
The Pathogenic Role of C-Reactive Protein in Diabetes-Linked Unstable Atherosclerosis

Abstract
1. Introduction
1.1. Atherosclerosis and Development of Unstable Plaques
1.2. Blood Sugar and Blocked Arteries: The Diabetes–Atherosclerosis Link
1.3. The Anatomy of an Inflammatory Marker (CRP) Linked to Cardiovascular Disease
2. CRP and hs-CRP: Molecular Sentinels in Cardiometabolic Disease
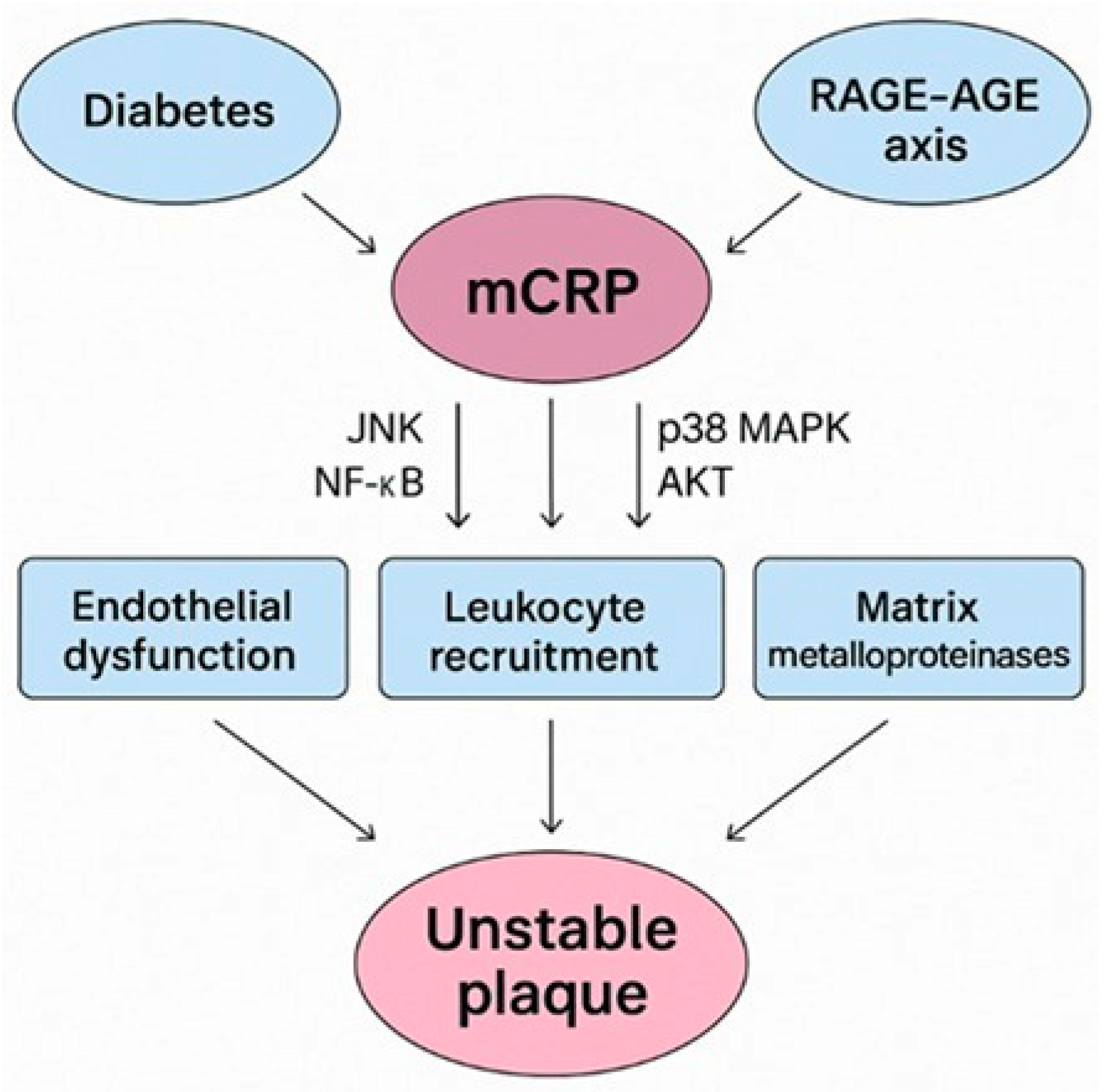
3. mCRP Prevalence in Diabetes-Driven Unstable Plaques
The RAGE-AGE-mCRP Axis in Diabetes
4. RAGE Signaling Pathway Aggravates Diabetes-Mediated Vascular Calcification
5. Conclusions
6. Future Directives: Targeting the mCRP–RAGE Axis: A Therapeutic Opportunity in Diabetes-Associated Plaque Instability
7. Limitations
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| ACS | Acute coronary syndrome |
| AGE–RAGE | Advanced glycation end products–receptor for advanced glycation end products |
| AGEs | Advanced glycation end products |
| AIx | Augmentation index |
| AKT | Activating protein kinase B |
| AMI | Acute myocardial infarction |
| BMI | Body mass index |
| BMP2 | Bone morphogenetic protein 2 |
| C1q | Complement 1q |
| C4BP | C4b-binding protein |
| CAD | Coronary artery disease |
| CD36 | Cluster of differentiation 36 |
| CHD | coronary heart disease |
| CKD | Chronic kidney disease |
| COX-2 | Cyclooxygenase-2 |
| CRP | C-reactive protein |
| CVD | Cardiovascular disease |
| DFU | Diabetes-related foot ulceration |
| DM | Diabetes mellitus |
| ECM | Extracellular matrix |
| eGFR | Estimated glomerular filtration rate |
| ELISA | Enzyme-linked immunosorbent assay |
| EndMT | Endothelial-to-mesenchymal transition |
| FAK | Gocal adhesion kinase |
| GPIIb-IIIa | IIb-IIIa glycoprotein |
| HbA1c | Hemoglobin A1c |
| HOMA-IR | Homeostasis model assessment of insulin resistance |
| hs-CRP | High-sensitivity C-reactive protein |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IL-1/IL-6/IL-8 | Interleukin 1/6/8 |
| IMT | Carotid intima-media thickness |
| JNK | The c-Jun N-terminal kinase |
| LDL-c | Low-density lipoprotein cholesterol |
| LTh | Lymphocyte T helper |
| LVEF | Left ventricular ejection fraction |
| MACE | Major adverse cardiovascular events |
| MCP-1 | Monocyte chemoattractant protein-1 |
| mCRP | Monomeric C-reactive protein |
| MMP | Matrix metalloproteinase |
| NETs | Neutrophil extracellular traps |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NOS | Nitric oxidative stress |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PAD | Peripheral artery disease |
| pCRP | Pentameric C-reactive protein |
| PECAM-1 | Platelet adhesion molecule-1 |
| PWV | Pulse wave velocity |
| rs1205 | Genetic polymorphism of the C-reactive protein (CRP) gene |
| SNPs | Single nucleotide polymorphisms |
| T1D | Type 1 diabetes |
| T2DM | Type 2 diabetes mellitus |
| TAVI | Transcatheter aortic valve implantation |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| VSMCs | Vascular smooth muscle cells |
