
Investigation of the Effect of C-Terminal Adjacent Phenylalanine Residues on Asparagine Deamidation by Quantum Chemical Calculations

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
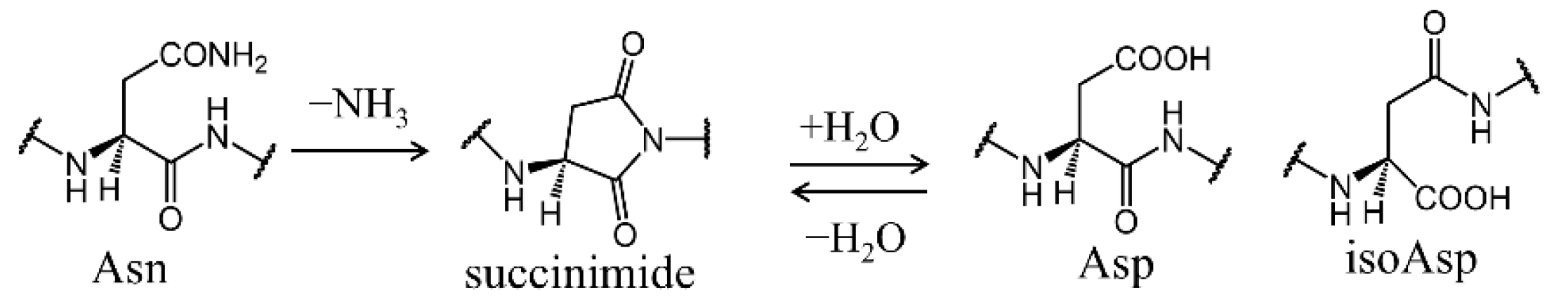
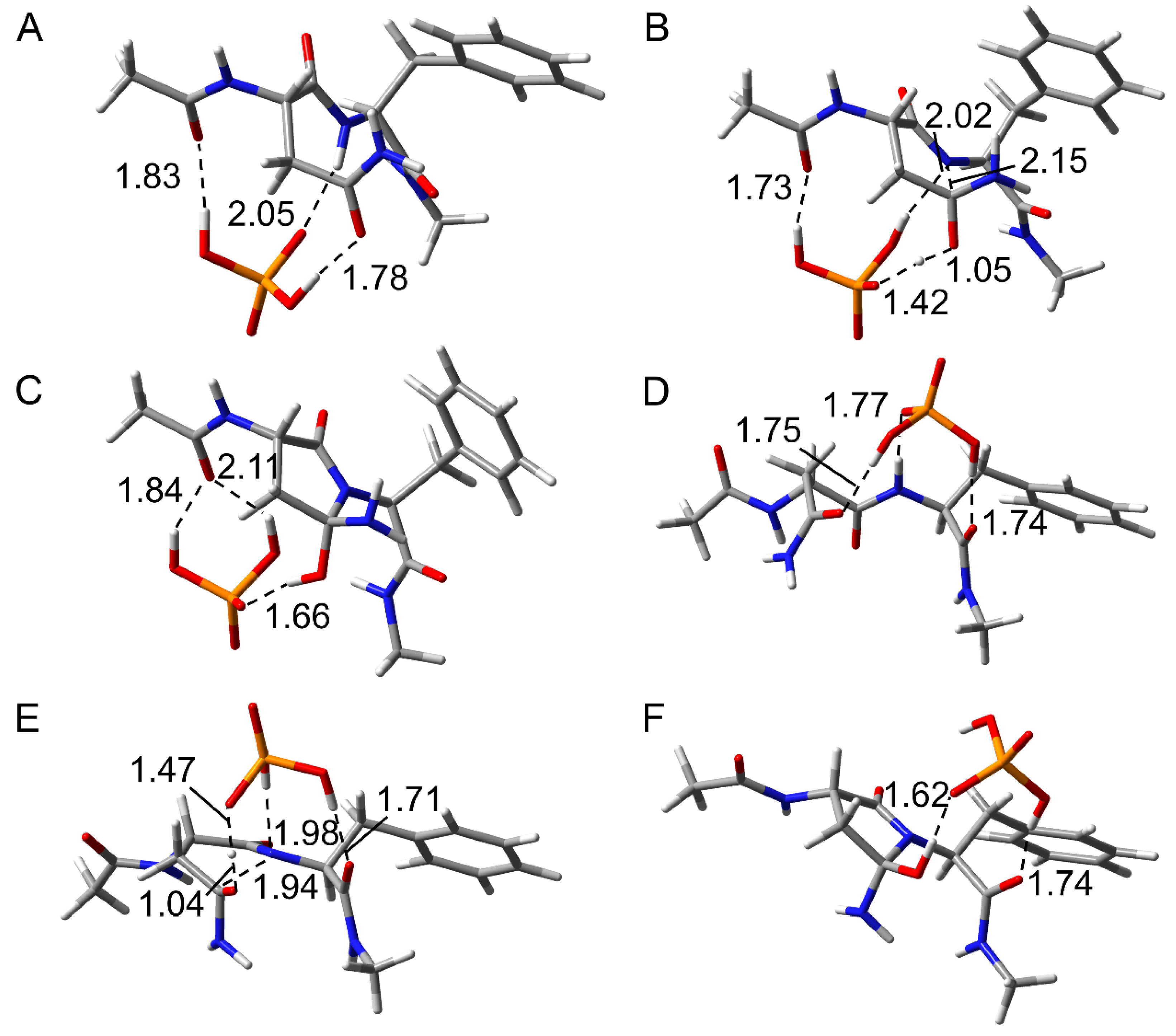
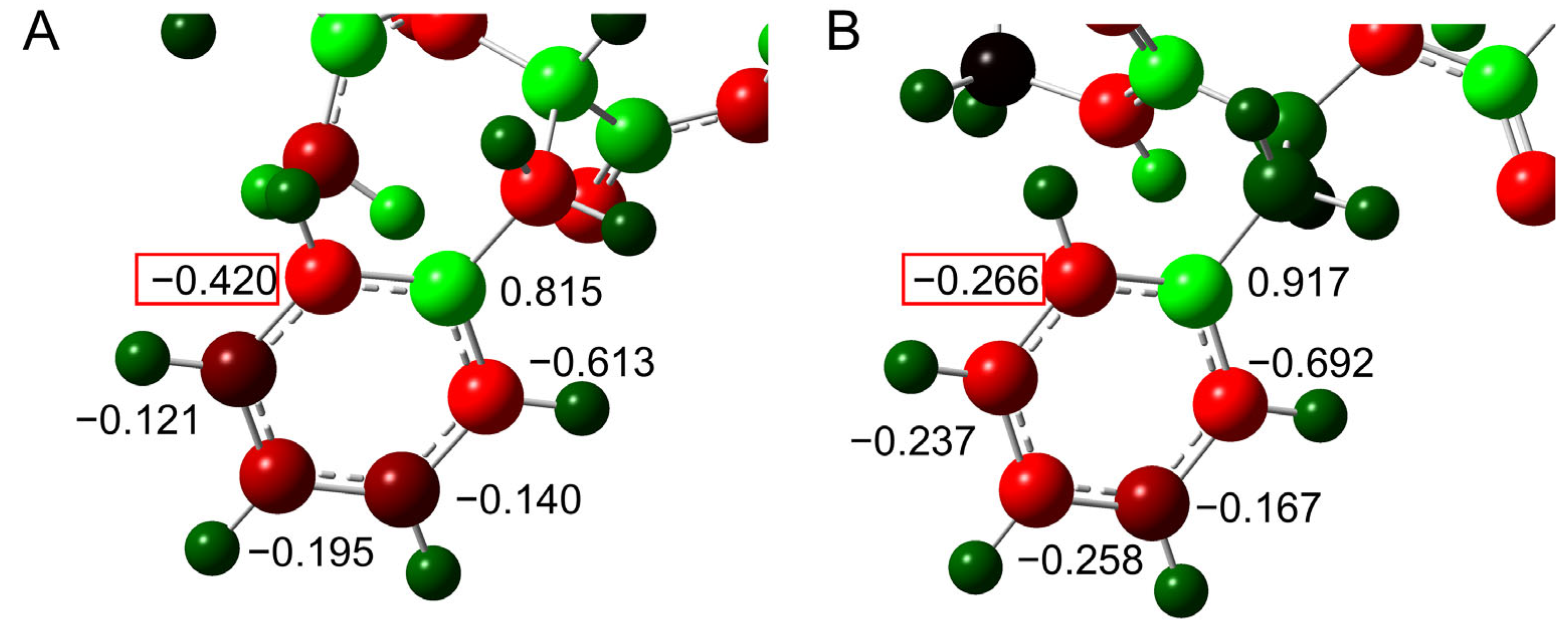
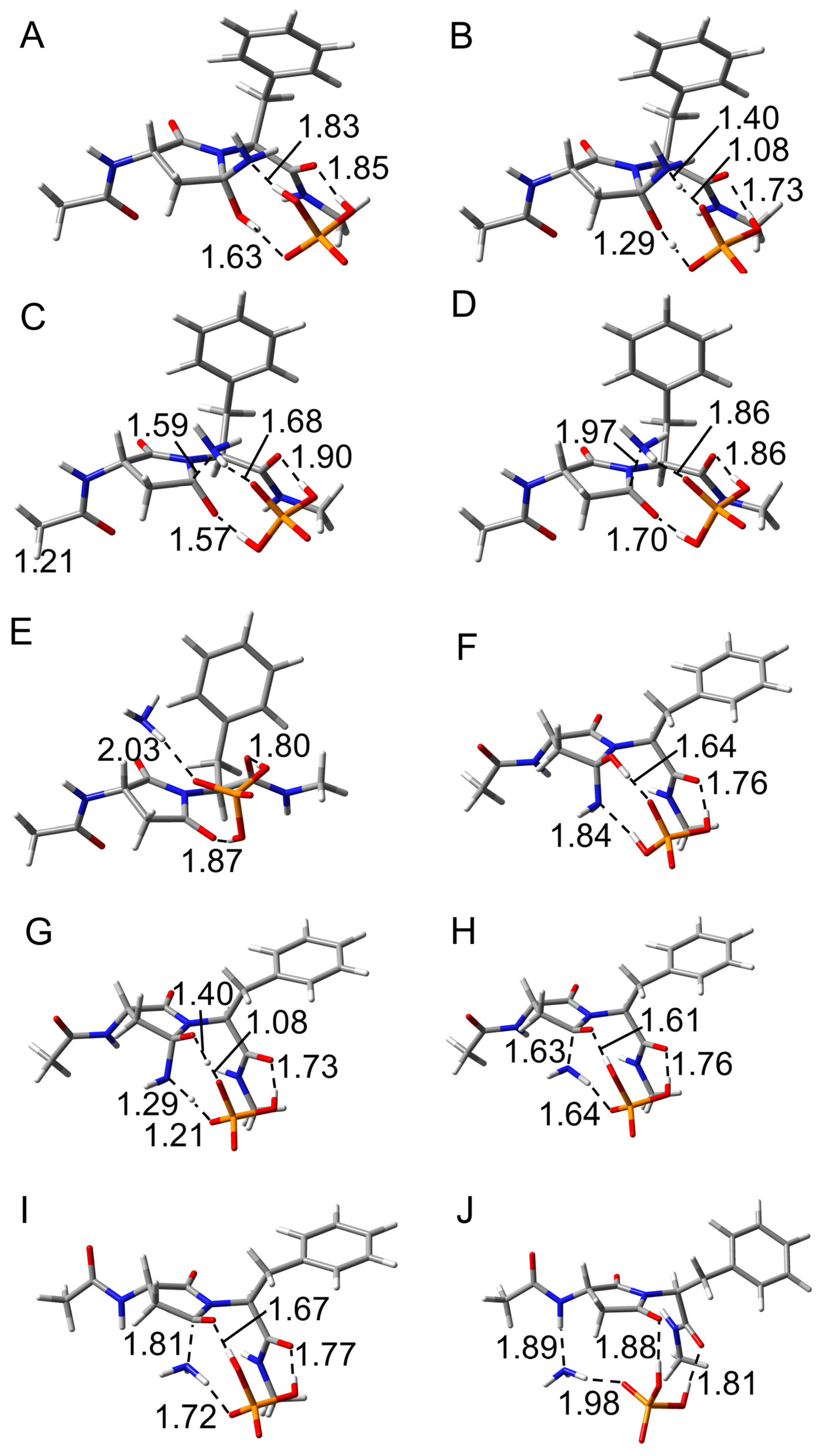
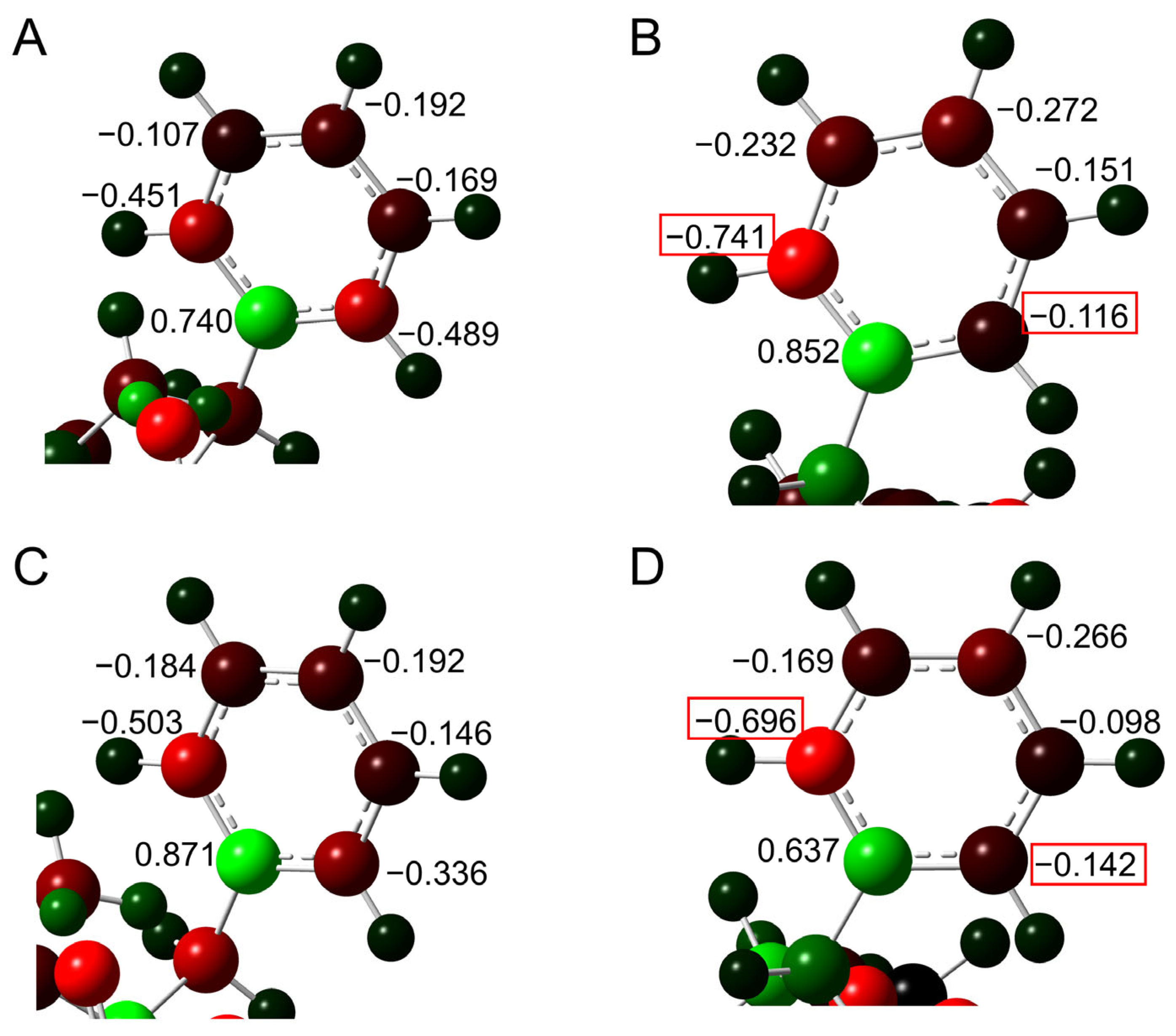
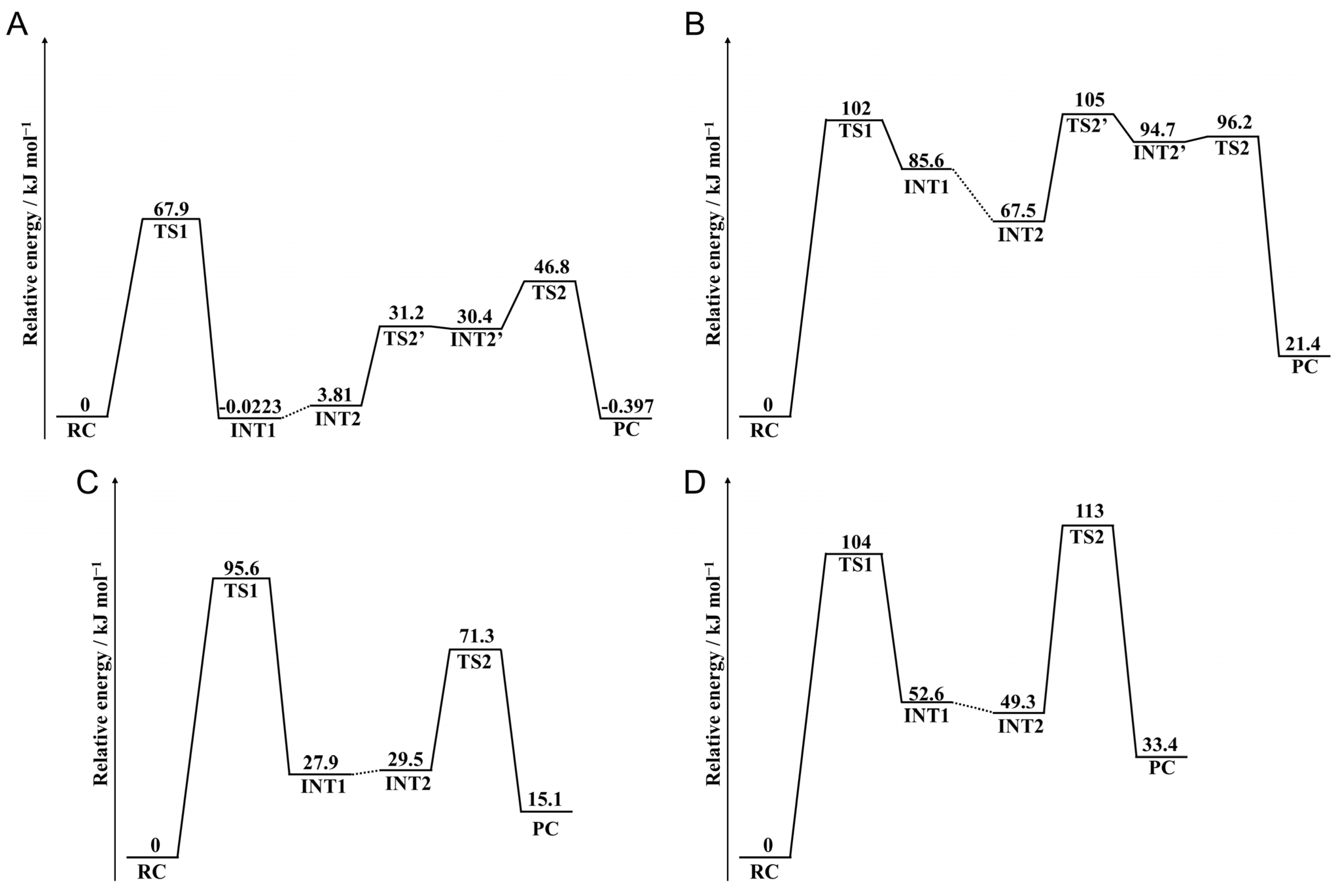
2.1. Cyclization Step
2.2. Deammoniation Step
2.3. Conformational Change Through the Deamidation
2.4. Barrier Heights
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RC | Reactant complex |
| TS | Transition state |
| INT | Intermediate |
| PC | Product complex |
References
- Lubsen, N.H.; Slingsby, C.; Tardieu, A. Ageing and Vision: Structure, Stability and Function of Lens Crystallins. Prog. Biophys. Mol. Biol. 2004, 86, 407–485. [Google Scholar] [CrossRef]
- Driessen, H.P.; Herbrink, P.; Bloemendal, H.; de Jong, W.W. Primary Structure of the Bovine β-crystallin Bp Chain. Internal Duplication and Homology with γ-Crystallin. Eur. J. Biochem. 1981, 121, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.; Tsui, L.C.; Gorin, M.; Breitman, M.L. Characterization of the Human β-crystallin Gene HuβA3/A1 Reveals Ancestral Relationships among the βγ-crystallin Superfamily. J. Biol. Chem. 1986, 261, 12420–12427. [Google Scholar] [CrossRef] [PubMed]
