
Combining CSF and Serum Biomarkers to Differentiate Mechanisms of Disability Worsening in Multiple Sclerosis

 , , , , , , , , , ,
, , , , , , , , , ,  , , , and add
Show full author list
, , , and add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics
2.2. Risk of RAW, Active, and Non-Active PIRA
2.3. Risk of IAW and naPIRA Across Different Biomarker Groups
- Triple Negative (NLGLM−): Low sNfL and sGFAP levels and absence of LS-OCMB.
- NLGLM+: Low sNfL and sGFAP levels, and the presence of LS-OCMB.
- NLGHM−: Low sNfL, high sGFAP, and absence of LS-OCMB.
- NLGHM+: Low sNfL, high sGFAP, and the presence of LS-OCMB.
- NHGLM−: High sNfL, low sGFAP, and absence of LS-OCMB.
- NHGHM−: High sNfL and sGFAP levels and absence of LS-OCMB.
- NHGLM+: High sNfL, low sGFAP, and the presence of LS-OCMB.
- Triple Positive (NHGHM+): High sNfL and sGFAP levels, with the presence of LS-OCMB.
2.4. CSF Cells and Soluble Factors Associated with Biomarkers
- sNfL low, LS-OCMB negative (NLM−).
- sNfL low, LS-OCMB positive (NLM+).
- sNfL high, LS-OCMB negative (NHM−).
- sNfL high, LS-OCMB positive (NHM+).
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Data Collection
4.3. CSF Analyses
4.4. Soluble Factors
4.5. MRI Protocols
4.6. Definitions
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| CD | Cluster of differentiation |
| CI | Confidence interval |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DMT | Disease-modifying treatments |
| EDSS | Expanded Disability Status Scale |
| ELISA | Enzyme-linked immunosorbent assay |
| FACS | Fluorescence-activated cell sorting |
| HR | Hazard ratio |
| IAW | Inflammatory-associated worsening |
| Ig | Immunoglobulin |
| ITMS | Intrathecal IgM synthesis |
| LS-OCMB | Lipid-specific IgM oligoclonal bands |
| mAb | Monoclonal antibodies |
| MS | Multiple sclerosis |
| MRI | Magnetic resonance imaging |
| OCB | Oligoclonal bands |
| PIRA | Progression independent of relapse activity |
| PM | Personalized medicine |
| PwMS | Patients with multiple sclerosis |
| RAW | Relapse-associated worsening |
| ROC | Receiver operating characteristic |
| sNfL | Serum neurofilament light chain |
| sGFAP | Serum glial fibrillary acidic protein |
| T-reg | Regulatory T cells |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recruitment of Patients |
|---|
| Hospital Universitario Ramón y Cajal (Madrid, Spain) |
| Hospital Universitario de Getafe (Madrid, Spain) |
| Hospital Universitario Gregorio Marañón (Madrid, Spain) |
| Hospital Universitario Príncipe de Asturias (Madrid, Spain) |
| Hospital Universitari Dr. Josep Trueta (Girona, Spain) |
| Hospital Clínic de Barcelona (Barcelona, Spain) |
| Hospital Universitari Vall d’Hebron (Barcelona, Spain) |
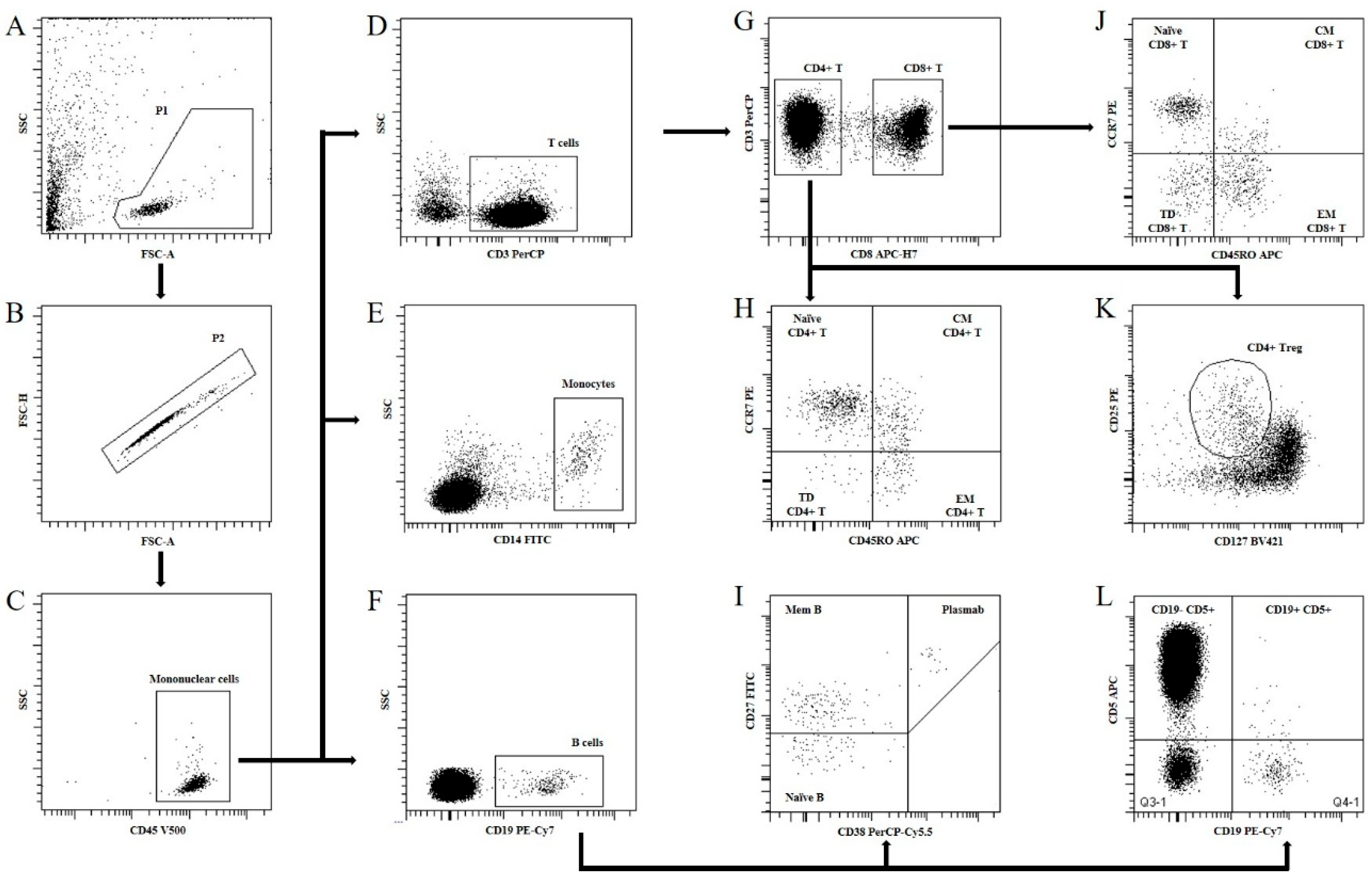
| Monoclonal Antibody | Fluorochrome Conjugated | Clone | Manufacturer | Catalog Number |