| vWF | von Willebrand Factor |
References
- Attiq, A.; Afzal, S.; Ahmad, W.; Kandeel, M. Hegemony of inflammation in atherosclerosis and coronary artery disease. Eur. J. Pharmacol. 2024, 966, 176338. [Google Scholar] [CrossRef] [PubMed]
- Eslava-Alcon, S.; Extremera-García, M.J.; González-Rovira, A.; Rosal-Vela, A.; Rojas-Torres, M.; Beltran-Camacho, L.; Sanchez-Gomar, I.; Jiménez-Palomares, M.; Alonso-Piñero, J.A.; Conejero, R.; et al. Molecular signatures of atherosclerotic plaques: An up-dated panel of protein related markers. J. Proteom. 2020, 221, 103757. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Waksman, R.; Merdler, I.; Case, B.C.; Waksman, O.; Porto, I. Targeting inflammation in atherosclerosis: Overview, strategy and directions. EuroIntervention 2024, 20, 32–44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeller, J.; Loseff-Silver, J.; Khoshmanesh, K.; Baratchi, S.; Lai, A.; Nero, T.L.; Roy, A.; Watson, A.; Dayawansa, N.; Sharma, P.; et al. Shear-Sensing by C-Reactive Protein: Linking Aortic Stenosis and Inflam-mation. Am. Heart Assoc. J. Circ. Res. 2024, 135, 1033–1047. [Google Scholar] [CrossRef]
- Drysdale, A.; Unsworth, A.J.; White, S.J.; Jones, S. The Contribution of Vascular Proteoglycans to Atherothrombosis: Clinical Implications. Int. J. Mol. Sci. 2023, 24, 11854. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Montanaro, M.; Cardellini, M.; Bonfiglio, R.; Anemona, L.; Urbano, N.; Bonanno, E.; Menghini, R.; Casagrande, V.; Martelli, E.; et al. High Sensitivity C-Reactive Protein Increases the Risk of Carotid Plaque Instability in Male Dyslipidemic Patients. Diagnostics 2021, 11, 2117. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: Molecular mechanisms and ther-apeutic strategies. Signal Transduct. Target Ther. 2023, 8, 152. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Allen, M.F.; Park, S.Y. Applying adipose tissue-derived stem cell therapies as a novel treatment for atherosclerotic plaque development: Importance of appropriate dosing. J. Mol. Cell. Cardiol. Plus 2024, 10, 100086. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Vlachakis, P.K.; Antoniadis, A.P.; Fragakis, N. Diabetes-Driven Atherosclerosis: Updated Mechanistic Insights and Novel Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 2196. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.S.R.; Dwivedi, G.; Fegan, P.G.; Game, F.; Hamilton, E.J. Unravelling the cardio-renal-metabolic-foot connection in people with diabe-tes-related foot ulceration: A narrative review. Cardiovasc. Diabetol. 2024, 23, 437. [Google Scholar] [CrossRef] [PubMed]