- Garlick, R.L.; Mazer, J.S.; Chylack, L.T., Jr.; Tung, W.H.; Bunn, H.F. Nonenzymatic glycation of human lens crystallin. Effect of aging and diabetes mellitus. J. Clin. Investig. 1984, 74, 1742–1749. [Google Scholar] [CrossRef] [PubMed]
- Slingsby, C.; Clout, N.J. Structure of the Crystallins. Eye 1999, 13, 395–402. [Google Scholar] [CrossRef]
- Moreau, K.L.; King, J.A. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol. Med. 2012, 18, 273–282. [Google Scholar] [CrossRef]
- Loh, D.; Reiter, R.J. Melatonin, ATP, and Cataracts: The Two Faces of Crystallin Phase Separation. Qeios 2024, D09YND. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Molecular clocks. Proc. Natl. Acad. Sci. USA 2001, 98, 944–949. [Google Scholar] [CrossRef]
- Midelfort, C.F.; Mehler, A.H. Deamidation in vivo of an asparagine residue of rabbit muscle aldolase. Proc. Natl. Acad. Sci. USA 1972, 69, 1816–1819. [Google Scholar] [CrossRef]
- Solstad, T.; Flatmark, T. Microheterogeneity of recombinant human phenylalanine hydroxylase as a result of nonenzymatic deamidations of labile amide containing amino acids. Effects on catalytic and stability properties. Eur. J. Biochem. 2000, 267, 6302–6310. [Google Scholar] [CrossRef]
- Capasso, S.; Salvadori, S. Effect of the three-dimensional structure on the deamidation reaction of ribonuclease A. J. Pept. Res. 1999, 54, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Cook, B.L.; Manson, S.R.; Niederhoff, R.A.; Langer, E.M.; Rosová, I.; Kulans, L.A.; Fu, X.; Weinberg, J.S.; Heinecke, J.W.; et al. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell 2002, 111, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Tiwari, A.; Liu, H. Asparagine deamidation in a complementarity determining region of a recombinant monoclonal antibody in complex with antigen. Anal. Chem. 2018, 90, 6998–7003. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm. Res. 1990, 7, 703–711. [Google Scholar] [CrossRef]
- Kaji, Y.; Oshika, T.; Takazawa, Y.; Fukayama, M.; Fujii, N. Accumulation of D-beta-aspartic acid-containing proteins in age-related ocular diseases. Chem. Biodivers. 2010, 7, 1364–1370. [Google Scholar] [CrossRef]
- Palmer, W.G.; Papaconstantinou, J. Aging of alpha-crystallins during development of the lens. Proc. Natl. Acad. Sci. USA 1969, 64, 404–410. [Google Scholar] [CrossRef]
- Takemoto, L.; Boyle, D. Deamidation of specific glutamine residues from alpha-A crystallin during aging of the human lens. Biochemistry 1998, 37, 13681–13685. [Google Scholar] [CrossRef]
- Lampi, K.J.; Wilmarth, P.A.; Murray, M.R.; David, L.L. Lens β-crystallins: The role of deamidation and related modifications in aging and cataract. Prog. Biophys. Mol. Biol. 2014, 115, 21–31. [Google Scholar] [CrossRef]
- Nilsson, M.R.; Driscoll, M.; Raleigh, D.P. Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: Implications for the study of amyloid formation. Protein Sci. 2002, 11, 342–349. [Google Scholar] [CrossRef]