|---|---|---|---|---|
| CD3 | PerCP | SK7 | BD Biosciences | 345766 |
| CD5 | APC | L17F12 | BD Biosciences | 345783 |
| CD8 | APC-H7 | SK1 | BD Biosciences | 641400 |
| CD14 | FITC | MφP9 | BD Biosciences | 345784 |
| CD19 | PE-Cy7 | SJ25C1 | BD Biosciences | 341113 |
| CD24 | PE | ML5 | BD Biosciences | 555428 |
| CD25 | PE | 2A3 | BD Biosciences | 341011 |
| CD27 | FITC | L128 | BD Biosciences | 340424 |
| CD38 | PE-Cy5 | HIT2 | BD Biosciences | 555461 |
| CD45 | V500 | 2D1 | BD Biosciences | 655873 |
| CD45RO | APC | UCHL1 | BD Biosciences | 559865 |
| CD127 | BV421 | HIL-7R-M21 | BD Biosciences | 562437 |
| CD197 | PE | 150503 | BD Biosciences | 560765 |
References
- Rotstein, D.; Montalban, X. Reaching an evidence-based prognosis for personalized treatment of multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Gaetani, L.; Centonze, D.; Hegen, H.; Kuhle, J.; Teunissen, C.E.; Tintoré, M.; Villar, L.M.; Willemse, E.A.; Zetterberg, H.; et al. Fluid biomarkers in multiple sclerosis: From current to future applications. Lancet Reg. Health-Eur. 2024, 44, 101009. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Häring, D.A.; Ganjgahi, H.; Ocampo, A.; Hatami, F.; Čuklina, J.; Aarden, P.; Dahlke, F.; Arnold, D.L.; Wiendl, H.; et al. How patients with multiple sclerosis acquire disability. Brain 2022, 145, 3147–3161. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Butzkueven, H.; Dhib-Jalbut, S.; Hobart, J.; Kobelt, G.; Pepper, G.; Sormani, M.P.; Thalheim, C.; Traboulsee, A.; Vollmer, T. Brain health: Time matters in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 9 (Suppl. S1), S5–S48. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Montalban, X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol. 2014, 13, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; Masjuan, J.; GonzálEz-Porqué, P.; Plaza, J.; SádAba, M.C.; Roldán, E.; Bootello, A.; Alvarez-Cermeño, J.C. Intrathecal IgM synthesis predicts the onset of new relapses and a worse disease course in MS. Neurology 2002, 59, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; Masjuan, J.; González-Porqué, P.; Plaza, J.; Sádaba, M.C.; Roldán, E.; Bootello, A.; Alvarez-Cermeño, J.C. Intrathecal IgM synthesis is a prognostic factor in multiple sclerosis. Ann. Neurol. 2002, 53, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; Sádaba, M.C.; Roldán, E.; Masjuan, J.; González-Porqué, P.; Villarrubia, N.; Espiño, M.; García-Trujillo, J.A.; Bootello, A.; Álvarez-Cermeño, J.C. Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J. Clin. Investig. 2005, 115, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Monreal, E.; de la Maza, S.S.; Costa-Frossard, L.; Walo-Delgado, P.; Zamora, J.; Fernández-Velasco, J.I.; Villarrubia, N.; Espiño, M.; Lourido, D.; Lapuente, P.; et al. Predicting Aggressive Multiple Sclerosis With Intrathecal IgM Synthesis Among Patients With a Clinically Isolated Syndrome. Neurol. Neuroimmunol. Neuroinflammation 2021, 8, e1047. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Meier, S.; Schaedelin, S.; Manouchehrinia, A.; Yaldizli, Ö.; Maceski, A.; Oechtering, J.; Achtnichts, L.; Conen, D.; Derfuss, T.; et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: A retrospective modelling and validation study. Lancet Neurol. 2022, 21, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Barro, C.; Healy, B.C.; Liu, Y.; Saxena, S.; Paul, A.; Polgar-Turcsanyi, M.; Guttmann, C.R.; Bakshi, R.; Kropshofer, H.; Weiner, H.L.; et al. Serum GFAP and NfL Levels Differentiate Subsequent Progression and Disease Activity in Patients with Progressive Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2022, 10, e200052. [Google Scholar] [CrossRef] [PubMed]
- Monreal, E.; Fernández-Velasco, J.I.; García-Sánchez, M.I.; de la Maza, S.S.; Llufriu, S.; Álvarez-Lafuente, R.; Casanova, B.; Comabella, M.; Ramió-Torrentà, L.; Martínez-Rodríguez, J.E.; et al. Association of Serum Neurofilament Light Chain Levels at Disease Onset With Disability Worsening in Patients With a First Demyelinating Multiple Sclerosis Event Not Treated With High-Efficacy Drugs. JAMA Neurol. 2023, 80, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Monreal, E.; Fernández-Velasco, J.I.; Álvarez-Lafuente, R.; de la Maza, S.S.; García-Sánchez, M.I.; Llufriu, S.; Casanova, B.; Comabella, M.; Martínez-Yélamos, S.; Galimberti, D.; et al. Serum biomarkers at disease onset for personalized therapy in multiple sclerosis. Brain 2024, 147, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.G.; Sampson, F.C.; Beard, S.M.; Tappenden, P. A review of the natural history and epidemiology of multiple sclerosis: Implications for resource allocation and health economic models. Health Technol. Assess. 2002, 6, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Laroni, A.; Weiner, H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2010, 221, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Tur, C.; Carbonell-Mirabent, P.; Cobo-Calvo, Á.; Otero-Romero, S.; Arrambide, G.; Midaglia, L.; Castilló, J.; Vidal-Jordana, Á.; Rodríguez-Acevedo, B.; Zabalza, A.; et al. Association of Early Progression Independent of Relapse Activity With Long-term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023, 80, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, A.; Stellmann, J.P.; Rico, A.; Boutiere, C.; Demortiere, S.; Durozard, P.; Zaaraoui, W.; Ranjeva, J.-P.; Pelletier, J.; Audoin, B. Active and non-active progression independent of relapse activity within the first 20 years of relapsing multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2024, 95, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Pfuhl, C.; Grittner, U.; Gieß, R.M.; Scheel, M.; Behrens, J.R.; Rasche, L.; Pache, F.C.; Wenzel, R.; Brandt, A.U.; Bellmann-Strobl, J.; et al. Intrathecal IgM production is a strong risk factor for early conversion to multiple sclerosis. Neurology 2019, 93, e1439–e1451. [Google Scholar] [CrossRef] [PubMed]