- Sibony, R.W.; Segev, O.; Dor, S.; Raz, I. Overview of oxidative stress and inflammation in diabetes. J. Diabetes 2024, 16, e70014. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, J.; Radovanovic, J.; Banjac, K.; Obradovic, M.; Essack, M.; Zafirovic, S.; Gluvic, Z.; Gojobori, T.; Isenovic, E.R. Role of C-Reactive Protein in Diabetic Inflammation. Mediat. Inflamm. 2022, 2022, 3706508. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Margolis, D.J.; Hofstad, O.; Feldman, H.I. Association between renal failure and foot ulcer or lower-extremity amputation in patients with diabetes. Diabetes Care 2008, 31, 1331–1336. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dasari, N.; Jiang, A.; Skochdopole, A.; Chung, J.; Reece, E.M.; Vorstenbosch, J.; Winocour, S. Updates in Diabetic Wound Healing, Inflammation, and Scarring. Semin. Plast. Surg. 2021, 35, 153–158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pastorello, Y.; Carare, R.O.; Banescu, C.; Potempa, L.; Di Napoli, M.; Slevin, M. Monomeric C-reactive protein: A novel biomarker predicting neurodegenerative disease and vascular dysfunction. Brain Pathol. 2023, 33, e13164. [Google Scholar] [CrossRef] [PubMed]
- Boncler, M.; Wu, Y.; Watala, C. The Multiple Faces of C-Reactive Protein—Physiological and Pathophysiological Implications in Cardiovascular Disease. Molecules 2019, 24, 2062. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, M.; Slevin, M.; Popa-Wagner, A.; Singh, P.; Lattanzi, S.; Divani, A.A. Monomeric C-Reactive Protein and Cerebral Hemorrhage: From Bench to Bedside. Front. Immunol. 2018, 9, 1921. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Melnikov, I.; Kozlov, S.; Pogorelova, O.; Tripoten, M.; Khamchieva, L.; Saburova, O.; Avtaeva, Y.; Zvereva, M.; Matroze, E.; Kuz-netsova, T.; et al. The monomeric C-reactive protein level is associated with the in-crease in carotid plaque number in patients with subclinical carotid atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 968267. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agrawal, A.; Wu, Y. Editorial: Biology of C-reactive protein. Front. Immunol. 2024, 15, 1445001. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Tang, B.; Liu, X.; Wu, X.; Wang, H.; Xu, D.; Guo, Y. Increased monomeric CRP levels in acute myocardial infarction: A possible new and specific biomarker for diagnosis and severity assessment of disease. Atherosclerosis 2015, 239, 343–349. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Kiefer, J.; Braig, D.; Loseff-Silver, J.; Potempa, L.A.; Eisenhardt, S.U.; Peter, K. Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front. Immunol. 2018, 9, 1351. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeinolabediny, Y.; Kumar, S.; Slevin, M. Monomeric C-Reactive Protein—A Feature of Inflammatory Disease Associated With Cardiovascular Pathophysiological Complications? In Vivo 2021, 35, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Siennicka, A. Association between microvesicles bearing monomeric C-reactive protein and platelet reactivity. Relation-ship with low response to antiplatelet drugs? J. Physiol. Pharmacol. 2021, 72. [Google Scholar] [CrossRef] [PubMed]