- Capasso, S. Estimation of the deamidation rate of asparagine side chains. J. Pept. Res. 2000, 55, 224–229. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, Z.W.; Robinson, B.R.; Robinson, A.L.; Robinson, J.A.; Robinson, M.L.; Robinson, A.B. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Res. 2004, 63, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.J.; Buchanan, A.; Andrews, J.; Chodorge, M.; Sridharan, S.; Mitchell, L.; Burmeister, N.; Kippen, A.D.; Vaughan, T.J.; Higazi, D.R.; et al. Rate of Asparagine Deamidation in a Monoclonal Antibody Correlating with Hydrogen Exchange Rate at Adjacent Downstream Residues. Anal. Chem. 2017, 89, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Manabe, N.; Kirikoshi, R. A Computational Study of the Mechanism of Succinimide Formation in the Asn-His Sequence: Intramolecular Catalysis by the His Side Chain. Molecules 2016, 21, 327. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, L. Deamidation of Asn-143 of gamma S crystallin from protein aggregates of the human lens. Curr. Eye Res. 2001, 22, 148–153. [Google Scholar] [CrossRef]
- Lapko, V.N.; Purkiss, A.G.; Smith, D.L.; Smith, J.B. Deamidation in human gamma S-crystallin from cataractous lenses is influenced by surface exposure. Biochemistry 2002, 41, 8638–8648. [Google Scholar] [CrossRef]
- Ray, N.J.; Hall, D.; Carver, J.A. Deamidation of N76 in human γS-crystallin promotes dimer formation. Biochim. Biophys. Acta 2016, 1860, 315–324. [Google Scholar] [CrossRef]