- Abdelhak, A.; Benkert, P.; Schaedelin, S.; Boscardin, W.J.; Cordano, C.; Oechtering, J.; Ananth, K.; Granziera, C.; Melie-Garcia, L.; Montes, S.C.; et al. Neurofilament Light Chain Elevation and Disability Progression in Multiple Sclerosis. JAMA Neurol. 2023, 80, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Scalfari, A.; Traboulsee, A.; Oh, J.; Airas, L.; Bittner, S.; Calabrese, M.; Dominguez, J.M.G.; Granziera, C.; Greenberg, B.; Hellwig, K.; et al. Smouldering-Associated Worsening in Multiple Sclerosis: An International Consensus Statement on Definition, Biology, Clinical Implications, and Future Directions. Ann. Neurol. 2024, 96, 826–845. [Google Scholar] [CrossRef] [PubMed]
- Oechtering, J.; Stein, K.; Schaedelin, S.A.; Maceski, A.M.; Orleth, A.; Meier, S.; Willemse, E.; Qureshi, F.; Heijnen, I.; Regeniter, A.; et al. Complement Activation Is Associated with Disease Severity in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2024, 11, e200212. [Google Scholar] [CrossRef] [PubMed]
- Oechtering, J.; Schaedelin, S.A.; Stein, K.; Maceski, A.M.; Melie-Garcia, L.; Benkert, P.; Cagol, A.; Leber, S.; Galbusera, R.; Ruberte, E.; et al. Aberrant Complement Activation Is Associated with Structural Brain Damage in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2025, 12, e200361. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.; García-Barragán, N.; Espiño, M.; Roldán, E.; Sádaba, M.; Gómez-Rial, J.; González-Porqué, P.; Álvarez-Cermeño, J. Influence of oligoclonal IgM specificity in multiple sclerosis disease course. Mult. Scler. J. 2007, 14, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Sádaba, M.C.; Tzartos, J.; Paíno, C.; García-Villanueva, M.; Álvarez-Cermeño, J.C.; Villar, L.M.; Esiri, M.M. Axonal and oligodendrocyte-localized IgM and IgG deposits in MS lesions. J. Neuroimmunol. 2012, 247, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Vasilenko, N.; Tieck, M.P.; Michel, T.; Schembecker, S.; Schwarz, P.; Guenther, A.; Ruschil, C.; Poli, S.; Ziemann, U.; Giede-Jeppe, A.; et al. In-depth analysis of serum antibodies against Epstein-Barr virus lifecycle proteins, and EBNA1, ANO2, GlialCAM and CRYAB peptides in patients with multiple sclerosis. Front. Immunol. 2024, 15, 1487523. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Zhang, S.-X.; Ma, X.-W.; Xue, Y.-L.; Gao, C.; Li, X.-Y.; Xu, A.-D. The proportion of peripheral regulatory T cells in patients with Multiple Sclerosis: A meta-analysis. Mult. Scler. Relat. Disord. 