altered Glutamatergic and Gamma-Aminobutyric Acid (GABA)ergic Neurotransmission in Social Anxiety Disorder (SAD). Int. J. Mol. Sci. 2025, 26, 6915. https://doi.org/10.3390/ijms26146915
Elsaid S, Wang R, Kloiber S, Desmond KL, Le Foll B. A Proton Magnetic Resonance Spectroscopy (1H MRS) Pilot Study Revealing Altered Glutamatergic and Gamma-Aminobutyric Acid (GABA)ergic Neurotransmission in Social Anxiety Disorder (SAD). International Journal of Molecular Sciences. 2025; 26(14):6915. https://doi.org/10.3390/ijms26146915
Chicago/Turabian StyleElsaid, Sonja, Ruoyu Wang, Stefan Kloiber, Kimberly L. Desmond, and Bernard Le Foll. 2025. "A Proton Magnetic Resonance Spectroscopy (1H MRS) Pilot Study Revealing Altered Glutamatergic and Gamma-Aminobutyric Acid (GABA)ergic Neurotransmission in Social Anxiety Disorder (SAD)" International Journal of Molecular Sciences 26, no. 14: 6915. https://doi.org/10.3390/ijms26146915
APA StyleElsaid, S., Wang, R., Kloiber, S., Desmond, K. L., & Le Foll, B. (2025). A Proton Magnetic Resonance Spectroscopy (1H MRS) Pilot Study Revealing Altered Glutamatergic and Gamma-Aminobutyric Acid (GABA)ergic Neurotransmission in Social Anxiety Disorder (SAD). International Journal of Molecular Sciences, 26(14), 6915. https://doi.org/10.3390/ijms26146915