- Molins, B.; Peña, E.; de la Torre, R.; Badimon, L. Monomeric C-reactive protein is prothrombotic and dissociates from circu-lating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc. Res. 2011, 92, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Tang, Y.L.; Xu, T.H.; Cheng, B. C-reactive protein: Structure, function, regulation, and role in clinical diseases. Front. Immunol. 2024, 15, 1425168. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Crawford, J.R.; Trial, J.; Nambi, V.; Hoogeveen, R.C.; Taffet, G.E.; Entman, M.L. Plasma Levels of Endothelial Microparticles Bearing Monomeric C-reactive Protein are Increased in Peripheral Artery Disease. J. Cardiovasc. Transl. Res. 2016, 9, 184–193. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krupinski, J.; Turu, M.M.; Martinez-Gonzalez, J.; Carvajal, A.; Juan-Babot, J.O.; Iborra, E.; Slevin, M.; Rubio, F.; Badimon, L. Endoge-nous expression of C-reactive protein is increased in active (ulcerated noncomplicated) human carotid artery plaques. Stroke 2006, 37, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Peña, E.; Arderiu, G.; Padró, T.; Slevin, M.; Vilahur, G.; Chiva-Blanch, G. C-Reactive Protein in Atherothrombosis and Angiogenesis. Front. Immunol. 2018, 9, 430. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Castro, A.R.; Silva, S.O.; Soares, S.C. The Use of High Sensitivity C-Reactive Protein in Cardiovascular Disease Detection. J. Pharm. Pharm. Sci. 2018, 21, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Moorthy, M.V.; Cook, N.R.; Rifai, N.; Lee, I.M.; Buring, J.E. Inflammation, Cholesterol, Lipoprotein(a), and 30-Year Cardiovascular Outcomes in Women. N. Engl. J. Med. 2024, 391, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, L.; Lv, H.; Wang, J.; Zhang, B.; Zhu, Y.; Zhang, X.; Zhu, H.; Zhou, X.; Xia, Y. Predictive value of high sensitivity C-reactive protein in three-vessel disease patients with and without type 2 diabetes. Cardiovasc. Diabetol. 2023, 22, 91. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, L.; Wang, J.; Ding, H.; Meng, S.; Zhang, X.; Lv, H.; Zhong, L.; Wu, J.; Xu, J.; Zhou, X.; et al. Long-term outcomes of medical therapy versus successful recanalisation for coronary chronic total occlusions in patients with and without type 2 diabetes mellitus. Cardiovasc. Diabetol. 2020, 19, 100, Erratum in Cardiovasc. Diabetol. 2021, 20, 31. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Paraskevaidis, I.; Kourek, C.; Tsougos, E. Chronic Coronary Artery Disease: Wall Disease vs. Lumenopathy. Biomolecules 2025, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, R.R.; Everett, B.M.; Perreault, L.; Michos, E.D. Heart Disease and Diabetes. In Diabetes in America [Internet]; Lawrence, J.M., Casagrande, S.S., Herman, W.H., Wexler, D.J., Cefalu, W.T., Eds.; National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): Bethesda, MD, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK597416/ (accessed on 1 April 2025).