2019, 28, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Libera, D.D.; Di Mitri, D.; Bergami, A.; Centonze, D.; Gasperini, C.; Grasso, M.G.; Galgani, S.; Martinelli, V.; Comi, G.; Avolio, C.; et al. T Regulatory Cells Are Markers of Disease Activity in Multiple Sclerosis Patients. PLoS ONE 2011, 6, e21386. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Maltos, M.; Treviño-Frenk, I.; García-González, H.; Rosetti, M.; Barriga-Maldonado, V.; Morales-Ramírez, F.; López-Hernández, D.; Rosetti, F.; Crispín, J. Identification of regulatory T cell molecules associated with severity of multiple sclerosis. Mult. Scler. J. 2020, 27, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Dybowski, S.; Torke, S.; Weber, M.S. Targeting B Cells and Microglia in Multiple Sclerosis with Bruton Tyrosine Kinase Inhibitors. JAMA Neurol. 2023, 80, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Cagol, A.; Lorscheider, J.; Tsagkas, C.; Benkert, P.; Yaldizli, Ö.; Kuhle, J.; Derfuss, T.; Sormani, M.P.; Thompson, A.; et al. Harmonizing Definitions for Progression Independent of Relapse Activity in Multiple Sclerosis. JAMA Neurol. 2023, 80, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Rodero-Romero, A.; Monreal, E.; Sainz-Amo, R.; Domínguez, J.M.G.; Villarrubia, N.; Veiga-González, J.L.; Fernández-Velasco, J.I.; Goicochea-Briceño, H.; Rodríguez-Jorge, F.; de la Maza, S.S.; et al. Establishing Normal Serum Values of Neurofilament Light Chains and Glial Fibrillary Acidic Protein Considering the Effects of Age and Other Demographic Factors in Healthy Adults. Int. J. Mol. Sci. 2024, 25, 7808. [Google Scholar] [CrossRef] [PubMed]


| Total (n = 535) | |
|---|---|
| Sex (female) | 372 (69.5) |
| Age at first symptom, y | 33.8 (27.2–42.2) |
| Age at serum analysis, y | 34.0 (27.5–42.5) |
| Time to analysis after first relapse, mo | 3.12 (0.59–6.23) |
| Topography of first relapse | |
| Optic nerve | 96 (17.9) |
| Brainstem | 124 (23.2) |
| Spinal cord | 219 (40.9) |
| Cerebral hemisphere | 59 (11.0) |
| Multifocal | 26 (4.9) |
| Paroxysmal symptoms | 11 (2.1) |
| EDSS at baseline | 1.5 (1–2) |
| T2 lesions at baseline | |
| 0–3 | 84 (15.7) |
| 4–9 | 142 (26.5) |
| 10–50 | 258 (48.2) |
| >50 | 51 (9.5) |
| Gadolinium-enhancing lesions | |
| Median (range) | 1 (0–45) |
| No. of patients with enhancing lesions (%) | 275/486 (56.6) |
| CSF data | |
| IgG oligoclonal bands | 489 (91.4) |
| IgM oligoclonal bands | 259 (48.4) |
| Lipid-specific IgM oligoclonal bands | 170 (31.8) |
| Serum biomarkers levels | |
| sNfL levels (pg/mL) | 11.1 (6.84–19.7) |
| sNfL z-score | 1.68 (0.44–2.58) |
| sGFAP levels (pg/mL) | 126.5 (90.9–181.5) |
| DMT use during follow-up 1 | |
| None | 89 (16.6) |
| Injectable/oral DMTs 2 | 380 (71.0) |
| Monoclonal antibodies 3 | 166 (31.0) |
| Time of follow-up, y | 7.05 (4.93–10.5) |
| Patients attaining 6-month CDW during follow-up 4 | |