- Pande, A.; Mokhor, N.; Pande, J. Deamidation of Human γS-Crystallin Increases Attractive Protein Interactions: Implications for Cataract. Biochemistry 2015, 54, 4890–4899. [Google Scholar] [CrossRef]
- Hains, P.G.; Truscott, R.J. Age-dependent deamidation of lifelong proteins in the human lens. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3107–3114. [Google Scholar] [CrossRef]
- Wilmarth, P.A.; Tanner, S.; Dasari, S.; Nagalla, S.R.; Riviere, M.A.; Bafna, V.; Pevzner, P.A.; David, L.L. Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: Does deamidation contribute to crystallin insolubility? J. Proteome Res. 2006, 5, 2554–2566. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Kurimoto, E.; Oda, A. Mechanisms of Deamidation of Asparagine Residues and Effects of Main-Chain Conformation on Activation Energy. Int. J. Mol. Sci. 2020, 21, 7035. [Google Scholar] [CrossRef]
- Kirikoshi, R.; Manabe, N.; Takahashi, O. Phosphate-Catalyzed Succinimide Formation from Asp Residues: A Computational Study of the Mechanism. Int. J. Mol. Sci. 2018, 19, 637. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nakayoshi, T.; Ishikawa, Y.; Kurimoto, E.; Oda, A. Computational Analysis of the Mechanism of Nonenzymatic Peptide Bond Cleavage at the C-Terminal Side of an Asparagine Residue. ACS Omega 2021, 6, 30078–30084. [Google Scholar] [CrossRef] [PubMed]