- Dehghan, A.; Kardys, I.; de Maat, M.P.; Uitterlinden, A.G.; Sijbrands, E.J.; Bootsma, A.H.; Stijnen, T.; Hofman, A.; Schram, M.T.; Witteman, J.C. Genetic variation, C-reactive protein levels, and incidence of diabetes. Diabetes 2007, 56, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.J.; Norrie, J.; Caslake, M.J.; Gaw, A.; Ford, I.; Lowe, G.D.; O’Reilly, D.S.; Packard, C.J.; Sattar, N.; West of Scotland Coronary Prevention Study. C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes 2002, 51, 1596–1600. [Google Scholar] [CrossRef] [PubMed]
- Thorand, B.; Löwel, H.; Schneider, A.; Kolb, H.; Meisinger, C.; Fröhlich, M.; Koenig, W. C-Reactive Protein as a Predictor for Incident Diabetes Mellitus Among Mid-dle-aged Men: Results From the MONICA Augsburg Cohort Study, 1984–1998. Arch. Intern. Med. 2003, 163, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Kiyohara, Y.; Kubo, M.; Ninomiya, T.; Wakugawa, Y.; Yonemoto, K.; Iwase, M.; Iida, M. Elevated C-Reactive Protein Is a Predictor of the Development of Diabetes in a General Japanese Population: The Hisayama Study. Diabetes Care 2005, 28, 2497–2500. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Deepa, R.; Velmurugan, K.; Premalatha, G. Association of C-reactive protein with body fat, diabetes and coronary artery disease in Asian Indians: The Chennai Urban Rural Epidemiology Study (CURES-6). Diabet. Med. 2005, 22, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Sandeep, S.; Deepa, M.; Gokulakrishnan, K.; Datta, M.; Deepa, R. A diabetes risk score helps identify metabolic syn-drome and cardiovascular risk in Indians-the Chennai Urban Rural Epidemiology Study (CURES-38). Diabetes Obes. Metab. 2007, 9, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Sander, D.; Schulze-Horn, C.; Bickel, H.; Gnahn, H.; Bartels, E.; Conrad, B. Combined effects of hemoglobin A1c and C-reactive protein on the progression of subclinical carotid atherosclerosis: The INVADE study. Stroke 2006, 37, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Samaropoulos, X.F.; Light, L.; Ambrosius, W.T.; Marcovina, S.M.; Probstfield, J.; Goff, D.C., Jr. The effect of intensive risk factor management in type 2 diabetes on inflammatory biomarkers. Diabetes Res. Clin. Pract. 2012, 95, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Kanmani, S.; Kwon, M.; Shin, M.K.; Kim, M.K. Association of C-Reactive Protein with Risk of Developing Type 2 Diabetes Mellitus, and Role of Obesity and Hypertension: A Large Population-Based Korean Cohort Study. Sci. Rep. 2019, 9, 4573. [Google Scholar] [CrossRef] [PubMed]
- Sharif, S.; Van der Graaf, Y.; Cramer, M.J.; Kapelle, L.J.; de Borst, G.J.; Visseren, F.L.J.; Westerink, J.; SMART Study Group. Low-grade inflammation as a risk factor for cardiovascular events and all-cause mortality in patients with type 2 diabetes. Cardiovasc. Diabetol. 2021, 20, 220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Soinio, M.; Marniemi, J.; Laakso, M.; Lehto, S.; Rönnemaa, T. High-sensitivity C-reactive protein and coronary heart disease mortality in patients with type 2 diabetes: A 7-year follow-up study. Diabetes Care 2006, 29, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Pivato, C.A.; Jones, D.; Cao, D.; Sartori, S.; Chiarito, M.; Nicolas, J.; Zhang, Z.; Beerkens, F.; Nardin, M.; Qiu, H.; et al. Prognostic Value of Baseline Inflammation in Diabetic and Nondiabetic Patients Undergoing Percutaneous Coronary Intervention. Can. J. Cardiol. 