| RAW | 86 (16.1) |
| ‘Active’ PIRA | 61 (11.4) |
| ‘Non-active’ PIRA | 72 (13.5) |
| Variables | RAW | Active PIRA | Non-Active PIRA | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | p | HR (95% CI) | p | HR (95% CI) | p | |
| c Statistics 0.82 | c Statistics 0.74 | c Statistics 0.82 | ||||
| Age at first relapse | 0.98 (0.96–1.00) | 0.05 | 1.03 (0.99–1.05) | 0.05 | 1.10 (1.07–1.13) | <0.001 |
| Sex (male) | 1.45 (0.91–2.32) | 0.12 | 1.26 (0.70–2.27) | 0.44 | 2.87 (1.69–4.88) | <0.001 |
| Baseline EDSS | 1.09 (0.82–1.44) | 0.55 | 0.71 (0.51–1.01) | 0.05 | 1.71 (1.30–2.25) | <0.001 |
| T2 lesion load | ||||||
| 0–3 (reference) | 1 | - | 1 | - | 1 | - |
| 4–9 | 1.93 (0.87–4.26) | 0.11 | 3.14 (0.90–10.9) | 0.07 | 1.03 (0.49–2.15) | 0.95 |
| 10–50 | 2.30 (1.08–4.88) | 0.03 | 3.48 (1.04–11.7) | 0.04 | 0.63 (0.31–1.28) | 0.20 |
| >50 | 3.98 (1.47–10.7) | 0.006 | 4.98 (1.23–20.2) | 0.02 | 1.23 (0.52–2.91) | 0.64 |
| sNfL z-score >1.5 | 2.12 (1.27–3.54) | 0.004 | 2.12 (1.17–3.86) | 0.01 | 1.44 (0.84–2.46) | 0.18 |
| High sGFAP levels 1 | 1.52 (0.97–2.40) | 0.07 | 1.51 (0.87–2.62) | 0.15 | 3.19 (1.84–5.34) | <0.001 |
| Lipid-specific IgM OCB | 2.15 (1.34–3.45) | 0.002 | 1.70 (0.96–3.02) | 0.07 | 0.82 (0.46–1.47) | 0.51 |
| Proportion of time of injectable/oral DMTs use 2 | 0.06 (0.03–0.11) | <0.001 | 0.38 (0.17–0.87) | 0.02 | 0.86 (0.38–1.96) | 0.72 |
| Proportion of time of mAb use 3 | 0.02 (0.01–0.07) | <0.001 | 0.09 (0.02–0.39) | 0.001 | 0.85 (0.26–2.73) | 0.78 |
| sNfL Low LS-OCMB (−) (n = 45) | sNfL Low LS-OCMB (+) (n = 16) | sNfL High LS-OCMB (−) (n = 36) | sNfL High LS-OCMB (+) (n = 25) | sGFAP Low (n = 75) | sGFAP High (n = 47) | |
|---|---|---|---|---|---|---|
| Lymphocyte subset | ||||||
| CD3+ T cells | 89.8 (86.8–91.8) | 90.8 (87.3–92.6) | 89.5 (87.6–90.7) | 87.2 (82.4–91.1) | 89.5 (86.6–91.2) | 89.1 (86.8–91.5) |
| CD4+ | 68.8 (61.8–74.4) | 67.6 (65.8–71.6) | 67.8 (63.0–73.6) | 62.3 (56.4–67.1) | 67.8 (61.5–71.9) | 66.8 (60.2–72.1) |
| Naive | 6.51 (5.20–8.48) | 7.22 (5.75–11.4) | 6.77 (4.23–8.80) | 1.11 (0.72–2.44) 1 | 5.77 (4.23–8.18) | 6.77 (5.16–8.84) |
| Central memory | 16.5 (13.6–26.4) | 30.3 (19.0–31.9) | 17.6 (11.9–27.6) | 11.2 (9.68–15.7) | 16.5 (9.90–26.6) | 19.9 (14.7–29.4) |
| Effector memory | 27.8 (24.6–37.7) | 26.6 (17.9–41.3) | 27.4 (22.1–34.0) | 40.2 (37.2–46.4) | 27.1 (24.7–37.7) | 27.4 (22.1–40.8) |
| Regulatory | 4.35 (3.20–5.80) | 5.69 (4.60–7.59) 1 | 3.98 (2.07–5.68) | 2.7 (2.25–4.38) | 4.33 (2.60–5.49) | 3.78 (2.25–6.12) |
| Terminally differentiated | 10.2 (5.63–17.9) | 7.27 (3.01–9.21) | 7.60 (6.14–15.3) | 5.40 (2.75–6.34) | 9.21 (4.50–15.3) | 6.27 (3.20–7.60) |
| CD8+ | 18.5 (15.5–21.6) | 19.3 (16.0–22.5) | 18.7 (15.8–23.5) | 20.3 (16.0–24.6) | 19.3 (16.1–23.0) | 18.7 (15.4–23.5) |
| Naive | 1.15 (0.82–2.09) | 2.02 (0.66–2.72) | 1.31 (1.18–2.68) | 1.18 (0.38–1.94) | 1.74 (0.80–2.42) | 1.30 (0.66–2.72) |