- Nakayoshi, T.; Kato, K.; Kurimoto, E.; Oda, A. Computational Studies on the Mechanisms of Nonenzymatic Intramolecular Cyclization of the Glutamine Residues Located at N-Termini Catalyzed by Inorganic Phosphate Species. ACS Omega 2020, 5, 9162–9170. [Google Scholar] [CrossRef] [PubMed]
- Nakayoshi, T.; Wanita, K.; Kato, K.; Kurimoto, E.; Oda, A. Computational analysis of nonenzymatic deamidation of asparagine residues catalysed by acetic acid. Mol. Phys. 2021, 119, e1827176. [Google Scholar] [CrossRef]
- Banerjee, A.; Saha, A.; Saha, B.K. Understanding the Behavior of π–π Interactions in Crystal Structures in Light of Geometry Corrected Statistical Analysis: Similarities and Differences with the Theoretical Models. Cryst. Growth Des. 2019, 19, 2245–2252. [Google Scholar] [CrossRef]
- Ciunik, Z.; Desiraju, G.R. Area correction of multi-atom–acceptor hydrogen bond frequency distributions. Chem. Commun. 2001, 703–704. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Shinohara, Y.; Kurimoto, E.; Oda, A.; Ishikawa, Y. Theoretical Studies on the Reaction Mechanism of Schiff Base Formation from Hexoses. J. Phys. Chem. B 2024, 128, 4952–4958. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Kurimoto, E.; Oda, A. Computational Studies on the Nonenzymatic Deamidation Mechanisms of Glutamine Residues. ACS Omega 2019, 4, 3508–3513. [Google Scholar] [CrossRef]
- Connolly, B.D.; Tran, B.; Moore, J.M.; Sharma, V.K.; Kosky, A. Specific catalysis of asparaginyl deamidation by carboxylic acids: Kinetic, thermodynamic, and quantitative structure-property relationship analyses. Mol. Pharm. 2014, 11, 1345–1358. [Google Scholar] [CrossRef]