2022, 38, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Mojiminiyi, O.A.; Abdella, N.; Moussa, M.A.; Akanji, A.O.; Al Mohammedi, H.; Zaki, M. Association of C-reactive protein with coronary heart disease risk factors in patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2002, 58, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bosevski, M.; Bosevska, G.; Stojanovska, L.; Apostolopoulos, V. CRP and fibrinogen imply clinical outcome of patients with Type-2 diabetes and coronary artery disease. Acta Biochim. Biophys. Sin. 2017, 49, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Dangwal, S.; Stratmann, B.; Bang, C.; Lorenzen, J.M.; Kumarswamy, R.; Fiedler, J.; Falk, C.S.; Scholz, C.J.; Thum, T.; Tschoepe, D. Im-pairment of Wound Healing in Patients With Type 2 Diabetes Mellitus Influences Circulating MicroRNA Patterns via Inflammatory Cytokines. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus-Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dungu, A.M.; Ryrsø, C.K.; Hegelund, M.H.; Jensen, A.V.; Kristensen, P.L.; Krogh-Madsen, R.; Ritz, C.; Faurholt-Jepsen, D.; Lindegaard, B. Diabetes Status, c-Reactive Protein, and Insulin Resistance in Community-Acquired Pneumonia-A Prospective Cohort Study. J. Clin. Med. 2023, 13, 245. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Casagrande, S.S.; Lawrence, J.M. Cardiovascular disease risk factors and their associations with inflammation among US adolescents: NHANES, 2015 to March 2020. BMJ Open Diabetes Res Care 2024, 12, e004148. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zha, Z.; Cheng, Y.; Cao, L.; Qian, Y.; Liu, X.; Guo, Y.; Wang, J. Monomeric CRP Aggravates Myocardial Injury After Myocardial Infarction by Polarizing the Macrophage to Pro-Inflammatory Phenotype Through JNK Signaling Pathway. J. Inflamm. Res. 2021, 14, 7053–7064. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, I.; Kozlov, S.; Okhota, S.; Saburova, O.; Avtaeva, Y.; Kuznetsova, T.; Guria, K.; Prokofieva, L.; Riazantseva, T.; Ji, S.R.; et al. Higher monomeric C-reactive protein levels are associated with premature coronary artery disease. Front. Immunol. 2025, 15, 1501125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Molins, B.; Peña, E.; Vilahur, G.; Mendieta, C.; Slevin, M.; Badimon, L. C-reactive protein isoforms differ in their effects on thrombus growth. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, H. Monomeric C-reactive protein affects cell injury and apoptosis through activation of p38 mito-gen-activated protein kinase in human coronary artery endothelial cells. Bosn. J. Basic Med. Sci. 2020, 20, 487–494. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bergen, K.; Mobarrez, F.; Jorneskog, G.; Wallen, H. High levels of endothelial and platelet microvesicles in patients with type 1 diabetes irrespective of microvascular complications. Thromb. Res. 2020, 196, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Pastorello, Y.; Manu, D.; Sawkulycz, X.; Caprio, V.; Banescu, C.; Dobreanu, M.; Potempa, L.; Di Napoli, M.; Slevin, M. mCRP-Induced Focal Adhesion Kinase-Dependent Monocyte Aggregation and M1 Polarization, Which Was Partially Blocked by the C10M Inhibitor. Int. J. Mol. Sci. 2024, 25, 3097. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reichert, S.; Triebert, U.; Santos, A.N.; Hofmann, B.; Schaller, H.G.; Schlitt, A.; Schulz, S. Soluble form of receptor for advanced glycation end products and incidence of new cardiovascular events among patients with cardiovascular disease. Atherosclerosis 2017, 266, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, S.H.; Liu, S.M.; Szmitko, P.E.; He, X.Q.; Fedak, P.W.; Verma, S. C-Reactive protein upregulates receptor for advanced glycation end products expression in human endothelial cells. Hypertension 2006, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, P.; Teppo, A.M.; Koistinen, H.A.; Koivisto, V.A. Concentration of the complement activation product, acyla-tion-stimulating protein, is related to C-reactive protein in patients with type 2 diabetes. Metabolism 2001, 50, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Chikazawa, M.; Shibata, T.; Hatasa, Y.; Hirose, S.; Otaki, N.; Nakashima, F.; Ito, M.; Machida, S.; Maruyama, S.; Uchida, K. Identification of C1q as a Binding Protein for Advanced Glycation End Products. Biochemistry 2016, 55, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, Y.; Shi, L.; Li, L.; Zhang, D.; Gong, Z.; Wu, Q. Activation and modulation of the AGEs-RAGE axis: Implications for inflammatory pathologies and therapeutic interventions—A review. Pharmacol. Res. 2024, 206, 107282. [Google Scholar] [CrossRef] [PubMed]
- Kennon, A.M.; Stewart, J.A., Jr. RAGE Differentially Altered in vitro Responses in Vascular Smooth Muscle Cells and Adventitial Fibroblasts in Diabetes-Induced Vascular Calcification. Front. Physiol. 2021, 12, 676727. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Park, S.; Lee, M.J.; Song, Y.R.; Han, S.H.; Kim, S.G.; Kang, S.W.; Choi, K.H.; Kim, H.J.; Yoo, T.H. Plasma levels of soluble recep-tor for advanced glycation end products (sRAGE) and proinflammatory ligand for RAGE (EN-RAGE) are associated with carotid atherosclerosis in patients with peritoneal dialysis. Atherosclerosis 2012, 220, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Lin, K.P.; Hsu, G.W.; Liu, K.L.; Guo, C.H. Altered Mineral Metabolism and Disequilibrium Between Calcification Promoters and Inhibitors in Chronic Hemodialysis Patients. Biol. Trace Elem. Res. 2020, 193, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Climie, R.E.; Alastruey, J.; Mayer, C.C.; Schwarz, A.; Laucyte-Cibulskiene, A.; Voicehovska, J.; Bianchini, E.; Bruno, R.M.; Charlton, P.H.; Grillo, A.; et al. Vascular ageing: Moving from bench towards bedside. Eur. J. Prev. Cardiol. 2023, 30, 1101–1117, Erratum in Eur. J. Prev. Cardiol. 2023, 30, 1165. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mozos, I.; Jianu, D.; Gug, C.; Stoian, D. Links between High-Sensitivity C-Reactive Protein and Pulse Wave Analysis in Middle-Aged Patients with Hypertension and High Normal Blood Pressure. Dis. Markers 2019, 2019, 2568069. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeller, J.; Cheung Tung Shing, K.S.; Nero, T.L.; McFadyen, J.D.; Krippner, G.; Bogner, B.; Kreuzaler, S.; Kiefer, J.; Horner, V.K.; Braig, D.; et al. A novel phosphocholine-mimetic inhibits a pro-inflammatory conformational change in C-reactive protein. EMBO Mol. Med. 2023, 15, e16236. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]


{kind=link}
{kind=link}