| Central memory | 0.62 (0.41–1.10) | 1.36 (0.86–4.17) | 1.03 (0.62–1.36) | 0.74 (0.53–1.50) | 0.84 (0.50–1.19) | 1.31 (0.62–4.17) |
| Effector memory | 6.57 (4.21–8.13) 2 | 7.81 (6.93–9.82) | 6.38 (3.14–9.27) 2 | 14.1 (10.2–16.0) | 6.61 (4.14–8.07) | 9.27 (6.38–10.7) |
| Terminally differentiated | 7.84 (6.03–11.7) | 5.81 (5.43–9.23) | 10.0 (3.65–12.3) | 7.99 (7.67–11.1) | 9.04 (6.84–13.0) | 5.69 (3.65–9.68) 4 |
| CD19+ B cells | 2.60 (1.95–4.30) | 3.63 (2.56–4.43) | 2.37 (1.45–4.00) | 4.25 (2.01–5.70) | 2.92 (1.91–4.43) | 2.40 (1.80–4.67) |
| CD5+ | 0.48 (0.20–0.80) | 0.60 (0.20–0.77) | 0.40 (0.20–0.75) | 0.85 (0.30–1.00) | 0.52 (0.20–0.90) | 0.44 (0.21–0.90) |
| CD5- | 2.10 (1.70–2.63) | 2.66 (1.80–3.28) | 1.70 (1.20–3.10) | 3.05 (1.67–4.70) | 2.17 (1.60–3.30) | 2.00 (1.30–3.90) |
| Memory | 1.80 (1.25–2.69) | 2.84 (2.38–4.41) | 1.65 (1.07–2.44) | 2.19 (1.50–4.96) | 2.00 (1.41–2.79) | 1.50 (1.04–2.80) |
| Plasmablasts | 0.43 (0.32–0.56) | 0.55 (0.26–0.70) | 0.42 (0.24–1.03) | 0.28 (0.10–0.48) | 0.48 (0.28–0.80) | 0.26 (0.19–0.59) |
| Monocytes | 2.71 (1.46–4.29) | 1.61 (1.30–2.70) | 3.20 (1.07–4.67) | 1.35 (0.80–2.78) | 2.40 (1.39–3.86) | 2.10 (0.93–4.13) |
| Soluble factors | ||||||
| NfL levels (pg/mL) | 577 (392–883.6) | 795 (472–1166) | 2680 (1311–4691) 1 | 3758 (1838–7575) 1 | 972 (576–2652) | 2274 (784–5083) 4 |
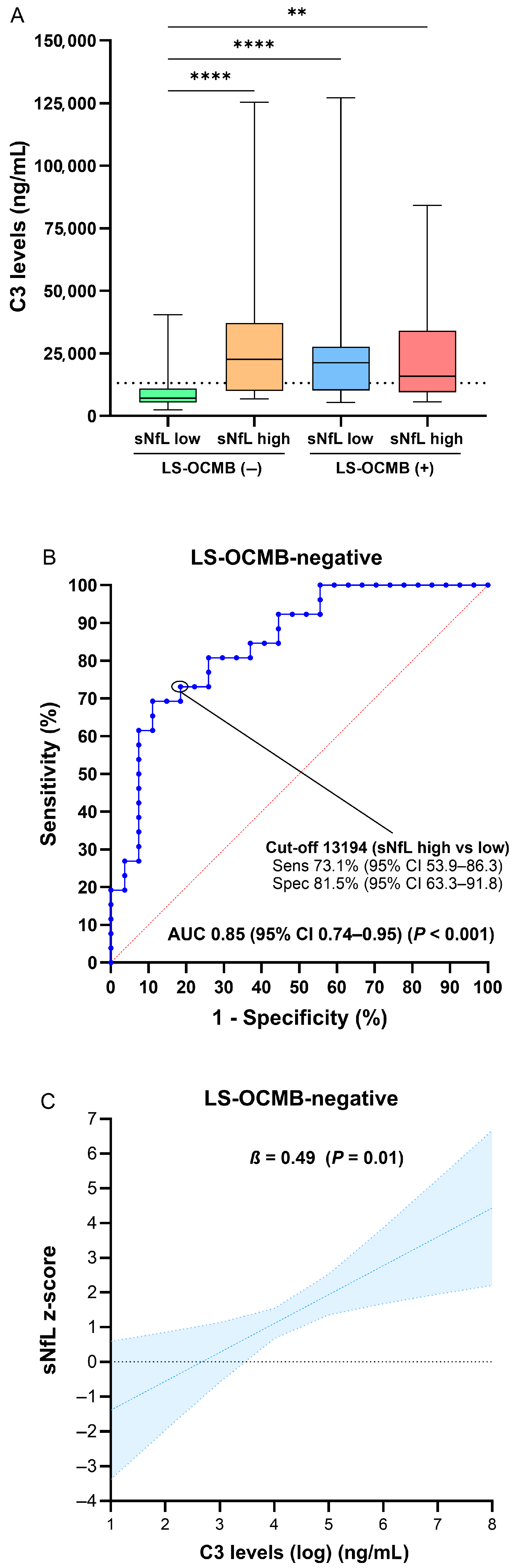
| C3 levels (ng/mL) | 7131 (5398–10,933) 1 | 12,016 (9244–25,129) | 14,121 (8610–21,003) | 12,178 (8387–20,800) | 15,101 (9212–25,838) | 21,065 (11,385–45,722) |
| C4 levels (ng/mL) | 899 (730–1107) 2 | 1189 (891.8–1699) | 1096 (826.8–1957) | 1257 (912–1629) | 2133 (1082–3545) | 2452 (1980–4594) 4 |
| IgG index | 0.81 (0.60–1.34) | 0.95 (0.71–1.38) | 0.84 (0.63–1.17) | 0.84 (0.68–1.32) | 0.84 (0.64–1.32) | 0.85 (0.64–1.23) |
| IgM index | 0.14 (0.08–0.23) 2,3 | 0.24 (0.15–0.47) | 0.13 (0.08–0.22) 2,3 | 0.24 (0.15–0.42) | 0.15 (0.09–0.28) | 0.16 (0.09–0.28) |
| Estimate (95% CI) | p Value | |
|---|---|---|
| Total (n = 108) | ||
| Intercept | 1.38 (−0.01–2.77) | 0.051 |
| Sex (male) | −0.12 (−0.70–0.47) | 0.69 |
| Age at sampling, y | 0.00 (−0.03–0.03) | 0.98 |
| Time to sampling, mo | −0.01 (−0.09–0.06) | 0.75 |
| CSF T-reg cells, % | −0.05 (−0.21–0.11) * | 0.53 |
| LS-OCMB (+) | 1.95 (0.67–3.24) * | 0.003 |
| LS-OCMB-positive (n = 39) | ||
| Intercept | 3.38 (1.43–5.33) | 0.001 |
| Sex (male) | −0.43 (−1.31–0.44) | 0.32 |
| Age at sampling, y | −0.01 (−0.06–0.05) | 0.80 |
| Time to sampling, mo | 0.04 (−0.06–0.14) | 0.43 |
| CSF T-reg cells, % | −0.36 (−0.55–[−0.16]) | 0.001 |
| LS-OCMB-negative (n = 69) | ||
| Intercept | 1.41 (−0.24–3.06) | 0.09 |
| Sex (male) | 0.09 (−0.70–0.88) | 0.82 |
| Age at sampling, y | 0.01 (−0.03–0.04) | 0.80 |
| Time to sampling, mo | −0.06 (−0.16–0.05) | 0.29 |