- Kingsley, C.N.; Brubaker, W.D.; Markovic, S.; Diehl, A.; Brindley, A.J.; Oschkinat, H.; Martin, R.W. Preferential and specific binding of human αB-crystallin to a cataract-related variant of γS-crystallin. Structure 2013, 21, 2221–2227. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Thorn, D.C.; Grosas, A.B.; Mabbitt, P.D.; Thorn, D.C.; Grosas, A.B.; Mabbitt, P.D.; Ray, N.J.; Jackson, C.J.; Carver, J.A. The Structure and Stability of the Disulfide-Linked γS-Crystallin Dimer Provide Insight into Oxidation Products Associated with Lens Cataract Formation. J. Mol. Biol. 2019, 431, 483–497. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Takahashi, O. A Computational DFT Study of the Stereoinversion of Succinimide Residues Formed in Proteins and Peptides Catalyzed by a Hydrogen Phosphate Ion: An Unsymmetrical SE1 Mechanism. Symmetry 2024, 16, 1369. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dihedral Angle | |||
|---|---|---|---|
| φ | ψ | χ | |
| RC | 61.8 | −139 | −166 |
| TS1 | 57.6 | −147 | 166 |
| INT1 | 52.6 | −144 | 147 |
| INT2 | 55.8 | −146 | 142 |
| TS2′ | 55.1 | −144 | 140 |
| INT3 | 55.6 | −144 | 142 |
| TS2 | 56.1 | −146 | 144 |
| PC | 56.3 | −142 | 136 |
| Dihedral Angle | |||
|---|---|---|---|
| φ | ψ | χ | |
| RC | −155 | −179 | 71.8 |
| TS1 | −168 | −138 | 98.0 |