| Study | Year of Publication | Cohort | Developed Diabetes | Association | Conclusion |
|---|---|---|---|---|---|
| The Women’s Health Study [38] | 2001 | 27,628 | 188 | CRP–IL-6–diabetes | Elevated levels of CRP and IL-6 predict the development of T2DM |
| The West of Scotland Coronary Prevention Study [39] | 2002 | 5245 | 127 | CRP–T2DM | CRP is an important independent predictor of diabetes development Low grade inflammation is connected to the diabetes pathogenesis |
| MONICA Augsburg Cohort Study [40] | 2003 | 2052 | 101 | CRP–diabetes | High CRP levels correspond to a 2.7 times higher risk of diabetes development Low grade inflammation is associated with increased T2DM risk |
| Hisayama study [41] | 2005 | 1759 | 131 | CRP–diabetes | Elevated CRP concentrations are a significant diabetes predictor, independent of obesity/insulin resistance |
| CURES-6 study [42] | 2005 | 26,001 | 150 | CRP–diabetes–CAD Body fat–diabetes–CAD | Diabetics with and without CAD had significantly higher CRP levels Hs-CRP levels increased with body fat and HbA1c increase Hs-CRP was strongly associated with CAD and diabetes |
| CURES-38 study [43] | 2007 | 2350 | 146 | Diabetes risk score–glucose intolerance | The diabetes risk score increases with increasing glucose intolerance The diabetes risk score is an effective indicator of metabolic syndrome and cardiovascular risk |
| INVADE study [44] | 2010 | 3534 | 882 | IMT progression–HbA1c and hsCRP | Hyperglycemia and inflammation are associated with an advanced early atherosclerosis progression and an increased risk of new vascular events in diabetic/nondiabetic subjects |
| The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial [45] | 2012 | 10,251 | 562 | Intensive glycemic control–lower hs-CRP | Intensive glycemic control leads to hs-CRP adjustment Adjusting BMI/waist circumference led to lower hs-CRP values |
| KoGES study [46] | 2019 | 22,946 | 278 | CRP, obesity, hypertension–T2DM | CRP is an independent risk determinant, or in combination with obesity and hypertension, of diabetes |
| Second Manifestations of ARTerial disease (SMART) [47] | 2021 | 1679 with diabetes | 650 with CV events | hs-CRP not associated with myocardial infarction, stroke, or vascular disease in T2DM | Low-grade inflammation (measured through hs-CRP) is an independent risk factor for vascular and all-cause mortality, but not for CV events in high risk T2DM subjects |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sibianu, M.; Slevin, M. The Pathogenic Role of C-Reactive Protein in Diabetes-Linked Unstable Atherosclerosis. Int. J. Mol. Sci. 2025, 26, 6855. https://doi.org/10.3390/ijms26146855
Sibianu M, Slevin M. The Pathogenic Role of C-Reactive Protein in Diabetes-Linked Unstable Atherosclerosis. International Journal of Molecular Sciences. 2025; 26(14):6855. https://doi.org/10.3390/ijms26146855
Chicago/Turabian StyleSibianu, Melania, and Mark Slevin. 2025. "The Pathogenic Role of C-Reactive Protein in Diabetes-Linked Unstable Atherosclerosis" International Journal of Molecular Sciences 26, no. 14: 6855. https://doi.org/10.3390/ijms26146855
APA StyleSibianu, M., & Slevin, M. (2025). The Pathogenic Role of C-Reactive Protein in Diabetes-Linked Unstable Atherosclerosis. International Journal of Molecular Sciences, 26(14), 6855. https://doi.org/10.3390/ijms26146855