| CSF T-reg cells, % | −0.06 (−0.23–0.11) | 0.51 |
| Estimate (95% CI) | p Value | |
|---|---|---|
| Total (n = 109) | ||
| Intercept | −4.85 (−10.2–[0.49]) | 0.08 |
| Sex (male) | 0.08 (−0.54–0.71) | 0.79 |
| Age at sampling, y | −0.01 (−0.04–0.02) | 0.57 |
| Time to sampling, mo | −0.05 (−0.14–0.04) | 0.27 |
| C3 levels (ng/mL) (per doubling) | 0.47 (0.11–0.84) * | 0.01 |
| LS-OCMB (+) | 8.39 (1.10–15.7) * | 0.03 |
| LS-OCMB-positive (n = 58) | ||
| Intercept | 3.38 (−1.83–8.60) | 0.20 |
| Sex (male) | 0.06 (−0.78–0.90) | 0.89 |
| Age at sampling, y | 0.02 (−0.03–0.06) | 0.52 |
| Time to sampling, mo | −0.05 (−0.17–0.06) | 0.35 |
| C3 levels (ng/mL) (per doubling) | −0.14 (−0.52–0.24) | 0.45 |
| LS-OCMB-negative (n = 51) | ||
| Intercept | −4.29 (−9.91–1.33) | 0.13 |
| Sex (male) | 0.13 (−0.85–1.11) | 0.79 |
| Age at sampling, y | −0.04 (−0.08–0.01) | 0.13 |
| Time to sampling, mo | −0.03 (−0.18–0.13) | 0.76 |
| C3 levels (ng/mL) (per doubling) | 0.49 (0.11–0.87) | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monreal, E.; Fernández-Velasco, J.I.; Sainz de la Maza, S.; Espiño, M.; Villarrubia, N.; Roldán-Santiago, E.; Aladro, Y.; Cuello, J.P.; Ayuso-Peralta, L.; Rodero-Romero, A.; et al. Combining CSF and Serum Biomarkers to Differentiate Mechanisms of Disability Worsening in Multiple Sclerosis. Int. J. Mol. Sci. 2025, 26, 6898. https://doi.org/10.3390/ijms26146898
Monreal E, Fernández-Velasco JI, Sainz de la Maza S, Espiño M, Villarrubia N, Roldán-Santiago E, Aladro Y, Cuello JP, Ayuso-Peralta L, Rodero-Romero A, et al. Combining CSF and Serum Biomarkers to Differentiate Mechanisms of Disability Worsening in Multiple Sclerosis. International Journal of Molecular Sciences. 2025; 26(14):6898. https://doi.org/10.3390/ijms26146898
Chicago/Turabian StyleMonreal, Enric, José Ignacio Fernández-Velasco, Susana Sainz de la Maza, Mercedes Espiño, Noelia Villarrubia, Ernesto Roldán-Santiago, Yolanda Aladro, Juan Pablo Cuello, Lucía Ayuso-Peralta, Alexander Rodero-Romero, and et al. 2025. "Combining CSF and Serum Biomarkers to Differentiate Mechanisms of Disability Worsening in Multiple Sclerosis" International Journal of Molecular Sciences 26, no. 14: 6898. https://doi.org/10.3390/ijms26146898
APA StyleMonreal, E., Fernández-Velasco, J. I., Sainz de la Maza, S., Espiño, M., Villarrubia, N., Roldán-Santiago, E., Aladro, Y., Cuello, J. P., Ayuso-Peralta, L., Rodero-Romero, A., Chico-García, J. L., Rodríguez-Jorge, F., Quiroga-Varela, A., Rodríguez-Martín, E., Pilo de la Fuente, B., Martín-Ávila, G., Martínez-Ginés, M. L., García-Domínguez, J. M., Rubio, L., ... Villar, L. M. (2025). Combining CSF and Serum Biomarkers to Differentiate Mechanisms of Disability Worsening in Multiple Sclerosis. International Journal of Molecular Sciences, 26(14), 6898. https://doi.org/10.3390/ijms26146898