| INT1 | −166 | −132 | 109 |
| INT2 | −162 | −112 | 98.0 |
| TS2′ | −167 | −108 | 94.7 |
| INT3 | −116 | −127 | 118 |
| TS2 | −117 | −127 | 118 |
| PC | −108 | −121 | 117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, K.; Asai, H.; Nakayoshi, T.; Mizuno, A.; Oda, A.; Ishikawa, Y. Investigation of the Effect of C-Terminal Adjacent Phenylalanine Residues on Asparagine Deamidation by Quantum Chemical Calculations. Int. J. Mol. Sci. 2025, 26, 6819. https://doi.org/10.3390/ijms26146819
Kato K, Asai H, Nakayoshi T, Mizuno A, Oda A, Ishikawa Y. Investigation of the Effect of C-Terminal Adjacent Phenylalanine Residues on Asparagine Deamidation by Quantum Chemical Calculations. International Journal of Molecular Sciences. 2025; 26(14):6819. https://doi.org/10.3390/ijms26146819
Chicago/Turabian StyleKato, Koichi, Haruka Asai, Tomoki Nakayoshi, Ayato Mizuno, Akifumi Oda, and Yoshinobu Ishikawa. 2025. "Investigation of the Effect of C-Terminal Adjacent Phenylalanine Residues on Asparagine Deamidation by Quantum Chemical Calculations" International Journal of Molecular Sciences 26, no. 14: 6819. https://doi.org/10.3390/ijms26146819
APA StyleKato, K., Asai, H., Nakayoshi, T., Mizuno, A., Oda, A., & Ishikawa, Y. (2025). Investigation of the Effect of C-Terminal Adjacent Phenylalanine Residues on Asparagine Deamidation by Quantum Chemical Calculations. International Journal of Molecular Sciences, 26(14), 6819. https://doi.org/10.3390/ijms26146819